Inhibited KdpFABC transitions into an E1 off-cycle state
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
KdpFABC is a bacterial potassium uptake transporter made up of a channel-like subunit (KdpA) and a P-type ATPase (KdpB). When potassium levels are low (< 2 mM), the transporter actively and selectively uptakes potassium, but must be switched off again to prevent excessive K+ accumulation. Although structures of KdpFABC have been determined before, the structural basis for inhibition by phosphorylation is unknown. Here, the authors have determined the structure of KdpABC in an arrested (off-state) that is in a distinct conformation from previously determined P-type ATPase structures. More detailed structural comparisons are needed to more convincingly show this, however, and the protein required to inhibit KdpABC by phosphorylation remains unknown. This paper will be of interest to researchers in the microbiology and transporter communities.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
- Reading List (BiophysicsColab)
Abstract
KdpFABC is a high-affinity prokaryotic K + uptake system that forms a functional chimera between a channel-like subunit (KdpA) and a P-type ATPase (KdpB). At high K + levels, KdpFABC needs to be inhibited to prevent excessive K + accumulation to the point of toxicity. This is achieved by a phosphorylation of the serine residue in the TGES 162 motif in the A domain of the pump subunit KdpB (KdpB S162-P ). Here, we explore the structural basis of inhibition by KdpB S162 phosphorylation by determining the conformational landscape of KdpFABC under inhibiting and non-inhibiting conditions. Under turnover conditions, we identified a new inhibited KdpFABC state that we termed E1P tight, which is not part of the canonical Post-Albers transport cycle of P-type ATPases. It likely represents the biochemically described stalled E1P state adopted by KdpFABC upon KdpB S162 phosphorylation. The E1P tight state exhibits a compact fold of the three cytoplasmic domains and is likely adopted when the transition from high-energy E1P states to E2P states is unsuccessful. This study represents a structural characterization of a biologically relevant off-cycle state in the P-type ATPase family and supports the emerging discussion of P-type ATPase regulation by such states.
Article activity feed
-

-

Author Response
Reviewer #2 (Public Review):
Silberberg et al. present a series of cryo-EM structures of the ATP dependent bacterial potassium importer KdpFABC, a protein that is inhibited by phosphorylation under high environmental K+ conditions. The aim of the study was to sample the protein's conformational landscape under active, non-phosphorylated and inhibited, phosphorylated (Ser162) conditions.
Overall, the study presents 5 structures of phosphorylated wildtype protein (S162-P), 3 structures of phosphorylated 'dead' mutant (D307N, S162-P), and 2 structures of constitutively active, non-phosphorylatable protein (S162A).
The true novelty and strength of this work is that 8 of the presented structures were obtained either under "turnover" or at least 'native' conditions without ATP, ie in the absence of any non-physiological …
Author Response
Reviewer #2 (Public Review):
Silberberg et al. present a series of cryo-EM structures of the ATP dependent bacterial potassium importer KdpFABC, a protein that is inhibited by phosphorylation under high environmental K+ conditions. The aim of the study was to sample the protein's conformational landscape under active, non-phosphorylated and inhibited, phosphorylated (Ser162) conditions.
Overall, the study presents 5 structures of phosphorylated wildtype protein (S162-P), 3 structures of phosphorylated 'dead' mutant (D307N, S162-P), and 2 structures of constitutively active, non-phosphorylatable protein (S162A).
The true novelty and strength of this work is that 8 of the presented structures were obtained either under "turnover" or at least 'native' conditions without ATP, ie in the absence of any non-physiological substrate analogues or stabilising inhibitors. The remaining 2 were obtained in the presence of orthovanadate.
Comparing the presented structures with previously published KdpFACB structures, there are 5 structural states that have not been reported before, namely an E1-P·ADP state, an E1-P tight state captured in the autoinhibited WT protein (with and without vanadate), and two different nucleotide-free 'apo' states and an E1·ATP early state.
Of these new states, the 'tight' states are of particular interest, because they appear to be 'off-cycle', dead end states. A novelty lies in the finding that this tight conformation can exist both in nucleotide-free E1 (as seen in the published first KdpFABC crystal structure), and also in the phosphorylated E1-P intermediate.
