Sex-biased regulatory changes in the placenta of native highlanders contribute to adaptive fetal development
Curation statements for this article:-
Curated by eLife

eLife assessment
This fundamental study reports differential expression of key genes in full-term placenta between Tibetans and Han Chinese at high elevations, which are more pronounced in the placenta of male fetus than in female fetus. The gene expression data were collected and analyzed using solid and validated methodology, although there is limited support for hypoxia-specific responses due to a lack of low-altitude samples. Several of the placental genes found in this study have been previously reported to show signatures of positive selection in Tibetans, pointing to a potential mechanism of how human populations adapt to high elevation by mitigating the negative effects of low oxygen on fetal growth. The work will be of interest to evolutionary and population geneticists as well as researchers working on human hypoxic response.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Compared with lowlander migrants, native Tibetans have a higher reproductive success at high altitude though the underlying mechanism remains unclear. Here, we compared the transcriptome and histology of full-term placentas between native Tibetans and Han migrants. We found that the placental trophoblast shows the largest expression divergence between Tibetans and Han, and Tibetans show decreased immune response and endoplasmic reticulum stress. Remarkably, we detected a sex-biased expression divergence, where the male-infant placentas show a greater between-population difference than the female-infant placentas. The umbilical cord plays a key role in the sex-biased expression divergence, which is associated with the higher birth weight of the male newborns of Tibetans. We also identified adaptive histological changes in the male-infant placentas of Tibetans, including larger umbilical artery wall and umbilical artery intima and media, and fewer syncytial knots. These findings provide valuable insights into the sex-biased adaptation of human populations, with significant implications for medical and genetic studies of human reproduction.
Article activity feed
-

-
-
-

Author response:
The following is the authors’ response to the previous reviews.
Public Review:
This manuscript by Yue et al. aims to understand the molecular mechanisms underlying the better reproductive outcomes of Tibetans at high altitude by characterizing the transcriptome and histology of full-term placenta of Tibetans and compare them to those Han Chinese at high elevations.
The approach is innovative, and the data collected are valuable for testing hypotheses regarding the contribution of the placenta to better reproductive success of populations that adapted to hypoxia. The authors identified hundreds of differentially expressed genes (DEGs) between Tibetans and Han, including the EPAS1 gene that harbors the strongest signals of genetic adaptation. The authors also found that such differential expression is more prevalent …
Author response:
The following is the authors’ response to the previous reviews.
Public Review:
This manuscript by Yue et al. aims to understand the molecular mechanisms underlying the better reproductive outcomes of Tibetans at high altitude by characterizing the transcriptome and histology of full-term placenta of Tibetans and compare them to those Han Chinese at high elevations.
The approach is innovative, and the data collected are valuable for testing hypotheses regarding the contribution of the placenta to better reproductive success of populations that adapted to hypoxia. The authors identified hundreds of differentially expressed genes (DEGs) between Tibetans and Han, including the EPAS1 gene that harbors the strongest signals of genetic adaptation. The authors also found that such differential expression is more prevalent and pronounced in the placentas of male fetuses than those of female fetuses, which is particularly interesting, as it echoes with the more severe reduction in birth weight of male neonates at high elevation observed by the same group of researchers (He et al., 2022).
This revised manuscript addressed several concerns raised by reviewers in last round. However, we still find the evidence for natural selection on the identified DEGs--as a group--to be very weak, despite more convincing evidence on a few individual genes, such as EPAS1 and EGLN1.
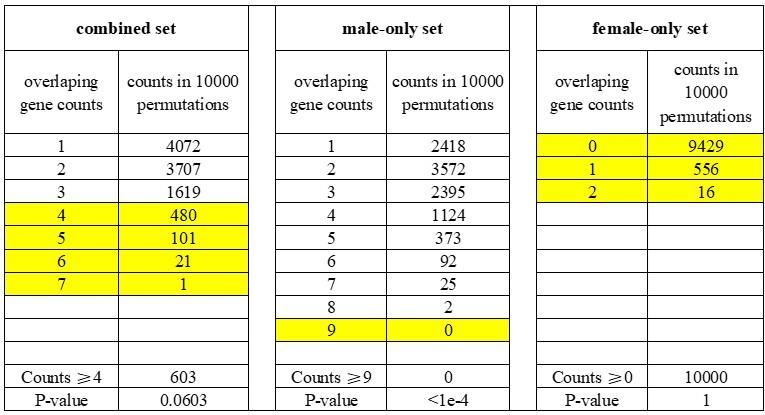
The authors first examined the overlap between DEGs and genes showing signals of positive selection in Tibetans and evaluated the significance of a larger overlap than expected with a permutation analysis. A minor issue related to this analysis is that the p-value is inflated, as the authors are counting permutation replicates with MORE genes in overlap than observed, yet the more appropriate way is counting replicates with EQUAL or MORE overlapping genes. Using the latter method of p-value calculation, the "sex-combined" and "female-only" DEGs will become non-significantly enriched in genes with evidence of selection, and the signal appears to solely come from male-specific DEGs. A thornier issue with this type of enrichment analysis is whether the condition on placental expression is sufficient, as other genomic or transcriptomic features (e.g., expression level, local sequence divergence level) may also confound the analysis.
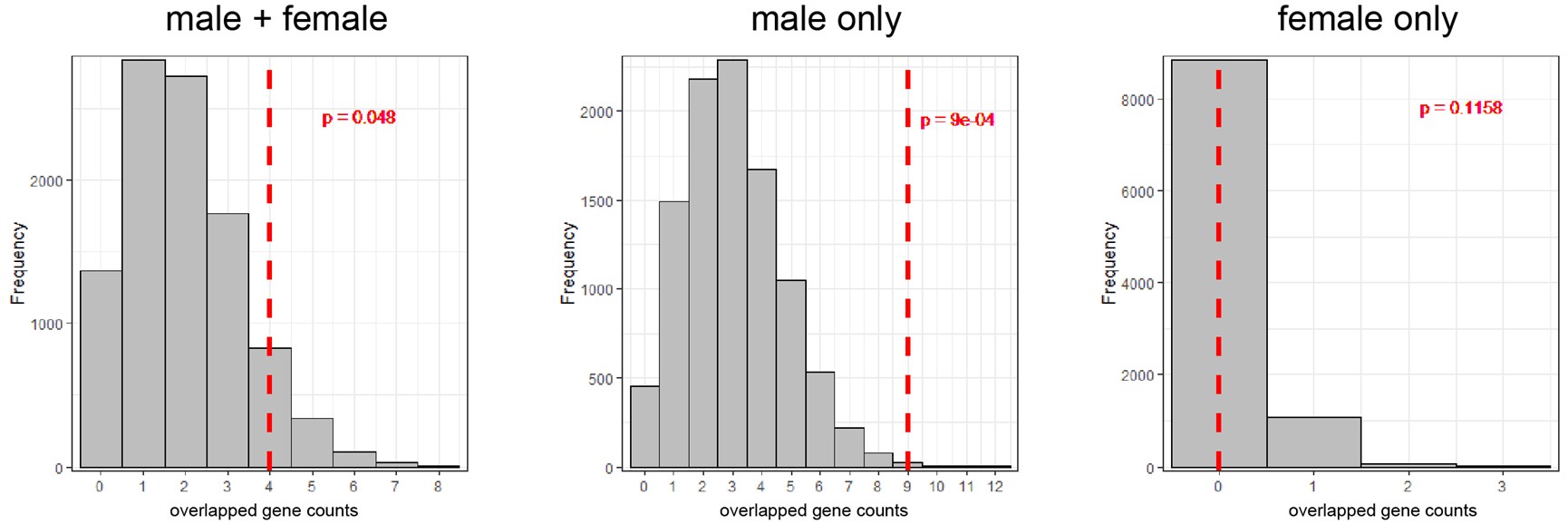
According to the suggested methods, we counted the replicates with equal or more overlapping genes than observed (≥4 for the “combined” set; ≥9 for the “male-only” set; ≥0 for the “female-only” set). We found that the overlaps between DEGs and TSNGs were significantly enriched only in the “male-only” set (p-value < 1e-4, counting 0 time from 10,000 permutations), but not in the “female-only” set (p-value = 1, counting 10,000 time from 10,000 permutations), or “combined” set (p-value = 0.0603, counting 603 time from 10,000 permutations) (see Table R1 below).
We updated this information in the revised manuscript, including Results, Methods, and Figure S9.
Author response table 1.
Permutation analysis of the overlapped genes between DEGs and TSNGs.
The authors next aimed to detect polygenic signals of adaptation of gene expression by applying the PolyGraph method to eQTLs of genes expressed in the placenta (Racimo et al 2018). This approach is ambitious but problematic, as the method is designed for testing evidence of selection on single polygenic traits. The expression levels of different genes should be considered as "different traits" with differential impacts on downstream phenotypic traits (such as birth weight). As a result, the eQTLs of different genes cannot be naively aggregated in the calculation of the polygenic score, unless the authors have a specific, oversimplified hypothesis that the expression increase of all genes with identified eQTL will improve pregnancy outcome and that they are equally important to downstream phenotypes. In general, PolyGraph method is inapplicable to eQTL data, especially those of different genes (but see Colbran et al 2023 Genetics for an example where the polygenic score is used for testing selection on the expression of individual genes).
We would recommend removal of these analyses and focus on the discussion of individual genes with more compelling evidence of selection (e.g., EPAS1, EGLN1).
According to the suggestion, we removed these analyses in the revised manuscript.
-
eLife assessment
This fundamental study reports differential expression of key genes in full-term placenta between Tibetans and Han Chinese at high elevations, which are more pronounced in the placenta of male fetus than in female fetus. The gene expression data were collected and analyzed using solid and validated methodology, although there is limited support for hypoxia-specific responses due to a lack of low-altitude samples. Several of the placental genes found in this study have been previously reported to show signatures of positive selection in Tibetans, pointing to a potential mechanism of how human populations adapt to high elevation by mitigating the negative effects of low oxygen on fetal growth. The work will be of interest to evolutionary and population geneticists as well as researchers working on human hypoxic response.
