A unique cell division protein critical for the assembly of the bacterial divisome
Curation statements for this article:-
Curated by eLife

eLife assessment:
This useful study shows that the essential Acinetobacter baumannii gene Aeg1 likely plays an key role in cell division. The strength of the work is the discovery that the depletion of Aeg1 leads to cell filamentation and that gain-of-function mutations in cell division genes FtsB and FtsL rescue the lethality of Aeg1 depletion. However, Aeg1's localization pattern and its requirement for other division proteins' localizations require further characterization of the functionality of fluorescent fusion proteins, fluorescence images of higher quality, and improvements in statistic qualifications, leaving the study' evidence for Aeg1's exact role in cell division incomplete at this time. In conclusion, the critical role of Aeg1 in the assembly of the A. baumannii divisome has yet to be established unambiguously.
This article has been Reviewed by the following groups

Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
- Evaluated articles (Arcadia Science)
Abstract
Identification of unique essential bacterial genes is important for not only the understanding of their cell biology but also the development of new antimicrobials. Here, we report a previously unrecognized core component of the Acinetobacter baumannii divisome. Our results reveal that the protein, termed Aeg1 interacts with multiple cell division proteins, including FtsN, which is required for components of the divisome to localize to the midcell. We demonstrate that the FtsA E202K and FtsB E65A mutants effectively bypassed the need of Aeg1 by A. baumannii , as did the activation variants FtsW M254I and FtsW S274G . Our results suggest that Aeg1 is a cell division protein that arrives at the division site to initiate cell division by recruiting FtsN, which activates FtsQLB and FtsA to induce the septal peptidoglycan synthase FtsWI. The discovery of the new essential cell division protein has provided a new target for the development of antibacterial agents.
Article activity feed
-

-
-
-

eLife assessment:
This useful study shows that the essential Acinetobacter baumannii gene Aeg1 likely plays an key role in cell division. The strength of the work is the discovery that the depletion of Aeg1 leads to cell filamentation and that gain-of-function mutations in cell division genes FtsB and FtsL rescue the lethality of Aeg1 depletion. However, Aeg1's localization pattern and its requirement for other division proteins' localizations require further characterization of the functionality of fluorescent fusion proteins, fluorescence images of higher quality, and improvements in statistic qualifications, leaving the study' evidence for Aeg1's exact role in cell division incomplete at this time. In conclusion, the critical role of Aeg1 in the assembly of the A. baumannii divisome has yet to be established unambiguously.
-
In this study, the authors confirm that one of the genes classified as essential in a Tn-mutagenesis study in A. baumannii, Aeg1, is, in fact, an essential gene. The strength of the work is that it discovered that the depletion of Aeg1 leads to cell filamentation and that activation mutations in various cell division genes can suppress the requirement for Aeg1. These results suggest that Aeg1 plays an important role in cell division. The work's weakness is that it lacks convincing evidence to define Aeg1's place or role in the divisome assembly pathway. It is unclear whether proteins are at the division site under the wildtype condition and when Aeg1 is depleted, and whether Aeg1 is indeed required for a set of division proteins to the division site.
Reviewer comments:
The revised manuscript partially addressed two of the three major …
In this study, the authors confirm that one of the genes classified as essential in a Tn-mutagenesis study in A. baumannii, Aeg1, is, in fact, an essential gene. The strength of the work is that it discovered that the depletion of Aeg1 leads to cell filamentation and that activation mutations in various cell division genes can suppress the requirement for Aeg1. These results suggest that Aeg1 plays an important role in cell division. The work's weakness is that it lacks convincing evidence to define Aeg1's place or role in the divisome assembly pathway. It is unclear whether proteins are at the division site under the wildtype condition and when Aeg1 is depleted, and whether Aeg1 is indeed required for a set of division proteins to the division site.
Reviewer comments:
The revised manuscript partially addressed two of the three major concerns from the previous assessment: (1) the functionality test of fluorescent fusion proteins using a spotting assay, and (2) membrane protein topology in the bacterial two-hybrid assays by constructing a C-terminal T25 fusion.
(1) In the spotting assay, all fluorescent fusion proteins rescued the growth of the corresponding deletion strain, which suggests these fusion proteins are functional. However, fluorescent images of these fusion proteins were diffusive, and only a few cells showed the expected midcell/membrane localization pattern for cell division proteins. This observation raised the concern that these fusion proteins may be cleaved in the middle, leading to the separation of the untagged fusion partner and diffusive fluorescent protein in the cytoplasm, which would explain the positive spotting rescue results. This phenomenon is commonly observed in other bacterial species. A western blot using an antibody targeting either the fluorescent protein or the fusion partner is widely used to examine whether the fusion protein is expressed at its full length.
(2) The authors constructed a C-terminal fusion of Aeg1 and showed that it still interacted with ZipA and FtsN. This result supports the authors' suggestion that the N-terminus of Aeg1 may not be the predicated membrane-targeting domain. Along the same line, the membrane topology of ZipA should also be considered. ZipA's N terminus is in the membrane facing the periplasm, and its C terminal domain is in the cytoplasm. Therefore, the PUT18C fusion will place the T18 domain of ZipA in the periplasm. All other division proteins' N termini are in the cytoplasm.