By EPR spectroscopy, the authors show that the nucleotide free 'tight' state readily converts into an active E1·ATP conformation when provided with nucleotide, leading to the conclusion that the E1-P·ADP state must be the true inhibitory species. This claim is supported by structural analysis supporting the hypothesis that the phosphorylation at Ser162 could stall the KdpB subunit in an E1P state unable to convert into E2P. This is further supported by the fact that the phosphorylated sample does not readily convert into an E2P state when exposed to vanadate, as would otherwise be expected.
The structures are of medium resolution (3.1 - 7.4 Å), but the key sites of nucleotide binding and/or phosphorylation are reasonably well supported by the EM maps, with one exception: in the 'E1·ATP early' state determined under turnover conditions, I find the map for the gamma phosphate of ATP not overly convincing, leaving the question whether this could instead be a product-inhibited, Mg-ADP bound E1 state resulting from an accumulation of MgADP under the turnover conditions used. Overall, the manuscript is well written and carefully phrased, and it presents interesting novel findings, which expand our knowledge about the conformational landscape and regulatory mechanisms of the P-type ATPase family.
We thank the reviewer for their comments and helpful insights. We have addressed the points as follows:
However in my opinion there are the following weaknesses in the current version of the manuscript:
- A lack of quantification. The heart of this study is the comparison of the newly determined KdpFABC structures with previously published ones (of which there are already 10). Yet, there are no RMSD calculations to illustrate the magnitude of any structural deviations. Instead, the authors use phrases like 'similar but not identical to', 'has some similarities', 'virtually identical', 'significant differences'. This makes it very hard to appreciate the true level of novelty/deviation from known structures.
This is a very valid point and we thank the reviewers for bringing it up. To provide a better overview and appreciation of conformational similarities and significant differences we have calculated RMSDs between all available structures of KdpFABC. They are summarised in the new Table 1 – Table Supplement 2. We have included individual rmsd values, whenever applicable and relevant, in the respective sections in the text and figures. We note that the RMSDs were calculated only between the cytosolic domains (KdpB N,A,P domains) after superimposition of the full-length protein on KdpA, which is rigid across all conformations of KdpFABC (see description in material and methods lines 1184-1191 or the caption to Table 1 – Table Supplement 2). We opted to not indicate the RMSD calculated between the full-length proteins, as the largest part of the complex does not undergo large structural changes (see Figure 1 – Figure Supplement 1, the transmembrane region of KdpB as well as KdpA, KdpC and KdpF show relatively small to no rearrangements compared to the cytosolic domains), and would otherwise obscure the relevant RMSD differences discussed here.
Also the decrease in EPR peak height of the E1 apo tight state between phosphorylated and non-phosphorylated sample - a key piece of supporting data - is not quantified.
EPR distance distributions have been quantified by fitting and integrating a gaussian distribution curve, and have been added to the corresponding results section (lines 523-542) and the methods section (lines 1230-1232).
- Perhaps as a consequence of the above, there seems to be a slight tendency towards overstatements regarding the novelty of the findings in the context of previous structural studies. The E1-P·ATP tight structure is extremely similar to the previously published crystal structure (5MRW), but it took me three reads through the paper and a structural superposition (overall RMSD less than 2Å), to realise that. While I do see that the existing differences, the two helix shifts in the P- and A- domains - are important and do probably permit the usage of the term 'novel conformation' (I don't think there is a clear consensus on what level of change defines a novel conformation), it could have been made more clear that the 'tight' arrangement of domains has actually been reported before, only it was not termed 'tight'.
As indicated above we have now included an extensive RMSD table between all available KdpFABC structures. To ensure a meaningful comparison, the rmsd are only calculated between the cytosolic domains after superimposition of the full-length protein on KdpA, as the transmembrane region of KdpFABC is largely rigid (see figure below panel B). However, we have to note that in the X-ray structure the transmembrane region of KdpB is displaced relative to the rest of the complex when compared to the arrangement found in any of the other 18 cryo-EM structures, which all align well in the TMD (see figure below panel C). These deviations make the crystal structure somewhat of an outlier and might be a consequence of the crystal packing (see figure below panel A). For completeness in our comparison with the X-Ray structure, we have included an RMSD calculated when superimposed on KdpA and additional RMSD that was calculated between structures when aligned on the TMD of KdpB (see figure below panel D,E). The reported RMSD that the reviewer mentiones of less than 2Å was probably obtained when superimposing the entire complex on each other (see figure below panel F). However, we do not believe that this is a reasonable comparison as the TMD of the complex is significantly displaced, which stands in strong contrast to all other RMSDs calculated between the rest of the structures where the TMD aligns well (see figure below panel B).