-
Joint Public Review:
This manuscript by Yue et al. aims to understand the molecular mechanisms underlying the better reproductive outcomes of Tibetans at high altitude by characterizing the transcriptome and histology of full-term placenta of Tibetans and compare them to those Han Chinese at high elevations.
The approach is innovative, and the data collected are valuable for testing hypotheses regarding the contribution of the placenta to better reproductive success of populations that adapted to hypoxia. The authors identified hundreds of differentially expressed genes (DEGs) between Tibetans and Han, including the EPAS1 gene that harbors the strongest signals of genetic adaptation. The authors also found that such differential expression is more prevalent and pronounced in the placentas of male fetuses than those of female fetuses, …
Joint Public Review:
This manuscript by Yue et al. aims to understand the molecular mechanisms underlying the better reproductive outcomes of Tibetans at high altitude by characterizing the transcriptome and histology of full-term placenta of Tibetans and compare them to those Han Chinese at high elevations.
The approach is innovative, and the data collected are valuable for testing hypotheses regarding the contribution of the placenta to better reproductive success of populations that adapted to hypoxia. The authors identified hundreds of differentially expressed genes (DEGs) between Tibetans and Han, including the EPAS1 gene that harbors the strongest signals of genetic adaptation. The authors also found that such differential expression is more prevalent and pronounced in the placentas of male fetuses than those of female fetuses, which is particularly interesting, as it echoes with the more severe reduction in birth weight of male neonates at high elevation observed by the same group of researchers (He et al., 2022).
Comments on latest version:
The revised manuscript has incorporated the suggested changes and weakened conclusions regarding natural selection. Limitations of the study are also clearly stated in the Discussion section.
-
-
Author response:
The following is the authors’ response to the previous reviews.
Reviewing Editor's comments:
There appears to be several mistakes/missing details in the additional statistical analyses reported in their response to Reviewer #'1 comments:
(1) Detecting differentially expressed genes (DEGs):
Reviewer #1 suggested adding an interaction term between sex and environment (ethnicity) in identifying DEGs. The authors performed ANCOVA analysis with sex and ethnicity as covariates (but not the interaction) and found sex explained more variance. This is not what the reviewer asked for, and the results do not help identify DEGs.
We understand the reviewer’s suggestion about identification of DEGs using sex × ethnicity interaction. However, we could not find an appropriate tool to make such analysis, though we have carefully …
Author response:
The following is the authors’ response to the previous reviews.
Reviewing Editor's comments:
There appears to be several mistakes/missing details in the additional statistical analyses reported in their response to Reviewer #'1 comments:
(1) Detecting differentially expressed genes (DEGs):
Reviewer #1 suggested adding an interaction term between sex and environment (ethnicity) in identifying DEGs. The authors performed ANCOVA analysis with sex and ethnicity as covariates (but not the interaction) and found sex explained more variance. This is not what the reviewer asked for, and the results do not help identify DEGs.
We understand the reviewer’s suggestion about identification of DEGs using sex × ethnicity interaction. However, we could not find an appropriate tool to make such analysis, though we have carefully searched it in the literature. It should be noted that the interaction analysis between sex and environment was only designed to study genotype data rather than gene expression data. Besides, considering that we have added multiple covariates in our DEG detection, adding an interaction term between sex and environment (ethnicity) in identifying DEGs make the formulation too complex to resolve using current tools. Alternatively, we have made a linear regression model to test the explanation of sex for DEG detection in the revision (see details below). We would appreciate if the reviewer could provide any available tools, or previous studies conducting interaction analysis for DEG identification.
(2) Overlap between DEGs and genes under positive selection in Tibetans (TSNGs)
The authors claimed that the overlaps are significantly enriched in "sex-combined" set (p=0.048) and "male-only" set (p=9e-4), but it seems that the authors calculated the p-values incorrectly. Based on the histogram shown in Fig 3R (left penal), at least 750 out of 10,000 permutations led to 4 genes in overlap and there are additional permutations with 5 or more genes in overlap, so the p-value for the sex-combined set cannot be 0.048. In addition, the permutation procedure is somewhat questionable: it is unclear whether randomly sampling 192 genes from the human genome is reasonable choice, without matching for relevant gene features.
As we explained in the response to Reviewer-1, we agree with the reviewer’s point that random sampling of genes in permutation should be extracted from genes expressed in each tissue rather than the entire genome. Based on this updated random sampling procedure, we redid the analysis, and our previous conclusions remain unchanged.
(3) Polygenic adaptation signal based on eQTL information:
The PolyGraph method is designed for highly polygenic traits with causal variants spread across the genome. However, the genetic architecture of the expression of a gene is much less polygenic with at most few cis- eQTLs per gene, so the PolyGraph model does not apply for expression of individual genes. On the other hand, eQTLs for different genes are associated with different "traits", so they cannot be simply aggregated together for PolyGraph analysis. Based on the Methods description, it is unclear how the authors ran the PolyGraph analysis on eQTLs practically and whether this practice is appropriate for detecting polygenic adaptation signal on gene expression.
We understand the reviewer’s concern on polygenic adaptation analysis. In this study, we tested whether the estimated polygenic scores from eQTLs (estimated using sums of allele frequencies at independent eQTLs weighted by their effect sizes) were significantly enriched in Tibetans compared to other populations. The detailed descriptions of polygenic test are provided in the response to Reviewer-1.
Reviewer #1 (Public Review):
The revised manuscript new presented 1) a permutation-based test for the significance of the overlap between DEGs and genes with positive selection signals in Tibetans, and 2) polygenic adaptation test for the eQTLs. I make my suggestions in detail as below:
Major Comments
(1) My previous concern regarding the DEG analysis remains unresolved. Although the authors agreed in their response that the difference between the male- and female-specific DEGs are insufficient to the difference between sex-combined and sex-specific DEGs (Figure S6). However, the results section still states the opposite pattern between males and females as a decisive reason for the difference (p. 9, lines 236-239). Again, I would like to recommend the authors to test alternative ways of analysis to boost statistical power for DEG detection other than simply splitting data into males and females and performing analysis in each subset. For example, the authors may consider utilizing gene by environment interaction analysis schemes here biological sex as an environmental factor.
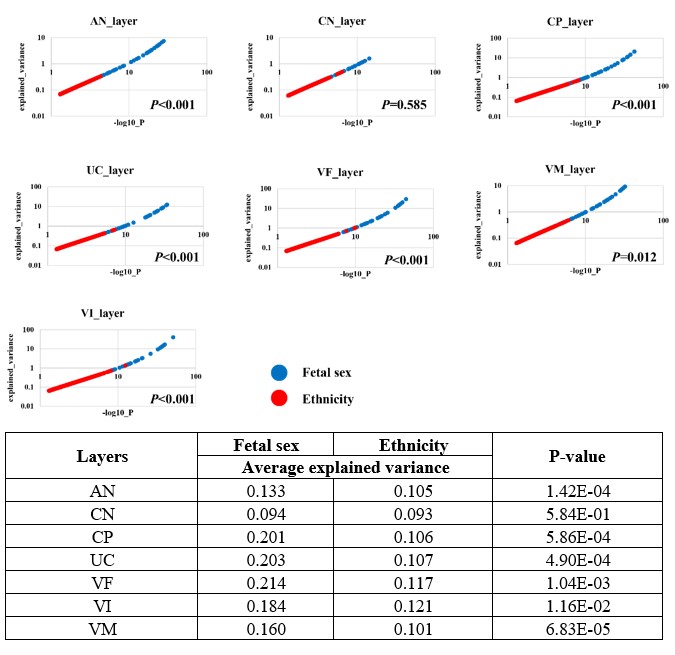
To evaluate the effect of gene expression of each layer by sex, we adopted two strategies: 1) to calculate the variance explained by sex from the expression data; 2) to evaluate the statistical significance of association between sex from the expression data.
Firstly, we observed a significantly higher variance explained by sex than by ethnicity in six layers of the placenta (see details in our previous response to reviewers).
Then, we performed a linear regression model to test whether gender affects the gene expression. For each gene, a linear regression model was made by using R glm function with sex as covariates: glm (gene expression ~ sex). We discovered 5,865 genes significantly associated with sex, and most of them were located on the sex chromosomes. We observed 62.63% genes overlapped with those genes with opposite differential directions between the sex-combined and the sex-specific analyses.
Considering the opposite direction of DEGs is likely only one of the explanations for the discrepancy between the sex-combined and the sex-specific DEGs, and there might be alternative mechanism for this phenomenon, we have tune down the description of this point in the revised manuscript:
“Considering 62.63% of DEGs (248/396) with an opposite direction of between-population expression divergence in males and females, respectively (Figure S6), we reckon that there might be other factors such as sample size or cell composition affecting the identification of DEGs, which could cancel out the differences in the sex-combined analysis.” (Page 9)
(2) Multiple testing schemes are still sub-optimal in some cases. Most of all, the p-values in the WGCNA analysis (p. 11), the authors corrected for the number of traits (n=12) after adjusting for the correlation between them. However, they did not mention whether they counted for the number of modules they tested at all (n=136 and 161 for males and females, respectively). Whether they account for the number of modules will make a substantial difference in the significance threshold, please incorporate and describe a proper multiple testing scheme for this analysis.
We understand the reviewer’s point. Indeed, for multiple testing schemes, we considered both the number of traits and the number of modules. For the number of modules, multiple testing correction is already imbedded in WGCNA, as described in the published studies (Li et al. 2018; Zeng et al. 2023).
(3) Evidence for natural selection on the observed DEG pattern is still weak and not properly described.