(3) Colocalization images did not show significant midcell localizations for each fluorescent protein; most cells showed diffusive cytoplasmic fluorescence. In all other species, midcell localization of cell division proteins is prominent in dividing cells, especially for early division proteins such as ZipA (at least 40-50% of cells show midcell bands). In A. baumannii, divisome localization timing may differ from other species, but this possibility needs to be established before the colocalization pattern is examined. Compounding this issue is that in Aeg1 depletion strains, some cells expressing ZipA, FtsB, FtsL, and FtsN fusions showed roughly regularly spaced puncta in long filamentous cells. It is hard to explain why this was observed if, under the WT condition, these fusions do not localize to the midcell. These results again raised concerns that these fusion proteins may not be functional and the observations are protein aggregates.
Besides these major issues, experimental observations did not support some claims in the main text. For example: (1) In the two-hybrid assay, only ZipA and FtsN showed significant interactions with Aeg1, as judged by the darkness of the blue spots. FtsL and FtsB showed pale spots. The quantified values accompanying this figure did not appear to agree with the image. (2) The spotting rescue assay showed that only FtsB-E56A and FtsA-E202K was able to bypass Aeg1 depletion (full dilution set comparable to that of Aeg1 complementation), but the main text claimed that FtsA-D124A and V144L, and FtsW-M254I and S274G also rescued the growth. These claims could be misleading.
-
Author response:
The following is the authors’ response to the previous reviews.
(1) The reviewers asked to clarify the BTH assay: The fused T25 and T18 domains must be in the cytoplasmic to complement successfully. The authors stated that the N terminus of Aeg1 transverses the membrane once, which means that the T25-Aeg1 will have T25 in the periplasm. However, T18C vector fusion with other division proteins will have T18C of ZipA in the periplasm (ZipA's N terminus is on the periplasmic side of the inner membrane) while that of FtsN in the cytoplasm (FtsN's N terminus is in the cytoplasm). As such, it isn't easy to understand why T25-Aeg1 showed positive results for both ZipA and FtsN. Note that FtsL, FtsB, and FtsI all have the same topology as FtsN but showed negative results. It is possible that these fusion proteins do not fold …
Author response:
The following is the authors’ response to the previous reviews.
(1) The reviewers asked to clarify the BTH assay: The fused T25 and T18 domains must be in the cytoplasmic to complement successfully. The authors stated that the N terminus of Aeg1 transverses the membrane once, which means that the T25-Aeg1 will have T25 in the periplasm. However, T18C vector fusion with other division proteins will have T18C of ZipA in the periplasm (ZipA's N terminus is on the periplasmic side of the inner membrane) while that of FtsN in the cytoplasm (FtsN's N terminus is in the cytoplasm). As such, it isn't easy to understand why T25-Aeg1 showed positive results for both ZipA and FtsN. Note that FtsL, FtsB, and FtsI all have the same topology as FtsN but showed negative results. It is possible that these fusion proteins do not fold correctly, and hence, the results cannot be interpreted directly. The authors did not address this concern but only cited that BTH is a commonly used assay for protein-protein interactions.
In response to the editor's comments and the concerns raised by the reviewer, we have performed two sets of Aeg1-T25 fusion experiments to determine whether the Aeg1 topology impacts protein interactions measured by bacterial two-hybrid (BTH) assays. In the first set of experiments, we fused the T25 domain to the N-terminus of Aeg1 and still observed strong binding of Aeg1 to ZipA and FtsN, respectively. Similar results were obtained from the second set of experiments in which the T25 domain was fused to the C-terminus of Aeg1.
These results indicate that the precise topology of Aeg1 does not significantly impact its ability to engage these binding partners. Aeg1 is predicted to harbor a single transmembrane domain, however, the precise location of this transmembrane segment differs in predictions made by different algorithms. The SMART Web site (1) predicted the transmembrane region to be located at the N-terminus of Aeg1 (7-29 aa). In contrast, Phobius, based on HMM (2, 3)suggested the transmembrane segment is situated more centrally within the Aeg1 protein (134-151 aa), and further proposed that the N-terminus may function as a signal peptide. This latter prediction also provides a potential explanation for the larger-than-expected molecular weight of the Aeg1 truncation mutant observed in the Western blot shown in Fig 1C. The removal of the putative signal peptide may have altered the protein structure, affecting its electrophoretic mobility. As a result, we are more inclined to favor the topology model for Aeg1 predicted by Phobius.
(2) It is still difficult to identify the midcell localization patterns of Aeg1 and other division proteins from microscopy images (Fig. 4C and Fig. 5A). In Fig 4C, only ZipA and Aeg1 formed clear, regular band-like colocalization patterns. Others formed irregular co-localized puncta along the cell length, different from the expected midcell localization patterns. Cells also appeared to be much longer than WT cells, suggesting cell division defects. The most likely reason for these aberrant localization patterns and filamentous cells is that GFP/mCherry-fusions of these division proteins are not functional and become dominant negative, interfering with proper cell division. The authors need to test the functionality of these fusion proteins before they can be used for imaging. (The authors also mislabeled Hoechst and the division protein GFP panels labels in this figure.)