From the resulting comparisons, we conclude that the E1P-tight and the X-Ray structure do have a certain similarity but are not identical. In particular not in the relative orientation of the cytosolic domains to the rest of the complex. We hope that including the RMSD in the text and separately highlighting the important features of the E1P tight state in the section “E1P tight is the consequence of an impaired E1P/E2P transition“ makes the story now more conclusive.
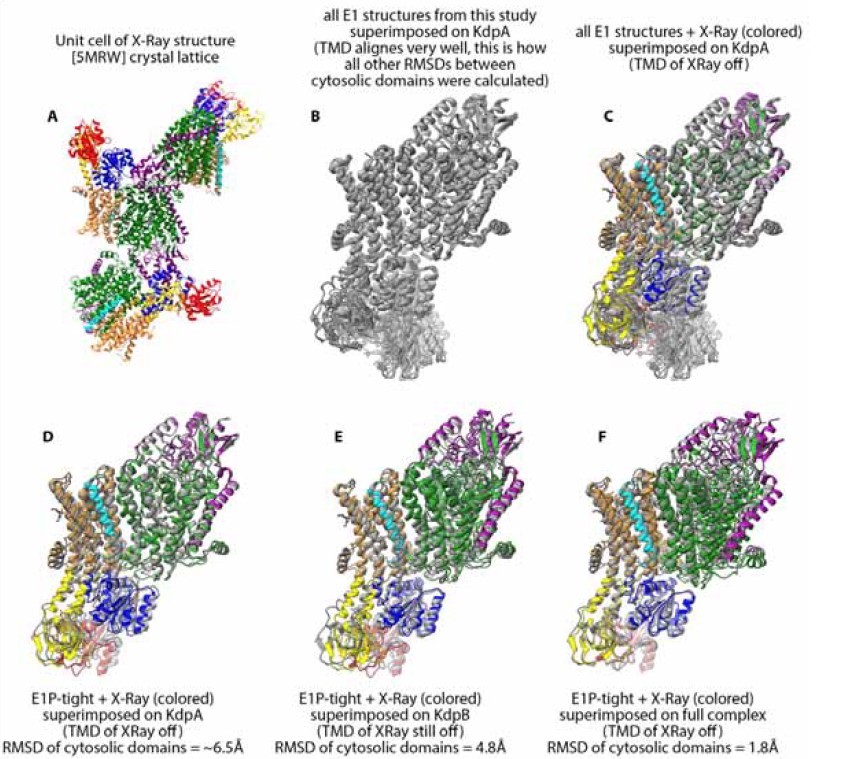
Likewise, the authors claim that they have covered the entire conformational cycle with their 10 structures, but this is actually not correct, as there is no representative of an E2 state or functional E1P state after ADP release.
This is correct, and we have adjusted the phrasing to “close to the entire conformational cycle” or “the entire KdpFABC conformational cycle except the highly transient E1P state after ADP release and E2 state after dephosphorylation.”
- A key hypothesis this paper suggests is that KdpFABC cannot undergo the transition from E1P tight to E2P and hence gets stuck in this dead end 'off cycle' state. To test this, the authors analysed an S162-P sample supplied with the E2P inducing inhibitor orthovanadate and found about 11% of particles in an E2P conformation. This is rationalised as a residual fraction of unphosphorylated, non-inhibited, protein in the sample, but the sample is not actually tested for residual unphosphorylated fraction or residual activity. Instead, there is a reference to Sweet et al, 2020. So the claim that the 11% E2P particles in the vanadate sample are irrelevant, whereas the 14% E1P tight from the turnover dataset are of key importance, would strongly benefit from some additional validation.