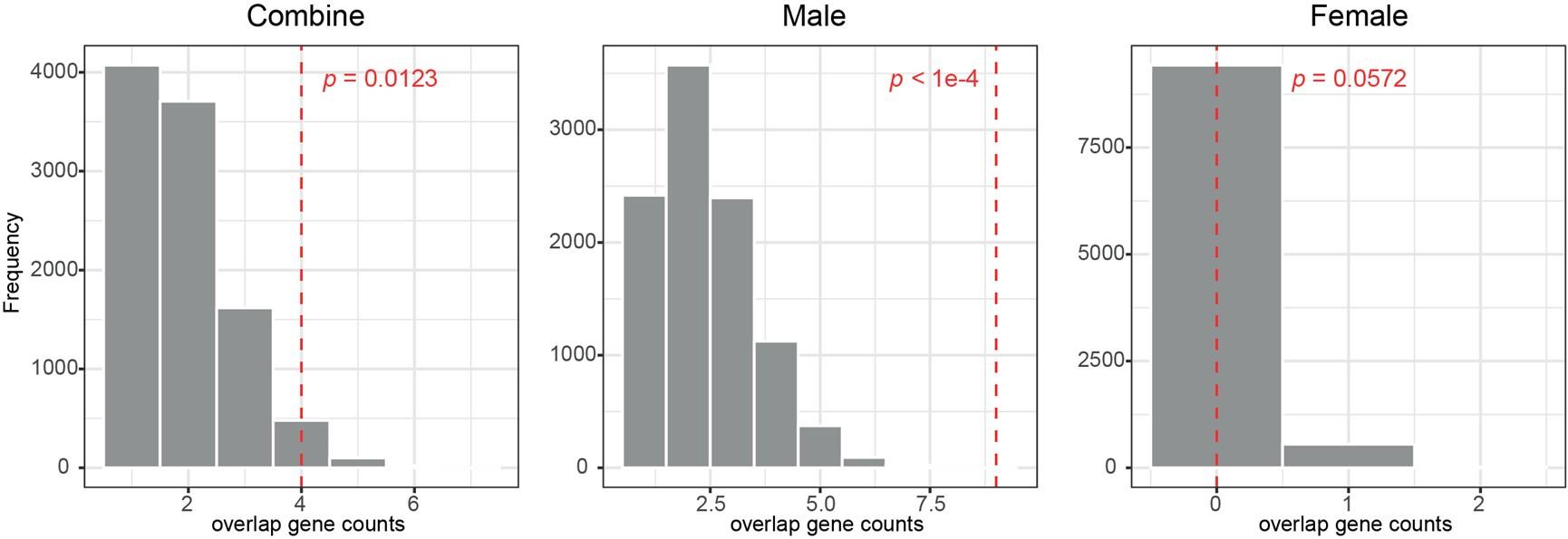
(1) For the overlap between DEGs and TSNGs, the authors introduced a permutation-based test, but used a total set of genes in the human genome as a comparison set (p. 25, lines 699-700). I believe that the authors should sample random sets of genes from those already expressed in each tissue to make a fair comparison.
We agree with the reviewer’s point that random sampling of genes in permutation should be extracted from genes expressed in each tissue, which is a fair comparison between the observed and the simulated counts of the overlapped genes.
Therefore, for each permutation, we randomly extracted 192 genes from all the placenta expressed genes identified from the seven layers (17,284 genes in total), and we overlapped them with DEGs of the three sets (female + male, female only, and male only) and counted the gene numbers. After 10,000 permutations, we constructed a null distribution for each set, and found that the overlaps between DEGs and TSNGs were significantly enriched in the “sex-combined” set (p-value = 0.0123) and the “male-only” set (p-value < 1e-4), but not in the “female-only” set (p-value = 0.0572) (Figure R1). This result suggests that the observed DEGs are significantly enriched in TSNGs when compared to the set of random sampling, especially for the DEGs from the “male-only” set.
Author response image 1.
The distribution of 10,000 permutation tests of counts of the overlapped genes between 192 TSNGs and the DEGs randomly selected from the expressed genes in the placenta. The red-dashed lines indicate the observed values based on the randomly selected DEGs.
(2) The entire polygraph analysis for polygenic adaptation is poorly described. The current version of the Methods does not clarify i) for which genes the eQTLs are discovered, 2) how the authors performed the eQTL analysis, iii) how the authors polarized the effect, and iv) how they set up a comparison between the eQTLs and the others.
Considering the RNA-seq data of placenta mostly represent the transcriptomes of the newborns according to our analysis on maternal-fetal compositions of each dissected layer, we conducted eQTL analysis using the fetal genotypes and the placental tissue gene expression data (TPM) using R package MatrixEQTL (https://github.com/andreyshabalin/MatrixEQTL), and the altitude and maternal age were taken as covariates. We take a window 1 Mb upstream and 1 Mb downstream around each SNP to select genes or expression probes to test. Associations between these SNP–gene combinations are calculated using linear model. This tool can distinguish local (cis-) and distant (trans) eQTLs. We performed separate corrections for multiple testing.
Finally, we detected 5,251 eQTLs (involving 319 eGenes), covering the SNPs significantly associated with gene expression (p-value < 5e-8). To identify the signatures of polygenic selection in Tibetans using eQTL information, we removed those SNPs in linkage disequilibrium (r2 > 0.2 in 1000 Genome Project) and obtained 176 independent eQTLs as input into PolyGraph (Racimo et al. 2018). QB (Racimo et al. 2018) and QX (Berg and Coop 2014) framework are used in Polygraph to determine whether the estimated polygenic scores exhibit more variance among populations than null expectation under genetic drift, by retrieving the summary statistics from the eQTL set.
In this study, we focused on testing whether the estimated polygenic scores from eQTLs (estimated using sums of allele frequencies at independent eQTLs weighted by their effect sizes) were significantly enriched in Tibetans compared to other populations. The significance was evaluated by comparing to 10,000 sets of the control SNPs. Each set of control SNPs was randomly drawn from the genomic SNPs, and contained an equal number of SNPs as the eQTLs matched one-to-one by minor allele frequency.
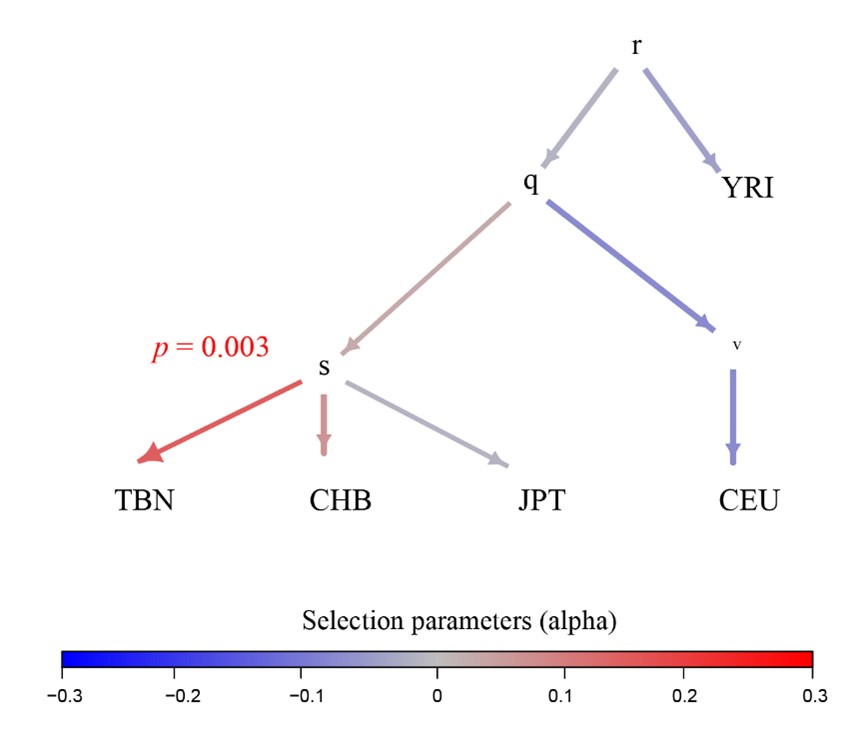
The PolyGraph result showed that Tibetans have a clear signature of polygenic selection on gene expression (Bonferroni-corrected p-value = 0.003, Figure S12). In other words, the frequency of alleles associated with gene expression (up-regulation or down-regulation) were specifically enriched in Tibetans, a signal of positive selection.
Minor comments (1) In Figure S1, the amount of variance explained by PC1 and PC2 need to be corrected. PC1 explains less variance than PC2 (0.11 vs 0.68%).
It was a typing error that mixed up the variances between PC1 and PC2. We have corrected it in the revised version.
(2) In the section "Sex-biased expression divergence ..." (p. 8), the authors are using the term "gender" instead of sex. Considering that they are talking about the biological sex of each infant, I believe that sex is a more appropriate term to be used than gender.
Following the reviewer’s suggestion, we rephrased “gender” as “sex” in the revised manuscript to describe the biological differences between females and males.
Reviewer #3 (Public Review):
More than 80 million people live at high altitude. This impacts health outcomes, including those related to pregnancy. Longer-lived populations at high altitudes, such as the Tibetan and Andean populations show partial protection against the negative health effects of high altitude. The paper by Yue sought to determine the mechanisms by which the placenta of Tibetans may have adapted to minimise the negative effect of high altitude on fetal growth outcomes. It compared placentas from pregnancies from Tibetans to those from the Han Chinese. It employed RNAseq profiling of different regions of the placenta and fetal membranes, with some follow-up of histological changes in umbilical cord structure and placental structure. The study also explored the contribution of fetal sex in these phenotypic outcomes.
A key strength of the study is the large sample sizes for the RNAseq analysis, the analysis of different parts of the placenta and fetal membranes, and the assessment of fetal sex differences.
A main weakness is that this study, and its conclusions, largely rely on transcriptomic changes informed by RNAseq. Changes in genes and pathways identified through bioinformatic analysis were not verified by alternate methods, such as by western blotting, which would add weight to the strength of the data and its interpretations. There is also a lack of description of patient characteristics, so the reader is unable to make their own judgments on how placental changes may link to pregnancy outcomes. Another weakness is that the histological analyses were performed on n=5 per group and were rudimentary in nature.
For the three weaknesses raised by the reviewer, here are our responses:
(1) Considering that our conclusions largely rely on the transcriptomic data, we agree with reviewer that more experiments are needed to validate the results from our transcriptomic data. However, this study was mainly aimed to provide a transcriptomic landscape of high-altitude placenta, and to characterize the gene-expression difference between native Tibetans and Han migrants. The molecular mechanism exploration is not the main task of this study, and more validation experiments are warranted in the future.