Thank you for raising this important point. To examine the functionality of the fluorescence protein fusion constructs, we have painstakingly performed conditional knockout of the genes of interest (zipA, ftsB, ftsL, and ftsN) in A. baumannii strains inducibly expressing the corresponding fusion protein. We found that these fluorescence protein fusions were able to fully rescue the growth of the mutant lacking the corresponding fts gene (Figure 4-figure supplement 1). Concurrently, we have also successfully knocked out the aeg1 gene under conditions in trans expression of an mCherry-Aeg1 fusion protein, which was able to effectively rescue the growth defects of the Δa_eg1_ mutant (Figure 4-figure supplement 1). We then introduced the functional fluorescence protein fusions into wild-type cells and observed the co-localization of Aeg1 with the relevant Fts proteins. The results showed that Aeg1 indeed co-localized with ZipA, FtsB, FtsL, and FtsN (Fig.4E, red arrows), but occasional non-co-localization was also observed (Fig.4E, white arrows).
We have utilized the functional fluorescence protein fusion constructs to analyze the localization of relevant Aeg1-interacting proteins in the Δaeg1 strain upon Aeg1 depletion. Our results showed that the depletion of Aeg1 indeed impacted the midcell localization of the several Aeg1-interacting Fts proteins.
References
(1) Letunic I, Khedkar S, Bork P. SMART: recent updates, new developments and status in 2020. Nucleic acids research. 2021;49:D458-d60.doi: 10.1093/nar/gkaa937.
(2) Käll L, Krogh A, Sonnhammer EL. A combined transmembrane topology and signal peptide prediction method. Journal of molecular biology. 2004;338:1027-36.doi: 10.1016/j.jmb.2004.03.016
(3) Käll L, Krogh A, Sonnhammer EL. Advantages of combined transmembrane topology and signal peptide prediction--the Phobius web server. Nucleic acids research. 2007;35:W429-32.doi: 10.1093/nar/gkm256
-

Thank you for your thorough work on the important bacterial pathogen Acinetobacter baumannii! It's absolutely essential to identify new targets, using modern approaches, that could aid us with better drug design. Regarding the essential protein you have identified (Aeg1), I was wondering if it may be a target for any Type VI Secretion effectors, or other types of injected effectors? Are you familiar with any known effectors that target proteins similar to Aeg1, or proteins performing a similar role in the divisome, either in Acinetobacter baumannii or in other bacteria? My thought is that any effectors targeting Aeg1 could help us understand a potentially viable way to inhibit this protein, or its function, with a drug. I know Acinetobacter baumannii does secrete/inject multiple different effectors and it may not be too surprising if …
Thank you for your thorough work on the important bacterial pathogen Acinetobacter baumannii! It's absolutely essential to identify new targets, using modern approaches, that could aid us with better drug design. Regarding the essential protein you have identified (Aeg1), I was wondering if it may be a target for any Type VI Secretion effectors, or other types of injected effectors? Are you familiar with any known effectors that target proteins similar to Aeg1, or proteins performing a similar role in the divisome, either in Acinetobacter baumannii or in other bacteria? My thought is that any effectors targeting Aeg1 could help us understand a potentially viable way to inhibit this protein, or its function, with a drug. I know Acinetobacter baumannii does secrete/inject multiple different effectors and it may not be too surprising if one of these effectors targets Aeg1, considering the essential role of this protein in cell division. Thank you so much for this important work!
-
-
Author Respose
The following is the authors’ response to the original reviews.
Reviewer #1 (Public Review):
The authors prepared several Acinetobacter baumannii strains from which an essential protein of known or unknown function can be depleted. They chose to study one of the proteins (AdvA) in more detail. AdvA is a known essential cell division protein that accumulates at cell division sites together with other such proteins. No clear homologs are present in model bacteria such as E.coli, and the precise role(s) of AdvA is still unclear. The authors rename AdvA here as Aeg1. The authors searched for suppressors of lethality caused by AdvA-depletion and recovered an allele of ftsA (E202K) that is capable of doing so. Based on similar superfission alleles previously recovered in other division genes in E.coli, they test several …
Author Respose
The following is the authors’ response to the original reviews.
Reviewer #1 (Public Review):
The authors prepared several Acinetobacter baumannii strains from which an essential protein of known or unknown function can be depleted. They chose to study one of the proteins (AdvA) in more detail. AdvA is a known essential cell division protein that accumulates at cell division sites together with other such proteins. No clear homologs are present in model bacteria such as E.coli, and the precise role(s) of AdvA is still unclear. The authors rename AdvA here as Aeg1. The authors searched for suppressors of lethality caused by AdvA-depletion and recovered an allele of ftsA (E202K) that is capable of doing so. Based on similar superfission alleles previously recovered in other division genes in E.coli, they test several mutant genes and find that certain alleles in ftsB, L and W can also suppress lethality of AdvA-minus cells.
In addition, the authors perform bacterial two-hybrid assays and protein sublocalization studies of AdvA and of other division proteins, but the results of these studies are either not new (confirming previous work) or not convincing.
We appreciate the vigor of this reviewer.