We have added an ATPase assay that shows the residual ATPase activity of WT KdpFABC compared to KdpFABS162AC, both purified from E. coli LB2003 cells, which is identical to the protein production and purification for the cryo-EM samples (see Figure 2-Suppl. Figure 5). The residual ATPase activity is ca. 14% of the uninhibited sample, which correlates with the E2-P fraction in the orthovanadate sample.
Reviewer #3 (Public Review):
The authors have determined a range of conformations of the high-affinity prokaryotic K+ uptake system KdpFABC, and demonstrate at least two novel states that shed further light on the structure and function of these elusive protein complexes.
The manuscript is well-written and easy to follow. The introduction puts the work in a proper context and highlights gaps in the field. I am however missing an overview of the currently available structures/states of KdpFABC. This could also be implemented in Fig. 6 (highlighting new vs available data). This is also connected to one of my main remarks - the lack of comparisons and RMSD estimates to available structures. Similarity/resemblance to available structures is indicated several times throughout the manuscript, but this is not quantified or shown in detail, and hence it is difficult for the reader to grasp how unique or alike the structures are. Linked to this, I am somewhat surprised by the lack of considerable changes within the TM domain and the overlapping connectivity of the K indicated in Table 1 - Figure Supplement 1. According to Fig. 6 the uptake pathway should be open in early E1 states, but not in E2 states, contrasting to the Table 1 - Figure Supplement 1, which show connectivity in all structures? Furthermore, the release pathway (to the inside) should be open in the E2-P conformation, but no release pathway is shown as K ions in any of the structures in Table 1 - Figure Supplement 1. Overall, it seems as if rather small shifts in-between the shown structures (are the structures changing from closed to inward-open)? Or is it only KdpA that is shown?
We thank the reviewer for their positive response and constructive criticisms. We have addressed these comments as follows:
The overview of the available structures has been implemented in Fig. 6, with the new structures from this study highlighted in bold.
RMSD values have been added to all comparisons, with a focus on the deviations of the cytosolic domains, which are most relevant to our conformational assignments and discussions.
To highlight the (comparatively small) changes in the TMD, we have expanded Table 1 - Figure Supplement 1 to include panels showing the outward-open half-channel in the E1 states with a constriction at the KdpA/KdpB interface and the inward-open half-channel in the E2 states. The largest observable rearrangements do however take place in the cytosolic domains. This is an absolute agreement with previous studies, which focused more on the transition occurring within the transmembrane region during the transport cycle (Stock et al, Nature Communication 2018; Silberberg et al, Nature Communication 2021; Sweet et al., PNAS 2021).
The ions observed in the intersubunit tunnel are all before the point at which the tunnel closes, explaining why there is no difference in this region between E1 and E2 structures. Moreover, as we discussed in our last publication (Silberberg, Corey, Hielkema et al., 2021, Nat. Comms.), the assignment of non-protein densities along the entire length of the tunnel is contentious and can only be certain in the selectivity filter of KdpA and the CBS of KdpB.
The release pathway from the CBS does not feature any defined K+ coordination sites, so ions are not expected to stay bound along this inward-open half-channel.
My second key remark concerns the "E1-P tight is the consequence of an impaired E1-P/E2-P transition" section, and the associated discussion, which is very interesting. I am not convinced though that the nucleotide and phosphate mimic-stabilized states (such as E1-P:ADP) represent the high-energy E1P state, as I believe is indicated in the text. Supportive of this, in SERCA, the shifts from the E1:ATP to the E1P:ADP structures are modest, while the following high-energy Ca-bound E1P and E2P states remain elusive (see Fig. 1 in PMID: 32219166, from 3N8G to 3BA6). Or maybe this is not what the authors claim, or the situation is different for KdpFABC? Associated, while I agree with the statement in rows 234-237 (that the authors likely have caught an off-cycle state), I wonder if the tight E1-P configuration could relate to the elusive high-energy states (although initially counter-intuitive as it has been caught in the structure)? The claims on rows 358-360 and 420-422 are not in conflict with such an idea, and the authors touch on this subject on rows 436-450. Can it be excluded that it is the proper elusive E1P state? If the state is related to the E1P conformation it may well have bearing also on other P-type ATPases and this could be expanded upon.