(2) For the lack of description of patient characteristics, actually, we provided three-level results on the placental changes of Tibetans: macroscopic phenotypes (higher placental weight and volume), histological phenotypes (larger umbilical vein walls and umbilical artery intima and media; lower syncytial knots/villi ratios) and transcriptomic phenotypes (DEG and differential modules). Combined with the previous studies, these placenta changes suggest a better reproductive outcome. For example, the placenta volume shows a significantly positive correlation with birth weight (R = 0.31, p-value = 2.5e-16), therefore, the larger placenta volume of Tibetans is beneficial to fetal development at high altitude. In addition, the larger umbilical vein wall and umbilical artery intima and media of Tibetans can explain their adaptation in preventing preeclampsia.
(3) For the sample size of histological analyses, we understand the reviewer’s concern that 5 vs. 5 samples are not very large in histological analyses. This is because it was difficult to collect high-altitude Han placenta samples, and we only got 13 Han samples, from which we selected 5 infant sex matched samples.
Minor point:
I feel the authors have responded well to the other reviewer comments. However, I am disappointed that the authors did not address my comment related to the validation of their RNAseq data. In particular, they failed to add new data that verifies and supports their RNAseq findings on pathways affected. This is imperative as their conclusions are based solely on the RNAseq analysis. The only other comment I have is that they should add a description of all abbreviations, including those in the supplementary information (like Table S12).
For experimental validation of transcriptome, we understand the concern of reviewer. However, as we mentioned before, this study was mainly aimed to provide a transcriptomic landscape of high-altitude placenta, the molecular mechanism exploration is not the main task of this study, and more validation experiments are warranted in the future. Actually, we have tune down the description of power from transcriptomic data for explanation of biological difference, and called for the further functional validations in the future:
“the transcriptome data is insufficient to explain the underlying molecular mechanisms of genetic adaptation in Tibetans. Future single-cell transcriptome analysis and functional validations of the candidate genes are warranted to reveal the responsible cell types and the molecular pathways.” (highlighted in Page 20)
For abbreviations of the manuscript, according to the reviewer’s suggestion, we added descriptions of all abbreviations of this study in corresponding position (Table S1 and S12).
References
Berg JJ, and Coop G (2014). A population genetic signal of polygenic adaptation. PLoS Genet 10(8): e1004412.
Li J, et al. (2018). Application of Weighted Gene Co-expression Network Analysis for Data from Paired Design. Sci Rep 8(1): 622.
Racimo F, Berg JJ, and Pickrell JK (2018). Detecting Polygenic Adaptation in Admixture Graphs. Genetics 208(4): 1565-1584.
Zeng JF, et al. (2023). Functional investigation and two-sample Mendelian randomization study of neuropathic pain hub genes obtained by WGCNA analysis. Frontiers in Neuroscience 17.
-
eLife assessment
This fundamental study reports differential expression of key genes in full-term placenta between Tibetans and Han Chinese at high elevations, which are more pronounced in the placentae of male than in female fetuses. If validated as functionally relevant, these results will help us understand how human populations adapt to high elevation by mitigating the negative effects of low oxygen on fetal growth. While the differential gene expression analyses are solid, the downstream analyses offer incomplete support for the connection to hypoxia-specific responses and adaptive genetic variation.
-
Joint Public Review:
This manuscript by Yue et al. aims to understand the molecular mechanisms underlying the better reproductive outcomes of Tibetans at high altitude by characterizing the transcriptome and histology of full-term placenta of Tibetans and compare them to those Han Chinese at high elevations.
The approach is innovative, and the data collected are valuable for testing hypotheses regarding the contribution of the placenta to better reproductive success of populations that adapted to hypoxia. The authors identified hundreds of differentially expressed genes (DEGs) between Tibetans and Han, including the EPAS1 gene that harbors the strongest signals of genetic adaptation. The authors also found that such differential expression is more prevalent and pronounced in the placentas of male fetuses than those of female fetuses, …
Joint Public Review:
This manuscript by Yue et al. aims to understand the molecular mechanisms underlying the better reproductive outcomes of Tibetans at high altitude by characterizing the transcriptome and histology of full-term placenta of Tibetans and compare them to those Han Chinese at high elevations.
The approach is innovative, and the data collected are valuable for testing hypotheses regarding the contribution of the placenta to better reproductive success of populations that adapted to hypoxia. The authors identified hundreds of differentially expressed genes (DEGs) between Tibetans and Han, including the EPAS1 gene that harbors the strongest signals of genetic adaptation. The authors also found that such differential expression is more prevalent and pronounced in the placentas of male fetuses than those of female fetuses, which is particularly interesting, as it echoes with the more severe reduction in birth weight of male neonates at high elevation observed by the same group of researchers (He et al., 2022).
This revised manuscript addressed several concerns raised by reviewers in last round. However, we still find the evidence for natural selection on the identified DEGs--as a group--to be very weak, despite more convincing evidence on a few individual genes, such as EPAS1 and EGLN1.
The authors first examined the overlap between DEGs and genes showing signals of positive selection in Tibetans and evaluated the significance of a larger overlap than expected with a permutation analysis. A minor issue related to this analysis is that the p-value is inflated, as the authors are counting permutation replicates with MORE genes in overlap than observed, yet the more appropriate way is counting replicates with EQUAL or MORE overlapping genes. Using the latter method of p-value calculation, the "sex-combined" and "female-only" DEGs will become non-significantly enriched in genes with evidence of selection, and the signal appears to solely come from male-specific DEGs. A thornier issue with this type of enrichment analysis is whether the condition on placental expression is sufficient, as other genomic or transcriptomic features (e.g., expression level, local sequence divergence level) may also confound the analysis.
The authors next aimed to detect polygenic signals of adaptation of gene expression by applying the PolyGraph method to eQTLs of genes expressed in the placenta (Racimo et al 2018). This approach is ambitious but problematic, as the method is designed for testing evidence of selection on single polygenic traits. The expression levels of different genes should be considered as "different traits" with differential impacts on downstream phenotypic traits (such as birth weight). As a result, the eQTLs of different genes cannot be naively aggregated in the calculation of the polygenic score, unless the authors have a specific, oversimplified hypothesis that the expression increase of all genes with identified eQTL will improve pregnancy outcome and that they are equally important to downstream phenotypes. In general, PolyGraph method is inapplicable to eQTL data, especially those of different genes (but see Colbran et al 2023 Genetics for an example where the polygenic score is used for testing selection on the expression of individual genes).
We would recommend removal of these analyses and focus on the discussion of individual genes with more compelling evidence of selection (e.g., EPAS1, EGLN1)
-
-
Author Response
The following is the authors’ response to the original reviews.
Reviewer #1:
In this manuscript, the authors investigate differences between Tibetans and Han Chinese at altitude in terms of placental transcriptomes during full-term pregnancy. Most importantly, they found that the inter-population differentiation is mostly male-specific and the observed direction of transcriptional differentiation seems to be adaptive at high altitude. In general, it is of great importance and provides new insights into the functional basis of Tibetan high-altitude adaptations, which so far have been mostly studied via population genetic measures only. More specifically, I firmly believe that we need more phenotype data (including molecular phenotypes such as gene expression data) to fully understand Tibetan adaptations to high …
Author Response
The following is the authors’ response to the original reviews.
Reviewer #1:
In this manuscript, the authors investigate differences between Tibetans and Han Chinese at altitude in terms of placental transcriptomes during full-term pregnancy. Most importantly, they found that the inter-population differentiation is mostly male-specific and the observed direction of transcriptional differentiation seems to be adaptive at high altitude. In general, it is of great importance and provides new insights into the functional basis of Tibetan high-altitude adaptations, which so far have been mostly studied via population genetic measures only. More specifically, I firmly believe that we need more phenotype data (including molecular phenotypes such as gene expression data) to fully understand Tibetan adaptations to high altitude, and this manuscript is a rare example of such a study. I have a few suggestions and/or questions with which I hope to improve the manuscript further, especially in terms of 1) testing if the observed DEG patterns are truly adaptive, and 2) how and whether the findings in this study can be linked to EPAS1 and EGLN1, the signature adaptation genes in Tibetans.
We appreciate the reviewer’s constructive comments. We have addressed these points and the details are discussed below.
Major Comments:
- The DEG analysis is the most central result in this manuscript, but the discrepancy between sex-combined and sex-specific DEGs is quite mind-boggling. For those that were differentially expressed in the sex-specific sets but not in the sex-combined one, the authors suggest an opposite direction of DE as an explanation (page 11, Figure S5). But Figure S5A does not show such a trend, showing that down-regulated genes in males are mostly not at all differentially expressed in females. Figure S5B does show such a trend, but it doesn't seem to be a dominant explanation. I would like to recommend the authors test alternative ways of analysis to boost statistical power for DEG detection other than simply splitting data into males and females and performing analysis in each subset. For example, the authors may consider utilizing gene-by-environment interaction analysis schemes here biological sex as an environmental factor.
We agree with reviewer that the opposite direction of DEGs is likely only one of the possible explanations for the discrepancy between the sex-combined and the sex-specific DEGs. We have toned down the description of this point in the revised manuscripts.
Following the suggestion of reviewer, we performed a ANCOVA analysis to evaluate the variance explained by sex from the expression data. For each gene, univariate comparisons of the average of gene expression between Tibetans and Han Chinese were made by using the ANCOVA test in R aov function with sex as covariates: aov (Expression ~ Ethnicity + Fetal sex). We observed a significantly higher variance explained by sex than by ethnicity in six layers of the placenta (except for the CN layer) (Author response image 1). For example, in the UC layer, fetal sex can explain ~0.203 variance, while the ethnicity explains ~0.107 variance (P-value = 4.9e-4). These results suggest a significant contribution of fetal sex for the observed variance of gene expression, consist with the observed sex-biased DEG patterns.
Author response image 1.
The ANCOVA results of the seven layers of placenta. The scatter plot shows the comparison of the explained variance (y-axis) and significance (x-axis, denoted by –log10(P-value)) between ethnicity (dots in red) and fetal sex (dots in blue). Each dot represents an investigated gene, and only genes with P<0.05 in significance are shown in the plots. The table is the summary statistics of the ANCOVA analysis.
- Please clarify how the authors handled multiple testing correction of p-values.