We agreed that the essentiality of AdvA/Aeg1 described in our submission is not new, we believed our work has firmly established its role as a cell division protein. The earlier work by the labs of Geisinger and Isberg labs (1) showed its essentiality and the cell morphology changes upon its depletion (Fig. 3 of ref. 1 in the end of this rebuttal letter). This protein was one of the many proteins addressed in their study and their results only suggests its role in cell division due to the close phenotypical relationships between AdvA/Aeg1 and genes associated with chromosome replication/segregation and cell division.
Reviewer #2 (Public Review):
In this study the authors confirm that one of the genes classified as essential in a Tn-mutagenesis study in A. baumannii is in fact an essential gene. It is also present in other closely related Gram-negative bacteria and the authors designated it Aeg1. Depletion of Aeg1 leads to cell filamentation and it appears that the requirement for Aeg1 can be suppressed by what appear to be activation mutations in various genes. Overall, it appears that Aeg1 is involved in cell division but many of the images suffer from poor quality - it may be due to conversion to PDF. One of the main issues is that depletion of Aeg1 is carried out for such long times (18 hr) (Fig. 2, 4 and 5). Depleting a cell division protein for such long times may have pleiotropic effects on cell physiology. A. baumannii grows quite fast and even with a small inoculum, cells will probably be in stationary phase. If Aeg1 is that essential cells should be quite filamentous 2-3 hours after Ara removal when they are still in exponential phase. Also, it would be better to see the recovery to small cells if cells are not grown such a long time before Ara is added back. Overall, Aeg1 is potentially interesting, but studies are needed to define its place in the assembly pathway for this to be published. What proteins are at the division site when Aeg1 is depleted and what proteins are required for Aeg1 to localize to the division site. These experiments should be done when cell are depleted of proteins for only 1 -2 hours.
We appreciate these insightful suggestions and have followed them to make necessary modifications in the revised manuscript, including:
1st, We have redone the experiment for Fig. 1C to obtain images of higher resolution.
2nd, We have more carefully examined the kinetics of the depletion of Aeg1-mCherry upon removal of the inducer arabinose from medium. We first evaluated the protein of Aeg1-mCherry at 2, 4, and 6 h after withdrawing arabinose and found that at the 2 h and 4 h time points mCherry-Aeg1was still readily detectable (Fig. S4). Importantly, we found that removal of arabinose for 6 h rendered Aeg1-mCherry undetectable in approximately 90% of the cells. We thus used the 6 h inducer depletion to examine the effects of Aeg1 depletion.
In experiments aiming to analyze the co-localization of Aeg1 with other core divisome proteins, cultures of strains derived from Δaeg1(PBAD::mCherry-Aeg1) harboring the GFP fusions were induced by ara for 16 h. The saturated bacterial cultures were then diluted into fresh LB broth without ara for 6 h to induce the elongation morphology. IPTG (0.25 mM) and ara (0.25%) were added to induce the expression of fusion proteins for 4 h before samples were processed for microscopic analysis. Our results indicate that Aeg1 colocalized with ZipA, FtsK, FtsL, FtsB, and FtsW (Fig. 4C), which is consistent with results from the protein interaction experiments using the bacterial two-hybrid assay.
To determine the impact of Aeg1 depletion on cellular localization of the several core cell divisome proteins. In cells in which Aeg1 had been depleted (by removing the inducer arabinose), all of the examined core division proteins displayed midcell mistargeting, including ZipA, FtsK, FtsB, FtsL, and FtsN (Fig. 5A).
Reviewer #1 (Recommendations For The Authors):
Specific remarks 1) The manuscript title is misleading in that the 'novel cell division protein' studied in this paper has already been identified as such, and studied in some detail, by the Geisinger and Isberg labs (refs 37 and 20).
We agreed with this point. Because of the data presented by Geisinger and Isberg labs (1) that demonstrated its essentiality and morphological changes upon its depletion (Fig. 3 in ref 1), we have changed the title to “A unique cell division protein critical for the assembly of the bacterial divisome”.
- The Isberg/Geisinger labs named this division protein AdvA in 2020 (ref 37). The authors of the present manuscript should follow this terminology, as there is no compelling reason to rename the protein Aeg1 here. It will only confuse the field.
We named this protein Aeg1 because we identified and named it before the work by Geisinger and Isberg labs (1) was published and this name has been used in all of our records. In addition, this is a part of our research exploring hypothetical essential genes in A. baumannii and we thus would like to keep the name in this manuscript.
- Membrane topology of AdvA? Line 103-104: The authors predict a single transmembrane domain in AdvA (Aeg1). However, reference 37 predicted two, and some prediction programs (e.g. CCTOP) predict three with the N-terminus periplasmic. A good understanding of the membrane topology of AdvA is important, if not only for the design of credible BACTH two-hybrid assays. Figure 6 indicates that the authors assume that the N-terminus of AdvA is periplasmic with the bulk of the protein cytoplasmic. But then they choose to use pKT25::AdvA for two-hybrid assays, which would place the CyaA T25 domain periplasmic as well. This should not yield faithful interaction data as both the T25 and T18 domains need to be cytoplasmic to restore CyaA activity.
The Bacterial Adenylate Cyclase-Based Two-Hybrid (BACTH) technique is a powerful tool for studying protein-protein interactions, especially those involving integral membrane or membrane-associated proteins. It overcomes the limitations of traditional two-hybrid systems by allowing the detection of interactions that occur within the membrane or in other difficult-to-study protein environments (2). This method has been successfully used to analyze the relationships among bacterial cell division proteins (e.g., ref 3 and 4). Furthermore,our results from bacterial two-hybrid and immunofluorescence techniques are consistent. As a result, the results presented here should be valid.