This a good point, particularly since the E1P·ADP state is the most populated state in our sample, which is also counterintuitive to “high-energy unstable state”. One possible explanation is that this state already has some of the E1-P strains (which we can see in the clash of D307-P with D518/D522), but the ADP and its associated Mg2+ in particular help to stabilize this. Once ADP dissociates and takes the Mg2+ with it, the full destabilization takes effect in the actual high-energy E1P state. Nonetheless, we consider it fair to compare the E1P tight with the E1P·ADP to look for electrostatic relaxation. We have clarified the sequence of events and our hypothesized role the ADP/Mg2+ have in stabilizing the E1P·ADP state that we can see (lines 609-619): “Moreover, a comparison of the E1P tight structure with the E1P·ADP structure, its most immediate precursor in the conformational cycle obtained, reveals a number of significant rearrangements within the P domain (Figure 5B,C). First, Helix 6 (KdpB538-545) is partially unwound and has moved away from helix 5 towards the A domain, alongside the tilting of helix 4 of the A domain (Figure 5B,C – arrow 2). Second, and of particular interest, are the additional local changes that occur in the immediate vicinity of the phosphorylated KdpBD307. In the E1P·ADP structure, the catalytic aspartyl phosphate, located in the D307KTG signature motif, points towards the negatively charged KdpBD518/D522. This strain is likely to become even more unfavorable once ADP dissociates in the E1P state, as the Mg2+ associated with the ADP partially shields these clashes. The ensuing repulsion might serve as a driving force for the system to relax into the E2 state in the catalytic cycle.”
We believe it is highly unlikely that the reported E1-P tight state represents an on-cycle high-energy E1P intermediate. For one, we observe a relaxation of electrostatic strains in this structure, in particular when compared to the obtained E1P ADP state. By contrast, the E1P should be the most energetically unfavourable state possible to ensure the rapid transition to the E2P state. As such, this state should be a transient state, making it less likely to be obtainable structurally as an accumulated state. Additionally, the association of the N domain with the A domain in the tight conformation, which would have to be reverted, would be a surprising intermediary step in the transition from E1P to E2P. Altogether, the here reported E1P tight state most likely represents an off-cycle state.
-
Evaluation Summary:
KdpFABC is a bacterial potassium uptake transporter made up of a channel-like subunit (KdpA) and a P-type ATPase (KdpB). When potassium levels are low (< 2 mM), the transporter actively and selectively uptakes potassium, but must be switched off again to prevent excessive K+ accumulation. Although structures of KdpFABC have been determined before, the structural basis for inhibition by phosphorylation is unknown. Here, the authors have determined the structure of KdpABC in an arrested (off-state) that is in a distinct conformation from previously determined P-type ATPase structures. More detailed structural comparisons are needed to more convincingly show this, however, and the protein required to inhibit KdpABC by phosphorylation remains unknown. This paper will be of interest to researchers in the microbiology and …
Evaluation Summary:
KdpFABC is a bacterial potassium uptake transporter made up of a channel-like subunit (KdpA) and a P-type ATPase (KdpB). When potassium levels are low (< 2 mM), the transporter actively and selectively uptakes potassium, but must be switched off again to prevent excessive K+ accumulation. Although structures of KdpFABC have been determined before, the structural basis for inhibition by phosphorylation is unknown. Here, the authors have determined the structure of KdpABC in an arrested (off-state) that is in a distinct conformation from previously determined P-type ATPase structures. More detailed structural comparisons are needed to more convincingly show this, however, and the protein required to inhibit KdpABC by phosphorylation remains unknown. This paper will be of interest to researchers in the microbiology and transporter communities.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
The paper has determined a considerable number of different structures and conformations by Cryo-EM, that describes the full conformational spectrum of the KdpFABC catalytic cycle. They also show by EPR that the non-phosphorylatable variant KdpBS162A variant was indeed arrested in the state observed by Cryo-EM.
Although they have been able to validate that the Cryo-EM structure of the off-cycle state is consistent with the conformational state probed by pulsed EPR, it is unclear what protein phosphorylates and then inactivates KdpFABC at higher K+ concentrations. As such, at present, it is not possible to fully comprehend the exact physiological conditions when the arrested state is formed.