There were three analyses involving multiple testing in this study: 1) for the differential expression analysis, we obtained the multiple corrected p-values by Benjamini-Hochberg FDR (false discovery rate) procedure; 2) for the GO enrichment analysis, we calculated the FDR-adjusted q-values from the overall p-values to correct for multiple testing.
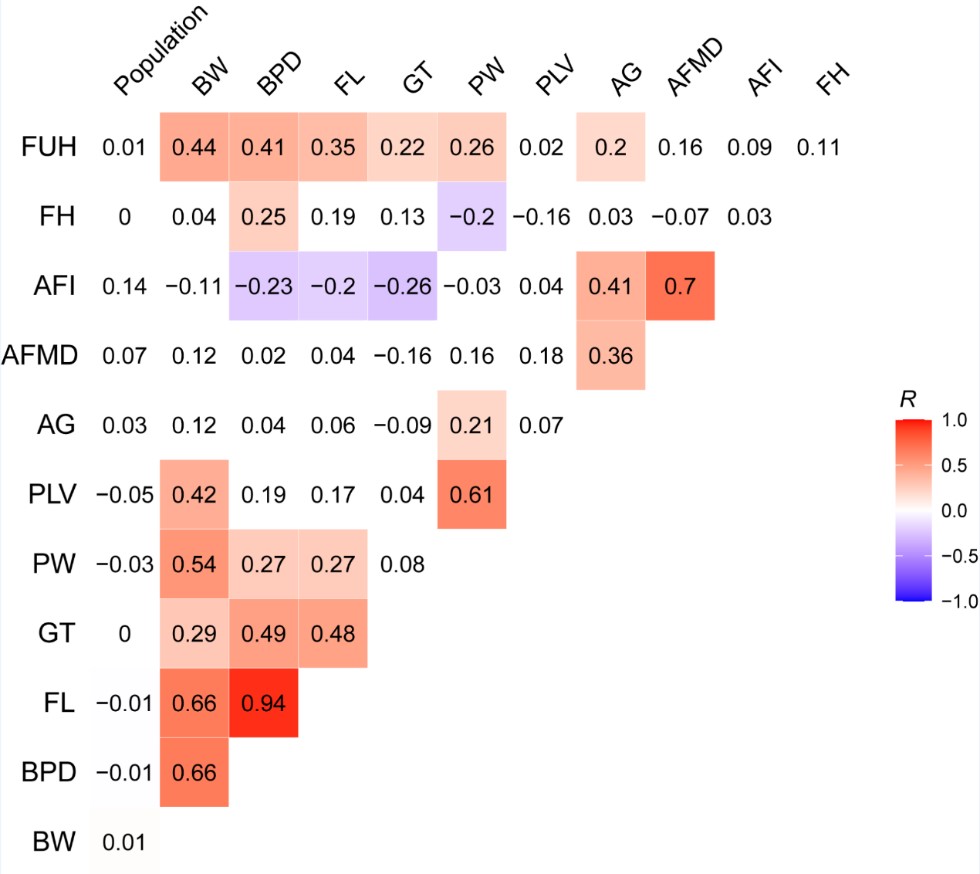
- for the WGCNA analysis, considering the 12 traits were involved, including population, birth weight (BW), biparietal diameter (BPD), femur length (FL), gestation time (GT), placental weight (PW), placental volume (PLV), abdominal girth (AG), amniotic fluid maximcon depth (AFMD), amniotic fluid (AFI), fetal heart rate (FH) and fundal height (FUH). We calculated a Bonferroni threshold (p-value = 0.05/the number of independent traits) using the correlation matrix of the traits to evaluate the significant modules. We estimated the number of independent traits among the 12 investigated traits was 4 (Author response image 2). Therefore, we used a more stringent significant threshold p-value = 0.0125 (0.05/4) as the final threshold to correct the multiple testing brought by multiple traits in our WGCNA analyses. We have updated this section based on the new threshold.
Author response image 2.
The correlation matrix of 12 traits involved in the WGCNA analysis. The correlation coefficients larger than 0.2 (or smaller than -0.2) are regarded as significant correlation and marked in gradient colors.
- The "natural selection acts on the placental DEGs ..." section is potentially misleading readers to assume that the manuscript reports evidence for positive selection on the observed DEG pattern between Tibetans and Han, which is not.
a) Currently the section simply describes an overlap between DEGs and a set of 192 genes likely under positive selection in Tibetans (TSNGs). The overlap is quite small, leading to only 13 genes in total (Figure 6). The authors are currently not providing any statistical measure of whether this overlap is significantly enriched or at the level expected for random sampling.
We understand the reviewer’s point that the observed gene counts overlapped between DEGs from the three sets (4 for female + male; 9 for male only and 0 for female only) with TSNGs should be tested using a statistical method. Therefore, we adopted permutation approach to evaluate the enrichment of the overlapped DEGs with TSNGs.
For each permutation, we randomly extracted 192 genes from the human genome, then overlapped with DEGs of the three sets (female + male; female only and male only) and counted the gene numbers. After 10,000 permutations, we constructed a null distribution for each set, and found that the overlaps between DEGs and TSNGs were significantly enriched in the “female + male” set (p-value = 0.048) and the “male only” set (p-value = 9e-4), but not in the “female only” set (p-value = 0.1158) (Author response image 3). This result suggests that the observed DEGs are significantly enriched in TSNGs when compared to random sampling, especially for the male DEGs. We added this analysis in the revised manuscript.
Author response image 3.
The distribution of 10,000 permutation tests of counts of the overlapped genes between DEGs and the 192 randomly selected genes in the genome. The red-dashed lines indicate the observed values based on the 192 TSNGs.
b) The authors are describing sets of DEGs that seem to affect important phenotypic changes in a consistent and adaptive direction. A relevant form of natural selection for this situation may be polygenic adaptation while the authors only consider strong positive selection at a single variant/gene level.
We agree with reviewer that polygenic adaptation might be a potential mechanism for DEGs to take effect on the adaptive phenotypes. Therefore, following the suggestion in the comment below, we conducted a polygenic adaptation analysis using eQTL information.
c) The manuscript is currently providing no eQTL information that can explain the differential expression of key genes. The authors can actually do this based on the genotype and expression data of the individuals in this study. Combining eQTL info, they can set up a test for polygenic adaptation (e.g., Berg and Coop; https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1004412). This will provide a powerful and direct test for the adaptiveness of the observed DEG pattern.
Following the reviewer’s suggestion, we employed the PolyGraph (Racimo et al., 2018) tool to identify the signatures of polygenic selection in Tibetans using eQTL information. We conducted eQTL analysis for the seven layers, and collected a set of 5,251 eQTLs, covering the SNPs associated with gene expression with a significanct p-value < 5e-8. To obtain a list of independent eQTLs, we removed those SNPs in linkage disequilibrium (r2 > 0.2 in 1000 Genome Project). Finally, we obtained 176 independent eQTLs. At the same time, we generated a set of 1,308,436 independent SNPs of Tibetans as the control panel. The PolyGraph result showed that Tibetans have a clear signature of polygenic selection on gene expression (Bonferroni-correction p-value = 0.003) (Author response image 4).
We have added this result in the revised manuscript (Figure S4), and added a detailed description of polygenic adaption in the Methods section.
Author response image 4.
Polygraphs for the eQTLs that show evidence for polygenic adaptation in the five-leaf tree built using the allele frequency data of 1001 Tibetans (Zheng et al. 2023) and 1000 Genome Project. The colors indicate the marginal posterior mean estimate of the selection parameter for variants associated with the gene expression. r, q, s and v in the tree nodes refer to the nodes in terminal branches and internal branches. TBN, Tibetans; CHB, Han Chinese in Beijing; JPT, Japanese in Tokyo, Japan; CEU, Northern Europeans from Utah; YRI, Yoruba in Ibadan, Nigeria.
- The manuscript is currently only minimally discussing how findings are linked to EPAS1 and EGLN1 genes, which show the hallmark signature of positive selection in Tibetans. In fact, the authors' group previously reported male-specific association between EPAS1 SNPs and blood hemoglobin level. Many readers will be intrigued to see a discussion about this point.
According to the reviewer’s suggestion, in the revised manuscript, we added a paragraph to discuss the relationship between our transcriptomic data and the two genes with strong selective signals, i.e. EPAS1 and EGLN1.
“As the gene with the strongest signal of natural selection in Tibetans, EPAS1 has been reported in numerus studies on its contribution to high altitude adaptation. In this study, we detected a significant expression reduction of EPAS1 in the Tibetan UC compared to the high-altitude Han. It was reported that the selected-for EPAS1 variants/haplotype were associated with lower hemoglobin levels in the Tibetan highlanders with a major effect (Beall et al., 2010; Peng et al., 2017), and the low hemoglobin concentration of Tibetans is causally associated with a better reproductive success (Cho et al., 2017). Therefore, we speculate that the selective pressure on EPAS1 is likely through its effect on hemoglobin, rather than directly on the reproductive traits. The down-regulation of EPAS1 in placentas likely reflects a blunted hypoxic response that may improve vasodilation of UC for better blood flow, and eventually leading to the higher BW in Tibetans (He et al., 2023). For EGLN1, another well-known gene in Tibetans, we detected between-population expression difference in the male UC layer, but not in other placental layers. Considering the known adaptation mechanism of EGLN1 is attributed to the two Tibetan-enriched missense mutations, the contribution of EGLN1 to the gene expression changes in the Tibetan UC is unexpected and worth to be explored in the future.”
Reviewer #2 (Public Review):
In this manuscript, the authors use newly-generated, large-scale transcriptomic data along with histological data to attempt to dissect the mechanisms by which individuals with Tibetan ancestry are able to mitigate the negative effects of high elevation on birth weight. They present detailed analyses of the transcriptomic data and find significant sex differences in the placenta transcriptome.
I have significant concerns about the conclusions that are presented. The analyses also lack the information necessary to evaluate their reliability.
The experimental design does not include a low elevation comparison and thus cannot be used to answer questions about how ancestry influences hypoxia responses and thus birthweight at high elevations. Importantly, because the placenta tissues (and trophoblasts specifically) are quickly evolving, there are a priori good reasons to expect to find population differences irrespective of adaptive evolution that might contribute to fetal growth protection. There are also significant details missing in the analyses that are necessary to substantiate and replicate the analyses presented.