- Strains and plasmids, Table S4 Far more detail is needed. a) Please provide complete genotypes of strains and, especially, of the plasmids used, including replication origin, antibiotic resistance markers, promoters, promoter repressors, inducible genes/fusions to be expressed, and the placement of genetic tags (T25, T18, XFP, Flag, etcetera).
We have added the information to Table S4.
b) In addition, provide details on how each strain/plasmid was constructed in the Methods section or as supplement. Currently, you only provide some details on one or two of the strains or plasmids.
We have added the necessary details about how the constructs and plasmids used in this study were made.
- Lines 114-129, Fig 2. AdvA is needed for cell division. a) Similar results were already described by refs 37 and 20, so this is merely confirmatory.
We revised the description accordingly.
b) Refs 37 and 20 should be referenced here, as well as in the section above where you find AdvA to be essential for viability on rich medium.
We have added the appropriate reference as suggested.
c) The micrographs in panel C are of poor quality. Consider higher magnification and resolution.
We have redone the experiments and images of higher resolution have been used in the revised manuscript.
- Lines 130-143, selection for suppressors of AdvA-depletion. I would expect quite a few mutations in araC repressor on the plasmid in this screen, rendering the promoter more constitutive (i.e. arabinose-independent). Did these not appear?
This is an interesting point. Unfortunately, we did not recover suppression mutants which mutations on araC or other elements of the BAD promoter. Given the complexity of AraC-mediated regulation (5), such mutants likely are rare or we did not screen enough candidates.
- Lines 173-178, Fig3E. Sublocalization of AdvA-mCherry. a) The micrographs in Fig. 3E are very poor and I can not see any specific localization, or barely any signal whatsoever, of the AdvA-mCherry fusion. Thus, this result is not convincing
We have replaced this image with a new one of higher-resolution.
b) In contrast, accumulation of an AdvA-GFP fusion at constriction sites was already clearly and convincingly shown in ref 37.
We have revised the text to reflect this fact.
c) So, this section needs convincing images, as well as a reference to ref 37.
We have added an image of higher resolution and revised the text accordingly. Thank you
- Lines 179-188, Fig4a-b. BACTH assays
a) As noted above (see point 3), the T25-AdvA fusion would likely place the T25 domain in the periplasm, casting doubt on the validity of these results.
b) Similarly, the T18-ZipA fusion would place the T18 domain in the periplasm, casting further doubt.
The Bacterial Adenylate Cyclase-Based Two-Hybrid (BACTH) technique is a powerful tool for studying protein-protein interactions, especially those involving integral membrane or membrane-associated proteins. It overcomes the limitations of traditional two-hybrid systems by allowing the detection of interactions that occur within the membrane or in other difficult-to-study protein environments (2). This method has been successfully used to analyze the relationships among bacterial cell division proteins (e.g., ref 3 and 4). Furthermore,our results from bacterial two-hybrid and immunofluorescence techniques are consistent. As a result, the results presented here should be valid.
- Lines 189-201, Fig4c, co-localization of proteins in AdvA-depleted filaments. These co-localization results are not convincing for several reasons:
a) None of the proteins accumulate in specific ring-like structures, as might be expected for ZipA, at least. One possible reason is that division rings are not made at all due to the partial depletion of AdvA in these cells. But another possible reason is that some or all the fusions are simply non-functional. Do any of these proteins (co-)localize to the septal ring in wt cells?
b) At least for the GFP-ZipA fusion, there is good reason to predict it is not functional, as correct membrane insertion of the fusion would place GFP in the periplasm. In E. coli this prevents GFP from becoming fluorescent in the first place. So the fluorescence seen here may reflect failure of the fusion to insert properly.
c) Another possible reason for rings being absent is that the fusions are massively overexpressed. The plasmids are multicopy, the BAD and TAC promoters are strong, and the used levels of inducers (Ara and IPTG) are high. How do fusion levels compare to that of native proteins? Perhaps some of the bright spots we see are inclusion bodies or other types of non-specific protein aggregates.
We appreciate these excellent suggestions and have carried out experiments to investigate the (co-)localization of these proteins at the septal ring in Δaeg1 cells under conditions of low-level inducers (Ara and IPTG) and reduced induction time.
Cultures of strains derived from Δaeg1(PBAD::mCherry-Aeg1) harboring the GFP fusions were induced by ara for 16 h, saturated bacterial cultures were then diluted into fresh LB broth without ara for 6 h to induce the elongation morphology. IPTG (0.2 mM) and ara (0.2%) were added to induce the expression of fusion proteins for 4 h before samples were processed for microscopic analysis. Consistent with results from the protein interaction experiments using the bacterial two-hybrid assay, Aeg1 colocalized with ZipA, FtsK, FtsL, FtsB, and FtsW (Fig. 4C). Thus, Aeg1 interacts with multiple core cell divisome proteins of A. baumannii.