-
Reviewer #2 (Public Review):
Silberberg et al. present a series of cryo-EM structures of the ATP dependent bacterial potassium importer KdpFABC, a protein that is inhibited by phosphorylation under high environmental K+ conditions. The aim of the study was to sample the protein's conformational landscape under active, non-phosphorylated and inhibited, phosphorylated (Ser162) conditions.
Overall, the study presents 5 structures of phosphorylated wildtype protein (S162-P), 3 structures of phosphorylated 'dead' mutant (D307N, S162-P), and 2 structures of constitutively active, non-phosphorylatable protein (S162A).
The true novelty and strength of this work is that 8 of the presented structures were obtained either under "turnover" or at least 'native' conditions without ATP, ie in the absence of any non-physiological substrate analogues or …
Reviewer #2 (Public Review):
Silberberg et al. present a series of cryo-EM structures of the ATP dependent bacterial potassium importer KdpFABC, a protein that is inhibited by phosphorylation under high environmental K+ conditions. The aim of the study was to sample the protein's conformational landscape under active, non-phosphorylated and inhibited, phosphorylated (Ser162) conditions.
Overall, the study presents 5 structures of phosphorylated wildtype protein (S162-P), 3 structures of phosphorylated 'dead' mutant (D307N, S162-P), and 2 structures of constitutively active, non-phosphorylatable protein (S162A).
The true novelty and strength of this work is that 8 of the presented structures were obtained either under "turnover" or at least 'native' conditions without ATP, ie in the absence of any non-physiological substrate analogues or stabilising inhibitors. The remaining 2 were obtained in the presence of orthovanadate.
Comparing the presented structures with previously published KdpFACB structures, there are 5 structural states that have not been reported before, namely an E1-P·ADP state, an E1-P tight state captured in the autoinhibited WT protein (with and without vanadate), and two different nucleotide-free 'apo' states and an E1·ATP early state.
Of these new states, the 'tight' states are of particular interest, because they appear to be 'off-cycle', dead end states. A novelty lies in the finding that this tight conformation can exist both in nucleotide-free E1 (as seen in the published first KdpFABC crystal structure), and also in the phosphorylated E1-P intermediate.By EPR spectroscopy, the authors show that the nucleotide free 'tight' state readily converts into an active E1·ATP conformation when provided with nucleotide, leading to the conclusion that the E1-P·ADP state must be the true inhibitory species. This claim is supported by structural analysis supporting the hypothesis that the phosphorylation at Ser162 could stall the KdpB subunit in an E1P state unable to convert into E2P. This is further supported by the fact that the phosphorylated sample does not readily convert into an E2P state when exposed to vanadate, as would otherwise be expected.
The structures are of medium resolution (3.1 - 7.4 Å), but the key sites of nucleotide binding and/or phosphorylation are reasonably well supported by the EM maps, with one exception: in the 'E1·ATP early' state determined under turnover conditions, I find the map for the gamma phosphate of ATP not overly convincing, leaving the question whether this could instead be a product-inhibited, Mg-ADP bound E1 state resulting from an accumulation of MgADP under the turnover conditions used.
Overall, the manuscript is well written and carefully phrased, and it presents interesting novel findings, which expand our knowledge about the conformational landscape and regulatory mechanisms of the P-type ATPase family.
However in my opinion there are the following weaknesses in the current version of the manuscript:
A lack of quantification. The heart of this study is the comparison of the newly determined KdpFABC structures with previously published ones (of which there are already 10). Yet, there are no RMSD calculations to illustrate the magnitude of any structural deviations. Instead, the authors use phrases like 'similar but not identical to', 'has some similarities', 'virtually identical', 'significant differences'. This makes it very hard to appreciate the true level of novelty/deviation from known structures.
Also the decrease in EPR peak height of the E1 apo tight state between phosphorylated and non-phosphorylated sample - a key piece of supporting data - is not quantified.Perhaps as a consequence of the above, there seems to be a slight tendency towards overstatements regarding the novelty of the findings in the context of previous structural studies. The E1-P·ATP tight structure is extremely similar to the previously published crystal structure (5MRW), but it took me three reads through the paper and a structural superposition (overall RMSD less than 2Å), to realise that. While I do see that the existing differences, the two helix shifts in the P- and A- domains - are important and do probably permit the usage of the term 'novel conformation' (I don't think there is a clear consensus on what level of change defines a novel conformation), it could have been made more clear that the 'tight' arrangement of domains has actually been reported before, only it was not termed 'tight'.