Although the datasets are ultimately valuable as reference sets, the absence of low elevation comparisons for Tibetans and Han Chinese individuals undermines the ability of the authors to assess whether differences observed between populations are linked to hypoxia responses or variation in the outcomes of interest (i.e., hypoxia-dependent fetal growth restriction).
We understand the reviewer’s concern about the lack of low-altitude comparison. For the placenta transcriptomic data, actually, we previously studied the comparison of placenta from high-altitude Tibetans and low-altitude Han Chinese, including 63 placentas of Tibetans living at Lhasa (elevation: 3650m) and 14 placentas of Han in Kunming (elevation: 1800m) (Peng et al. 2017). The main finding was that in general, the expression profiles are similar between the high-altitude Tibetans and the low-altitude Han. In particular, most high-altitude Tibetans have a similar level of EPAS1 expression in the placenta as the lowlander Han Chinese, a reflection of Tibetans’ adaptation at altitude. In other words, (Peng et al. 2017). In this study, we observed a significant down-regulation of EPAS1 in the Tibetan UC when compared to Han Chinese living at the same high altitude. Therefore, the observed differences between Tibetans and Han Chinese placenta at high altitude are due to the adaptation of Tibetans.
For phenotypic data, we made a systematical comparison of reproductive outcomes in our previous studies (He et al., 2023; He et al., 2022). We proved that polygenic adaptation of reproduction in Tibetans tends to reduce the chance of preterm birth and eliminate the restriction on fetal development at high altitude. Compared to the high-altitude Han Chinese migrants, the high-altitude Tibetans exhibit a less birth weight reduction and infant mortality induced by hypoxia, similar with the lowland Han Chinese as reference.
In summary, although we cannot make combination analysis with our high-altitude data and the published low-altitude data because of batch effect and difference of sampling strategy, we obtained more supportive evidence for the adaptation of placenta expression regulation in Tibetans. To be objective, we have discussed the limitation of the lack of lowlander placenta data in the Discussion section.
The authors attempt to tackle this phenotypic association by looking for correlations between gene networks (WGCNA) and individual genes with birthweight and other measurements collected at birth. I have some reservations about this approach with only two groups (i.e., missing the lowland comparison), but it is further problematic that the authors do not present data demonstrating that there are differences in birthweight or any other traits between the populations in the samples they collected.
Throughout, I thus find conclusions about the adaptive value and hypoxia-responses made by the authors to be unsubstantiated and/or the data to be inadequate. There are also a gratuitous number of speculative statements about mechanisms by which differential gene expression leads to the protection of birthweight that are not evaluated and thus cannot be substantiated by the data presented.
As currently presented and discussed, these results thus can only be used to evaluate population differences and tissue-specific variation therein.
We understand the reviewer’s point that the observed differences of gene expression between Tibetan natives and Han immigrants living at high altitude might be explained by ancestral divergence, rather than hypoxia-associated response and genetic adaptation of native Tibetans.
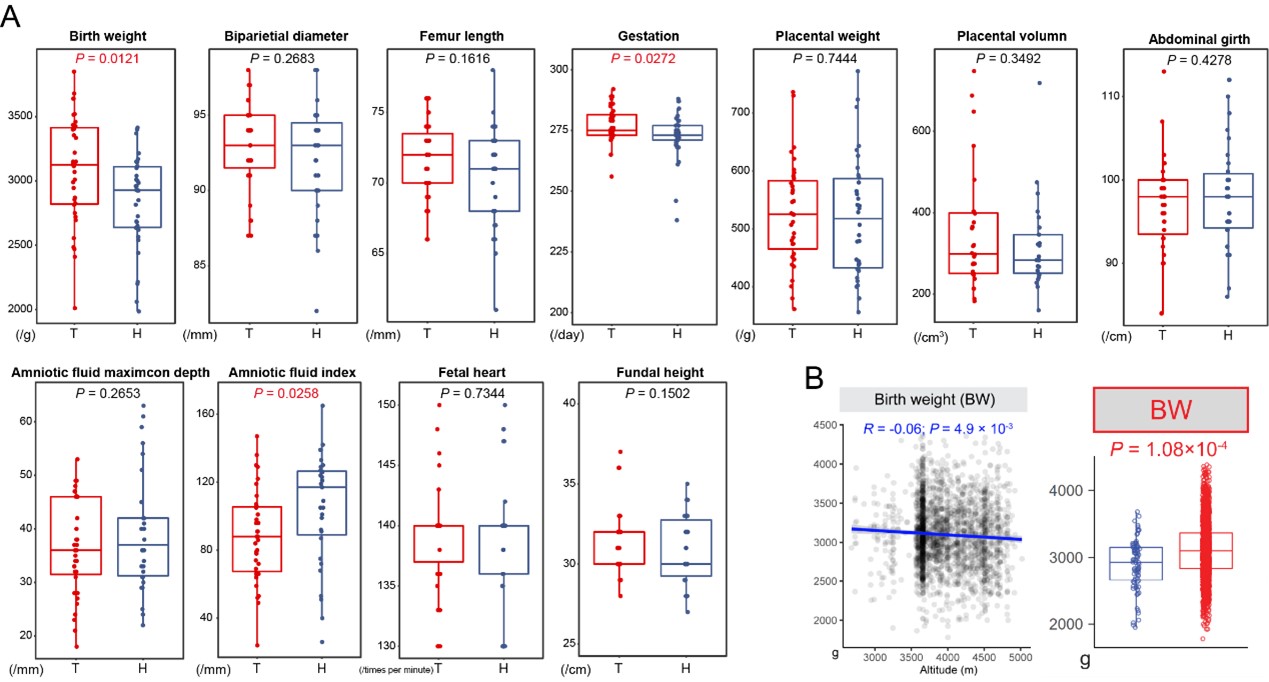
Firstly, we conclude that Tibetans have a better reproductive outcome, not only based on the two highlander groups living at the same altitude, but also relied on the change direction compared to the lowland level. For example, we observed a significant higher BW in Tibetans than Han migrants in our dataset (35 Tibetans vs. 34 Han: p-value = 0.012) (Author response image 5), and in a larger dataset (He et al. 2023) (1,317 Tibetans vs. 87 Han: p-value = 1.1e-6), suggesting an adaptation of Tibetans because BW decreases with the increase of altitude. The logic was the same to the other traits. Following the suggestion of reviewer, we added these phenotype comparisons in the revised manuscripts. The detailed information of the investigated samples and the statistic results were also added as supplementary tables in the revised version.
For the WGCNA, we agree with the reviewer that the detected modules both showing significant correlation with population and other reproductive traits cannot be fully explained by adaptation of Tibetans. Therefore, we tuned down the description of this section and added other possible explanations, such as population differences, in the discussion.
Author response image 5.
Comparison of 11 reproductive traits between Tibetans and Han immigrants. (A) comparison based on the dataset of this study (35 Tibetans vs. 34 Han); (B) correlation between BW and altitude (left panel) and comparison analysis based on the larger sample size (the data were retrieved from (He et al., 2023)). Univariate comparisons of the average of each trait cross population were made by using the ANCOVA test in R aov function with fetal sex and maternal age as covariates.
There is also some important methodological information missing that makes it difficult or impossible to assess the quality of the underlying data and/or reproduce the analyses, further limiting the potential impact of these data:
- Transcriptome data processing and analyses: RNA quality information is not mentioned (i.e., RIN). What # of reads are mapped to annotated regions? How many genes were expressed in each tissue (important for contextualizing the # of DE genes reported - are these a significant proportion of expressed genes or just a small subset?).
According to the reviewer’s suggestion, we added more information about transcriptome data processing and analyses in the revised Methods and Results:
“After RNA extraction, we assessed the RNA integrity and purity using 1% agarose gel electrophoresis. The RIN value of extracted RNA was 7.56 ± 0.71.”
“In total, 10.6 billion reads were mapped to the annotated regions, and 17,283 genes express in all the investigated placenta.”
“We identified 579 differentially expressed genes (DEGs) between Tibetans and Han, accounting for 3.4% of the total number of expressed genes.”
- The methods suggest that DE analyses were run using data that were normalized prior to reading them into DESeq2. DESeq2 has an internal normalization process and should not be used on data that was already normalized. Please clarify how and when normalization was performed.
Actually, we made raw read count matrix as input file when conducting differential analysis using DESeq2, rather than using the normalized data. We have updated our description in the method section of the revised manuscript.
- For enrichment analyses, the background gene set (all expressed genes? all genes in the genome? or only genes expressed in the tissue of interest?) has deterministic effects on the outcomes. The background sets are not specified for any analyses.
Actually, we utilized the genes expressed in placenta as the background gene set for enrichment analyses. The genes with more than two transcripts per million transcripts (TPM) were regarded as an expressed gene, which is commonly used criteria for RNA-seq data.
- In the WGCNA analysis, P-values for correlations of modules with phenotype data (birthweight etc.) should be corrected for multiple testing (i.e., running the module correlation for each outcome variables) and p.adjust used to evaluate associations to limit false positives given the large number of correlations being run.
As we explained in response to comment#2 of Reviwer-1, we used a more stringent significant threshold of p-value = 0.0125 (0.05/4) as the final threshold to correct the multiple testing brought by multiple traits in the WGCNA analysis.
- The plots for umbilical histological data (Fig 5 C) contain more than 5 points, but the use of replicate sections is not specified. If replicate sections were used, the authors should control for non-independence of replicate sections in their analyses (i.e., random effects model).
We did not use replicate sections. Figure 5C shows the umbilical artery intima and media. Because each human umbilical cord includes two umbilical arteries, the 5 vs. 5 individual comparison generates 10 vs. 10 umbilical artery comparison. To be clearer, we added an explanation in the revised manuscript.
On more minor notes:
There is significant and relevant published data on sex differences and hypoxia in rodents (see Cuffe et al 2014, "Mid- to late-term hypoxia in the mouse alters placental morphology, glucocorticoid regulatory pathways, and nutrient transporters in a sex-specific manner" and review by Siragher and Sferuzzi-Perro 2021, "Placental hypoxia: What have we learnt from small animal models?"), and historical work reporting sex differences in placental traits associated with high elevation adaptation in Andeans (series of publications by Moira Jackson in the late 1980s, reviewed in Wilsterman and Cheviron 2021, "Fetal growth, high altitude, and evolutionary adaptation: A new perspective").