In cells of the wild-type A. baumannii strain, we have observed cell elongation upon overexpression of FtsL, FtsB, FtsW, or FtsN. This raises concerns regarding the physiological relevance of the results obtained in wild-type cells. Of note, the phenotype of cell elongation following overexpression of division proteins has been observed in Escherichia coli by several groups (6-11).
- Lines 202-214, Fig5a, localization of division proteins in AdvA-depleted filaments. These localization results are not convincing for the same reasons outlined above (see point 9).
a) Do any of the fusions localize correctly under similar expression conditions, but in normally dividing cells?
In wild-type A. baumannii cells, cell elongation occurs upon overexpression of FtsL, FtsB, FtsW or FtsN, which raises the concern that the results from the suggested experiments may not physiologically relevant.
b) Even the regular structures seen with GFP-FtsZ do not resemble rings, but appear more like blobs. Perhaps fixation with glutaraldehyde would preserve structures better?
We have followed the suggestion to use glutaraldehyde fixation for cell fixation. The new images have been used in the revised manuscript.
- Other points:
a) Line 97, Fig1. Is AdvA essential on minimal medium (~ slow growth) as well?
We have performed this experiment. Yes, AdvA/Aeg1 is essential for A. baumannii growth in the Vogel-Bonner minimal medium with succinate (VBS) as the sole carbon source (12) (Fig S1).
b) Fig1. What residues are actually missing (or replaced?) in the delta-TM version of AdvA?
We have added the information, residues 1-23 have been removed.
c) Fig1D. Also, the delta-TM version of HA-AdvA runs slower than HA-AdvA itself. Why?
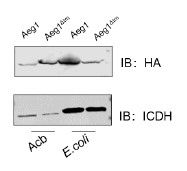
We have also been puzzled by this phenomenon that full-length AdvA/Aeg1 migrated faster than the delta-TM mutant. Interestingly, this discrepancy did not occur when the proteins were expressed in E. coli (see Author response image 1). We do not have a good explanation for this phenomenon.
Author response image 1.
The expression of the Aeg1 and Aeg1∆TM in A. baumannii and E. coli. Total proteins resolved by SDS-PAGE was probed by immunoblotting with the HA-specific antibody. The metabolic enzyme isocitrate dehydrogenase (ICDH) was probed as a loading control. Similar results were obtained in three independent experiments.
d) Lines 159, 165 and elsewhere. The mutation in E. coli is actually FtsA(R286W), not Q286W.
We have corrected this error. Thank you!
e) Line 161. These alleles of ftsA should be referenced properly: ref 33 for I143L and ref 29 for E124A.
We have made the correction. Thank you!
f) Line 692, you incorrectly switched the two CyaA domains here.
We have corrected this error.
g) Fig4b. Is 'none' a vector control (pUT18C-Flag)?
We have specified the control, it is the vector pUT18C-Flag.
h) Lines 727-729. I don't understand this sentence. Please explain.
We have revised this sentence.
Reviewer #2 (Recommendations For The Authors):
Line 159 and Fig. 2 Panel D. I am not sure that this panel should be in the paper for two reasons: 1) FtsA from E. coli and A. baumannii are only 50% identical and its not clear that one can make corresponding mutations and expect similar behavior. FtsA* from E. coli is R286W not Q286W. R286 does not appear to be conserved in A. baumannii. Also, what you label as Q286 appears to be Q285. Please check. 2) the alleles that are tested in this panel do not rescue the deletion of Aeg1. This may be due to the instability of the mutant proteins. It would be better to characterize the mutant that you have isolated - is it a superfission mutation; that is does it produce small cells in a strain that contains WT Aeg1?
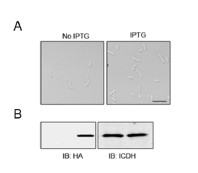
Thank you! We have more carefully examined the relevant sites in these proteins. We did not observe the small cell phenotype when FtsAE202K was overexpressed in WT strains (please see Author response image 2).
Author response image 2
The overexpression of FtsAE202K did not cause a small cell phenotype in A. baumannii. Bacterial strains derived from WT (Ptac::FtsAE202K) grown in LB broth overnight were diluted into fresh medium with the inducer and the cultures were induced with IPTG for 4 h prior to being processed for imaging (A). Total proteins were resolved by SDS-PAGE and proteins transferred onto nitrocellulose membranes were detected by immunoblotting with the HA-specific antibody. ICDH was probed as a loading control (B, right panels). Images were representatives of three parallel cultures. Bar, 10 µm.
The images in Fig. 3, Panel C are quite poor (perhaps the original images [not PDF] are better). It is difficult to see the localization.
We have redone the experiments and replaced the images with ones of higher resolution.
Fig. 4. Panel C. This is an effort to show that Aeg1 colocalizes with known cell division proteins. Since in Fig. 3, panel C it is claimed that Aeg1 localizes to the division site, them it must colocalize with known division proteins. Doing the long term depletion of Aeg1 is likely causing artefacts. The localization of proteins seems very erratic. A better experiment would be to express the GFP fusions to the known proteins and then deplete Aeg1 and see what happens. Does depletion of Aeg1 prevent the localization of FtsZ, FtsK or FtsN? Another important question is if one of the known cell division proteins is depleted does Aeg1 localize to division sites. Since it is speculated that Aeg1 interacts with ZipA and FtsN, these proteins could be depleted and see if Aeg1 localizes.