Likewise, the authors claim that they have covered the entire conformational cycle with their 10 structures, but this is actually not correct, as there is no representative of an E2 state or functional E1P state after ADP release.
- A key hypothesis this paper suggests is that KdpFABC cannot undergo the transition from E1P tight to E2P and hence gets stuck in this dead end 'off cycle' state. To test this, the authors analysed an S162-P sample supplied with the E2P inducing inhibitor orthovanadate and found about 11% of particles in an E2P conformation. This is rationalised as a residual fraction of unphosphorylated, non-inhibited, protein in the sample, but the sample is not actually tested for residual unphosphorylated fraction or residual activity. Instead, there is a reference to Sweet et al, 2020. So the claim that the 11% E2P particles in the vanadate sample are irrelevant, whereas the 14% E1P tight from the turnover dataset are of key importance, would strongly benefit from some additional validation.
-
Reviewer #3 (Public Review):
The authors have determined a range of conformations of the high-affinity prokaryotic K+ uptake system KdpFABC, and demonstrate at least two novel states that shed further light on the structure and function of these elusive protein complexes.
The manuscript is well-written and easy to follow. The introduction puts the work in a proper context and highlights gaps in the field. I am however missing an overview of the currently available structures/states of KdpFABC. This could also be implemented in Fig. 6 (highlighting new vs available data). This is also connected to one of my main remarks - the lack of comparisons and RMSD estimates to available structures. Similarity/resemblance to available structures is indicated several times throughout the manuscript, but this is not quantified or shown in detail, and …
Reviewer #3 (Public Review):
The authors have determined a range of conformations of the high-affinity prokaryotic K+ uptake system KdpFABC, and demonstrate at least two novel states that shed further light on the structure and function of these elusive protein complexes.
The manuscript is well-written and easy to follow. The introduction puts the work in a proper context and highlights gaps in the field. I am however missing an overview of the currently available structures/states of KdpFABC. This could also be implemented in Fig. 6 (highlighting new vs available data). This is also connected to one of my main remarks - the lack of comparisons and RMSD estimates to available structures. Similarity/resemblance to available structures is indicated several times throughout the manuscript, but this is not quantified or shown in detail, and hence it is difficult for the reader to grasp how unique or alike the structures are. Linked to this, I am somewhat surprised by the lack of considerable changes within the TM domain and the overlapping connectivity of the K indicated in Table 1 - Figure Supplement 1. According to Fig. 6 the uptake pathway should be open in early E1 states, but not in E2 states, contrasting to the Table 1 - Figure Supplement 1, which show connectivity in all structures? Furthermore, the release pathway (to the inside) should be open in the E2-P conformation, but no release pathway is shown as K ions in any of the structures in Table 1 - Figure Supplement 1. Overall, it seems as if rather small shifts in-between the shown structures (are the structures changing from closed to inward-open)? Or is it only KdpA that is shown?
My second key remark concerns the "E1-P tight is the consequence of an impaired E1-P/E2-P transition" section, and the associated discussion, which is very interesting. I am not convinced though that the nucleotide and phosphate mimic-stabilized states (such as E1-P:ADP) represent the high-energy E1P state, as I believe is indicated in the text. Supportive of this, in SERCA, the shifts from the E1:ATP to the E1P:ADP structures are modest, while the following high-energy Ca-bound E1P and E2P states remain elusive (see Fig. 1 in PMID: 32219166, from 3N8G to 3BA6). Or maybe this is not what the authors claim, or the situation is different for KdpFABC? Associated, while I agree with the statement in rows 234-237 (that the authors likely have caught an off-cycle state), I wonder if the tight E1-P configuration could relate to the elusive high-energy states (although initially counter-intuitive as it has been caught in the structure)? The claims on rows 358-360 and 420-422 are not in conflict with such an idea, and the authors touch on this subject on rows 436-450. Can it be excluded that it is the proper elusive E1P state? If the state is related to the E1P conformation it may well have bearing also on other P-type ATPases and this could be expanded upon.
-