We thank the reviewer for the constructive comments on literature review. We have cited and discussed them in the revised manuscript.
Reviewer #3 (Public Review):
More than 80 million people live at high altitude. This impacts health outcomes, including those related to pregnancy. Longer-lived populations at high altitudes, such as the Tibetan and Andean populations show partial protection against the negative health effects of high altitude. The paper by Yue sought to determine the mechanisms by which the placenta of Tibetans may have adapted to minimise the negative effect of high altitude on fetal growth outcomes. It compared placentas from pregnancies from Tibetans to those from the Han Chinese. It employed RNAseq profiling of different regions of the placenta and fetal membranes, with some follow-up of histological changes in umbilical cord structure and placental structure. The study also explored the contribution of fetal sex in these phenotypic outcomes.
A key strength of the study is the large sample sizes for the RNAseq analysis, the analysis of different parts of the placenta and fetal membranes, and the assessment of fetal sex differences.
A main weakness is that this study, and its conclusions, largely rely on transcriptomic changes informed by RNAseq. Changes in genes and pathways identified through bioinformatic analysis were not verified by alternate methods, such as by western blotting, which would add weight to the strength of the data and its interpretations. There is also a lack of description of patient characteristics, so the reader is unable to make their own judgments on how placental changes may link to pregnancy outcomes. Another weakness is that the histological analyses were performed on n=5 per group and were rudimentary in nature.
For the weakness raised by the reviewer, here are our responses:
(1) Considering that our conclusions largely rely on the transcriptomic data, we agree with reviewer that more experiments are needed to validate the results from our transcriptomic data. However, this study was mainly aimed to provide a transcriptomic landscape of high-altitude placenta, and to characterize the gene-expression difference between native Tibetans and Han migrants. The molecular mechanism exploration is not the main task of this study, and more validation experiments are warranted in the future.
(2) For the lack of description of patient characteristics, actually, we provided three level results on the placental changes of Tibetans: macroscopic phenotypes (higher placental weight and volume), histological phenotypes (larger umbilical vein walls and umbilical artery intima and media; lower syncytial knots/villi ratios) and transcriptomic phenotypes (DEG and differential modules). Combined with the previous studies, these placenta changes suggest a better reproductive outcome. For example, the placenta volume shows a significantly positive correlation with birth weight (R = 0.31, p-value = 2.5e-16), therefore, the larger placenta volume of Tibetans is beneficial to fetal development at high altitude. In addition, the larger umbilical vein wall and umbilical artery intima and media of Tibetans can explain their adaptation in preventing preeclampsia.
(3) For the sample size of histological analyses, we understand the reviewer’s concern that 5 vs. 5 samples are not large in histological analyses. This is because it was difficult to collect high-altitude Han placenta samples, and we only got 13 Han samples, from which we selected 5 infant sex matched samples.
References
Beall, C.M., Cavalleri, G.L., Deng, L.B., Elston, R.C., Gao, Y., Knight, J., Li, C.H., Li, J.C., Liang, Y., McCormack, M., et al. (2010). Natural selection on EPAS1 (HIF2 alpha) associated with low hemoglobin concentration in Tibetan highlanders. P Natl Acad Sci USA 107, 11459-11464.
Cho, J.I., Basnyat, B., Jeong, C., Di Rienzo, A., Childs, G., Craig, S.R., Sun, J., and Beall, C.M. (2017). Ethnically Tibetan women in Nepal with low hemoglobin concentration have better reproductive outcomes. Evol Med Public Health 2017, 82-96. He, Y., Guo, Y., Zheng, W., Yue, T., Zhang, H., Wang, B., Feng, Z., Ouzhuluobu, Cui, C., Liu, K., et al. (2023). Polygenic adaptation leads to a higher reproductive fitness of native Tibetans at high altitude. Curr Biol.
He, Y., Li, J., Yue, T., Zheng, W., Guo, Y., Zhang, H., Chen, L., Li, C., Li, H., Cui, C., et al. (2022). Seasonality and Sex-Biased Fluctuation of Birth Weight in Tibetan Populations. Phenomics 2, 64-71.
Peng, Y., Cui, C., He, Y., Ouzhuluobu, Zhang, H., Yang, D., Zhang, Q., Bianbazhuoma, Yang, L., He, Y., et al. (2017). Down-Regulation of EPAS1 Transcription and Genetic Adaptation of Tibetans to High-Altitude Hypoxia. Mol Biol Evol 34, 818-830.
Racimo, F., Berg, J.J., and Pickrell, J.K. (2018). Detecting Polygenic Adaptation in Admixture Graphs. Genetics 208, 1565-1584.
-
eLife assessment
This fundamental study reports differential expression of key genes in full-term placenta between Tibetans and Han Chinese at high elevations, which are more pronounced in the placentae of male than in female fetuses. If validated as functionally relevant, these results will help us understand how human populations adapt to high elevation by mitigating the negative effects of low oxygen on fetal growth. At this time, while the differential gene expression analyses are solid, the downstream analyses offer incomplete support for the connection to hypoxia-specific responses and adaptive genetic variation.
-
Reviewer #1 (Public Review):
The revised manuscript new presented 1) a permutation-based test for the significance of the overlap between DEGs and genes with positive selection signals in Tibetans, and 2) polygenic adaptation test for the eQTLs. I make my suggestions in detail as below:
Major Comments
1. My previous concern regarding the DEG analysis remains unresolved. Although the authors agreed in their response that the difference between the male- and female-specific DEGs are insufficient to the difference between sex-combined and sex-specific DEGs (Figure S6). However, the results section still states the opposite pattern between males and females as a decisive reason for the difference (p. 9, lines 236-239). Again, I would like to recommend the authors to test alternative ways of analysis to boost statistical power for DEG …
Reviewer #1 (Public Review):
The revised manuscript new presented 1) a permutation-based test for the significance of the overlap between DEGs and genes with positive selection signals in Tibetans, and 2) polygenic adaptation test for the eQTLs. I make my suggestions in detail as below:
Major Comments
1. My previous concern regarding the DEG analysis remains unresolved. Although the authors agreed in their response that the difference between the male- and female-specific DEGs are insufficient to the difference between sex-combined and sex-specific DEGs (Figure S6). However, the results section still states the opposite pattern between males and females as a decisive reason for the difference (p. 9, lines 236-239). Again, I would like to recommend the authors to test alternative ways of analysis to boost statistical power for DEG detection other than simply splitting data into males and females and performing analysis in each subset. For example, the authors may consider utilizing gene by environment interaction analysis schemes here biological sex as an environmental factor.
2. Multiple testing schemes are still sub-optimal in some cases. Most of all, the p-values in the WGCNA analysis (p. 11), the authors corrected for the number of traits (n=12) after adjusting for the correlation between them. However, they did not mention whether they counted for the number of modules they tested at all (n=136 and 161 for males and females, respectively). Whether they account for the number of modules will make a substantial difference in the significance threshold, please incorporate and describe a proper multiple testing scheme for this analysis.
3. Evidence for natural selection on the observed DEG pattern is still weak and not properly described.
- For the overlap between DEGs and TSNGs, the authors introduced a permutation-based test, but used a total set of genes in the human genome as a comparison set (p. 25, lines 699-700). I believe that the authors should sample random sets of genes from those already expressed in each tissue to make a fair comparison.
- The entire polygraph analysis for polygenic adaptation is poorly described. The current version of the Methods does not clarify i) for which genes the eQTLs are discovered, 2) how the authors performed the eQTL analysis, iii) how the authors polarized the effect, and iv) how they set up a comparison between the eQTLs and the others.
-
Reviewer #2 (Public Review):
More than 80 million people live at high altitude. This impacts health outcomes, including those related to pregnancy. Longer-lived populations at high altitudes, such as the Tibetan and Andean populations show partial protection against the negative health effects of high altitude. The paper by Yue sought to determine the mechanisms by which the placenta of Tibetans may have adapted to minimise the negative effect of high altitude on fetal growth outcomes. It compared placentas from pregnancies from Tibetans to those from the Han Chinese. It employed RNAseq profiling of different regions of the placenta and fetal membranes, with some follow-up of histological changes in umbilical cord structure and placental structure. The study also explored the contribution of fetal sex in these phenotypic outcomes.
A key …
Reviewer #2 (Public Review):
More than 80 million people live at high altitude. This impacts health outcomes, including those related to pregnancy. Longer-lived populations at high altitudes, such as the Tibetan and Andean populations show partial protection against the negative health effects of high altitude. The paper by Yue sought to determine the mechanisms by which the placenta of Tibetans may have adapted to minimise the negative effect of high altitude on fetal growth outcomes. It compared placentas from pregnancies from Tibetans to those from the Han Chinese. It employed RNAseq profiling of different regions of the placenta and fetal membranes, with some follow-up of histological changes in umbilical cord structure and placental structure. The study also explored the contribution of fetal sex in these phenotypic outcomes.
A key strength of the study is the large sample sizes for the RNAseq analysis, the analysis of different parts of the placenta and fetal membranes, and the assessment of fetal sex differences.
A main weakness is that this study, and its conclusions, largely rely on transcriptomic changes informed by RNAseq. Changes in genes and pathways identified through bioinformatic analysis were not verified by alternate methods, such as by western blotting, which would add weight to the strength of the data and its interpretations. There is also a lack of description of patient characteristics, so the reader is unable to make their own judgments on how placental changes may link to pregnancy outcomes. Another weakness is that the histological analyses were performed on n=5 per group and were rudimentary in nature.
-
-
eLife assessment
This study reports the fundamental finding of differential expression of key genes in full-term placenta between Tibetans and Han Chinese at high elevations, which are more pronounced in the placentas of male fetuses than in female ones. If confirmed, these results will help us understand how human populations adapt to high elevation by mitigating the negative effects of low oxygen on fetal growth. However, although the differential gene expression reported is solid, the downstream analyses offer only incomplete support for its connection to hypoxia-specific responses, adaptive genetic variation, and pregnancy outcomes.