We greatly appreciate your insightful suggestions. We have carefully redone these experiments as follows: Each of the testing strains was grown in LB broth with ara overnight prior to being diluted into fresh medium without ara for 6 h to induce the elongation morphology. IPTG (0.25 mM) and ara (0.25%) were added to induce the expression of fusion proteins for 4 h before samples were processed for microscopic analysis. Consistent with results from the protein interaction experiments using the bacterial two-hybrid assay, we observed that Aeg1 colocalized with ZipA, FtsK, FtsL, FtsB, or FtsW (Fig. 4C).
In cells not expressing Aeg1, all of the examined core division proteins including FtsZ, FtsK, and FtsN displayed midcell mistargeting, (Fig. 5A).
As for the localization of Aeg1 upon depleting ZipA or FtsN, this is an ongoing project in our lab. Such information is beyond the scope of this manuscript.
Fig. 5. Panel A. again the images are not of good quality. Also, why deplete for 18 hrs. This is too long.
We have redone these experiments and images of higher resolution are now used in the revised manuscript. After extensive test, we have chosen to use a 6-h depletion, which gave us the window to observe the phenotype (Fig. 5A).
Line 25. Change 'so' to 'as'
Corrected as suggested. Thank you!
Line 28. "Induces' to 'induce'
We have made the suggested correction. Thank you!
Line 43. Change 'of' to 'with'
Corrected as suggested. Thank you!
Line 74. Change 'determine' to 'test'
Corrected as suggested. Thank you!
Line 89. Delete 'of the'
We have made the suggested correction. Thank you!
Line 102. Some strains of E. coli? Does that mean there are strains that do not contain Aeg1? What are they?
Yes, this is indeed the case, the common strains of E. coli derived from strain K12 does not have a discernable homolog of aeg1. This gene is present in some clinic E. coli isolates (e.g. HAY5567682, HBI862710, HAY5567682, MDD9849866, EFE8345364, and KAE9874289).
Line 112. Note this TM domain has a rare topology as it is similar to ZipA. Please mention that this is a Type 1b.
We have made the suggested revision. Thank you!
Reference:
Geisinger E, Mortman NJ, Dai Y, Cokol M, Syal S, Farinha A, et al. Antibiotic susceptibility signatures identify potential antimicrobial targets in the Acinetobacter baumannii cell envelope. Nature communications. 2020;11:4522.doi: 10.1038/s41467-020-18301-2
Karimova G, Gauliard E, Davi M, Ouellette SP, Ladant D. Protein-Protein Interaction: Bacterial Two-Hybrid. Methods in molecular biology (Clifton, NJ). 2017;1615:159-76.doi: 10.1007/978-1-4939-7033-9_13
Karimova G, Dautin N, Ladant D. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid analysis. Journal of bacteriology. 2005;187:2233-43.doi: 10.1128/jb.187.7.2233-2243.2005
Boldridge WC, Ljubetič A, Kim H, Lubock N, Szilágyi D, Lee J, et al. A multiplexed bacterial two-hybrid for rapid characterization of protein-protein interactions and iterative protein design. Nature communications. 2023;14:4636.doi: 10.1038/s41467-023-38697-x
Schleif R. AraC protein, regulation of the l-arabinose operon in Escherichia coli, and the light switch mechanism of AraC action. FEMS microbiology reviews. 2010;34:779-96.doi: 10.1111/j.1574-6976.2010.00226.x
Addinall SG, Cao C, Lutkenhaus J. FtsN, a late recruit to the septum in Escherichia coli. Molecular microbiology. 1997;25:303-9.doi: 10.1046/j.1365-2958.1997.4641833.x
Pichoff S, Lutkenhaus J. Identification of a region of FtsA required for interaction with FtsZ. Molecular microbiology. 2007;64:1129-38.doi: 10.1111/j.1365-2958.2007.05735.x
Du S, Henke W, Pichoff S, Lutkenhaus J. How FtsEX localizes to the Z ring and interacts with FtsA to regulate cell division. Molecular microbiology. 2019;112:881-95.doi: 10.1111/mmi.14324
Park KT, Du S, Lutkenhaus J. Essential Role for FtsL in Activation of Septal Peptidoglycan Synthesis. mBio. 2020;11.doi: 10.1128/mBio.03012-20
Barre FX, Aroyo M, Colloms SD, Helfrich A, Cornet F, Sherratt DJ. FtsK functions in the processing of a Holliday junction intermediate during bacterial chromosome segregation. Genes & development. 2000;14:2976-88.doi: 10.1101/gad.188700
Cameron TA, Vega DE, Yu C, Xiao H, Margolin W. ZipA Uses a Two-Pronged FtsZ-Binding Mechanism Necessary for Cell Division. mBio. 2021;12:e0252921.doi: 10.1128/mbio.02529-21
Vogel HJ, Bonner DM. Acetylornithinase of Escherichia coli: partial purification and some properties. The Journal of biological chemistry. 1956;218:97-106.doi:
-
eLife assessment
This study is useful in identifying several gain-of-function division variants that can suppress the elongated cell division defect phenotype caused by the depletion of Age1 (or AdvA as named in other studies), a known essential cell division protein in Acinetobacter baumannii. However, characterizations of AdvA's localization patterns and its interactions with other divisome proteins are incomplete due to the lack of (1) functional characterizations of fluorescent fusion proteins, (2) considerations for membrane protein topology in the bacterial two hybrid assay, and (3) lack of high-quality fluorescence images for the co-localization studies. The results do not yet support the major claim that Age1 plays a critical role in the assembly of the A. baumannii divisome.
-
Joint Public Review:
In this study the authors confirm that one of the genes classified as essential in a Tn-mutagenesis study in A. baumannii is in fact an essential gene. It is also present in other closely related Gram negative bacteria and the authors designated it Aeg1.
The strength of the work is that it discovered that the depletion of Aeg1 leads to cell filamentation and that the requirement for Aeg1 can be suppressed by activation mutations in various cell division genes. These results suggest that Aeg1 plays an important role in cell division.
The weakness of the work is that it lacks convincing evidence to define Aeg1 place or role in the divisome assembly pathway. It is unclear what proteins are at the division site when Aeg1 is depleted and what proteins are required for Aeg1 to localize to the division site.
-
-
eLife assessment
This useful study identifies several gain-of-function division protein variants that can suppress the lethality caused by depletion of Aeg1/AdvA in Acinetobacter baumannii, but characterization and description of AdvA in its localization patterns and interactions with other divisome proteins are inadequate. Since a role of AdvA in cell division has been shown before, the claim that a "novel cell division protein" has been identified is incorrect. The evidence provided also does not support the claim that AdvA plays a critical role in the assembly of the A. baumannii divisome.
-
Reviewer #1 (Public Review):
The authors prepared several Acinetobacter baumannii strains from which an essential protein of known or unknown function can be depleted. They chose to study one of the proteins (AdvA) in more detail. AdvA is a known essential cell division protein that accumulates at cell division sites together with other such proteins. No clear homologs are present in model bacteria such as E.coli, and the precise role(s) of AdvA is still unclear. The authors rename AdvA here as Aeg1. The authors searched for suppressors of lethality caused by AdvA-depletion and recovered an allele of ftsA (E202K) that is capable of doing so. Based on similar superfission alleles previously recovered in other division genes in E.coli, they test several mutant genes and find that certain alleles in ftsB, L and W can also suppress …
Reviewer #1 (Public Review):
The authors prepared several Acinetobacter baumannii strains from which an essential protein of known or unknown function can be depleted. They chose to study one of the proteins (AdvA) in more detail. AdvA is a known essential cell division protein that accumulates at cell division sites together with other such proteins. No clear homologs are present in model bacteria such as E.coli, and the precise role(s) of AdvA is still unclear. The authors rename AdvA here as Aeg1. The authors searched for suppressors of lethality caused by AdvA-depletion and recovered an allele of ftsA (E202K) that is capable of doing so. Based on similar superfission alleles previously recovered in other division genes in E.coli, they test several mutant genes and find that certain alleles in ftsB, L and W can also suppress lethality of AdvA-minus cells.
In addition, the authors perform bacterial two-hybrid assays and protein sublocalization studies of AdvA and of other division proteins, but the results of these studies are either not new (confirming previous work) or not convincing.
-
Reviewer #2 (Public Review):
In this study the authors confirm that one of the genes classified as essential in a Tn-mutagenesis study in A. baumannii is in fact an essential gene. It is also present in other closely related Gram negative bacteria and the authors designated it Aeg1. Depletion of Aeg1 leads to cell filamentation and it appears that the requirement for Aeg1 can be suppressed by what appear to be activation mutations in various genes. Overall, it appears that Aeg1 is involved in cell division but many of the images suffer from poor quality - it may be due to conversion to PDF. One of the main issues is that depletion of Aeg1 is carried out for such long times (18 hr) (Fig. 2, 4 and 5). Depleting a cell division protein for such long times may have pleiotropic effects on cell physiology. A. baumannii grows quite fast and …
Reviewer #2 (Public Review):
In this study the authors confirm that one of the genes classified as essential in a Tn-mutagenesis study in A. baumannii is in fact an essential gene. It is also present in other closely related Gram negative bacteria and the authors designated it Aeg1. Depletion of Aeg1 leads to cell filamentation and it appears that the requirement for Aeg1 can be suppressed by what appear to be activation mutations in various genes. Overall, it appears that Aeg1 is involved in cell division but many of the images suffer from poor quality - it may be due to conversion to PDF. One of the main issues is that depletion of Aeg1 is carried out for such long times (18 hr) (Fig. 2, 4 and 5). Depleting a cell division protein for such long times may have pleiotropic effects on cell physiology. A. baumannii grows quite fast and even with a small inoculum, cells will probably be in stationary phase. If Aeg1 is that essential cells should be quite filamentous 2-3 hours after Ara removal when they are still in exponential phase. Also, it would be better to see the recovery to small cells if cells are not grown such a long time before Ara is added back. Overall, Aeg1 is potentially interesting but studies are needed to define its place in the assembly pathway. What proteins are at the division site when Aeg1 is depleted and what proteins are required for Aeg1 to localize to the division site. These experiments should be done when cell are depleted of proteins for only 1 -2 hours.
-