-
Reviewer #1 (Public Review):
In this manuscript, the authors investigate differences between Tibetans and Han Chinese at altitude in terms of placental transcriptomes during full-term pregnancy. Most importantly, they found that the inter-population differentiation is mostly male-specific and the observed direction of transcriptional differentiation seems to be adaptive at high altitude. In general, it is of great importance and provides new insights into the functional basis of Tibetan high-altitude adaptations, which so far have been mostly studied via population genetic measures only. More specifically, I firmly believe that we need more phenotype data (including molecular phenotypes such as gene expression data) to fully understand Tibetan adaptations to high altitude, and this manuscript is a rare example of such a study. I have a …
Reviewer #1 (Public Review):
In this manuscript, the authors investigate differences between Tibetans and Han Chinese at altitude in terms of placental transcriptomes during full-term pregnancy. Most importantly, they found that the inter-population differentiation is mostly male-specific and the observed direction of transcriptional differentiation seems to be adaptive at high altitude. In general, it is of great importance and provides new insights into the functional basis of Tibetan high-altitude adaptations, which so far have been mostly studied via population genetic measures only. More specifically, I firmly believe that we need more phenotype data (including molecular phenotypes such as gene expression data) to fully understand Tibetan adaptations to high altitude, and this manuscript is a rare example of such a study. I have a few suggestions and/or questions with which I hope to improve the manuscript further, especially in terms of 1) testing if the observed DEG patterns are truly adaptive, and 2) how and whether the findings in this study can be linked to EPAS1 and EGLN1, the signature adaptation genes in Tibetans.
Major Comments:
1. The DEG analysis is the most central result in this manuscript, but the discrepancy between sex-combined and sex-specific DEGs is quite mind-boggling. For those that were differentially expressed in the sex-specific sets but not in the sex-combined one, the authors suggest an opposite direction of DE as an explanation (page 11, Figure S5). But Figure S5A does not show such a trend, showing that down-regulated genes in males are mostly not at all differentially expressed in females. Figure S6B does show such a trend, but it doesn't seem to be a dominant explanation. I would like to recommend the authors test alternative ways of analysis to boost statistical power for DEG detection other than simply splitting data into males and females and performing analysis in each subset. For example, the authors may consider utilizing gene-by-environment interaction analysis schemes here biological sex as an environmental factor.
2. Please clarify how the authors handled multiple testing correction of p-values.
3. The "natural selection acts on the placental DEGs ..." section is potentially misleading readers to assume that the manuscript reports evidence for positive selection on the observed DEG pattern between Tibetans and Han, which is not.
a) Currently the section simply describes an overlap between DEGs and a set of 192 genes likely under positive selection in Tibetans (TSNGs). The overlap is quite small, leading to only 13 genes in total (Figure 6). The authors are currently not providing any statistical measure of whether this overlap is significantly enriched or at the level expected for random sampling.
b) The authors are describing sets of DEGs that seem to affect important phenotypic changes in a consistent and adaptive direction. A relevant form of natural selection for this situation may be polygenic adaptation while the authors only consider strong positive selection at a single variant/gene level.
c) The manuscript is currently providing no eQTL information that can explain the differential expression of key genes. The authors can actually do this based on the genotype and expression data of the individuals in this study. Combining eQTL info, they can set up a test for polygenic adaptation (e.g. Berg and Coop; https://journals.plos.org/plosgenetics/article?id=10.1371/journal.pgen.1004412). This will provide a powerful and direct test for the adaptiveness of the observed DEG pattern.4. The manuscript is currently only minimally discussing how findings are linked to EPAS1 and EGLN1 genes, which show the hallmark signature of positive selection in Tibetans. In fact, the authors' group previously reported male-specific association between EPAS1 SNPs and blood hemoglobin level. Many readers will be intrigued to see a discussion about this point.
-
Reviewer #2 (Public Review):
In this manuscript, the authors use newly-generated, large-scale transcriptomic data along with histological data to attempt to dissect the mechanisms by which individuals with Tibetan ancestry are able to mitigate the negative effects of high elevation on birth weight. They present detailed analyses of the transcriptomic data and find significant sex differences in the placenta transcriptome.
I have significant concerns about the conclusions that are presented. The analyses also lack the information necessary to evaluate their reliability.
The experimental design does not include a low elevation comparison and thus cannot be used to answer questions about how ancestry influences hypoxia responses and thus birthweight at high elevations. Importantly, because the placenta tissues (and trophoblasts …
Reviewer #2 (Public Review):
In this manuscript, the authors use newly-generated, large-scale transcriptomic data along with histological data to attempt to dissect the mechanisms by which individuals with Tibetan ancestry are able to mitigate the negative effects of high elevation on birth weight. They present detailed analyses of the transcriptomic data and find significant sex differences in the placenta transcriptome.
I have significant concerns about the conclusions that are presented. The analyses also lack the information necessary to evaluate their reliability.
The experimental design does not include a low elevation comparison and thus cannot be used to answer questions about how ancestry influences hypoxia responses and thus birthweight at high elevations. Importantly, because the placenta tissues (and trophoblasts specifically) are quickly evolving, there are a priori good reasons to expect to find population differences irrespective of adaptive evolution that might contribute to fetal growth protection. There are also significant details missing in the analyses that are necessary to substantiate and replicate the analyses presented.
Although the datasets are ultimately valuable as reference sets, the absence of low elevation comparisons for Tibetans and Han Chinese individuals undermines the ability of the authors to assess whether differences observed between populations are linked to hypoxia responses or variation in the outcomes of interest (i.e., hypoxia-dependent fetal growth restriction).
The authors attempt to tackle this phenotypic association by looking for correlations between gene networks (WGCNA) and individual genes with birthweight and other measurements collected at birth. I have some reservations about this approach with only two groups (i.e., missing the lowland comparison), but it is further problematic that the authors do not present data demonstrating that there are differences in birthweight or any other traits between the populations in the samples they collected.
Throughout, I thus find conclusions about the adaptive value and hypoxia-responses made by the authors to be unsubstantiated and/or the data to be inadequate. There are also a gratuitous number of speculative statements about mechanisms by which differential gene expression leads to the protection of birthweight that are not evaluated and thus cannot be substantiated by the data presented.
As currently presented and discussed, these results thus can only be used to evaluate population differences and tissue-specific variation therein.
There is also some important methodological information missing that makes it difficult or impossible to assess the quality of the underlying data and/or reproduce the analyses, further limiting the potential impact of these data:
1. Transcriptome data processing and analyses: RNA quality information is not mentioned (i.e., RIN). What # of reads are mapped to annotated regions? How many genes were expressed in each tissue (important for contextualizing the # of DE genes reported - are these a significant proportion of expressed genes or just a small subset?).
2. The methods suggest that DE analyses were run using data that were normalized prior to reading them into DESeq2. DESeq2 has an internal normalization process and should not be used on data that was already normalized. Please clarify how and when normalization was performed.
3. For enrichment analyses, the background gene set (all expressed genes? all genes in the genome? or only genes expressed in the tissue of interest?) has deterministic effects on the outcomes. The background sets are not specified for any analyses.
4. In the WGCNA analysis, P-values for correlations of modules with phenotype data (birthweight etc.) should be corrected for multiple testing (i.e., running the module correlation for each outcome variables) and p.adjust used to evaluate associations to limit false positives given the large number of correlations being run.
6. The plots for umbilical histological data (Fig 5 C) contain more than 5 points, but the use of replicate sections is not specified. If replicate sections were used, the authors should control for non-independence of replicate sections in their analyses (i.e., random effects model).On more minor notes:
There is significant and relevant published data on sex differences and hypoxia in rodents (see Cuffe et al 2014, "Mid- to late-term hypoxia in the mouse alters placental morphology, glucocorticoid regulatory pathways, and nutrient transporters in a sex-specific manner" and review by Siragher and Sferuzzi-Perro 2021, "Placental hypoxia: What have we learnt from small animal models?"), and historical work reporting sex differences in placental traits associated with high elevation adaptation in Andeans (series of publications by Moira Jackson in the late 1980s, reviewed in Wilsterman and Cheviron 2021, "Fetal growth, high altitude, and evolutionary adaptation: A new perspective"). -
Reviewer #3 (Public Review):
More than 80 million people live at high altitude. This impacts health outcomes, including those related to pregnancy. Longer-lived populations at high altitudes, such as the Tibetan and Andean populations show partial protection against the negative health effects of high altitude. The paper by Yue sought to determine the mechanisms by which the placenta of Tibetans may have adapted to minimise the negative effect of high altitude on fetal growth outcomes. It compared placentas from pregnancies from Tibetans to those from the Han Chinese. It employed RNAseq profiling of different regions of the placenta and fetal membranes, with some follow-up of histological changes in umbilical cord structure and placental structure. The study also explored the contribution of fetal sex in these phenotypic outcomes.
A key …
Reviewer #3 (Public Review):
More than 80 million people live at high altitude. This impacts health outcomes, including those related to pregnancy. Longer-lived populations at high altitudes, such as the Tibetan and Andean populations show partial protection against the negative health effects of high altitude. The paper by Yue sought to determine the mechanisms by which the placenta of Tibetans may have adapted to minimise the negative effect of high altitude on fetal growth outcomes. It compared placentas from pregnancies from Tibetans to those from the Han Chinese. It employed RNAseq profiling of different regions of the placenta and fetal membranes, with some follow-up of histological changes in umbilical cord structure and placental structure. The study also explored the contribution of fetal sex in these phenotypic outcomes.
A key strength of the study is the large sample sizes for the RNAseq analysis, the analysis of different parts of the placenta and fetal membranes, and the assessment of fetal sex differences.
A main weakness is that this study, and its conclusions, largely rely on transcriptomic changes informed by RNAseq. Changes in genes and pathways identified through bioinformatic analysis were not verified by alternate methods, such as by western blotting, which would add weight to the strength of the data and its interpretations. There is also a lack of description of patient characteristics, so the reader is unable to make their own judgments on how placental changes may link to pregnancy outcomes. Another weakness is that the histological analyses were performed on n=5 per group and were rudimentary in nature.
-
