Rab12 is a regulator of LRRK2 and its activation by damaged lysosomes
Curation statements for this article:-
Curated by eLife

eLife assessment
This valuable study shows that Rab12 is required for LRRK2 activation. However, while some of the data are compelling, some claims, especially the ones related to LRRK2's membrane association are not supported. Addressing discrepancies between figures (pointed out by reviewers) and re-writing certain sections will greatly improve this manuscript.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Leucine-rich repeat kinase 2 (LRRK2) variants associated with Parkinson’s disease (PD) and Crohn’s disease lead to increased phosphorylation of its Rab substrates. While it has been recently shown that perturbations in cellular homeostasis including lysosomal damage can increase LRRK2 activity and localization to lysosomes, the molecular mechanisms by which LRRK2 activity is regulated have remained poorly defined. We performed a targeted siRNA screen to identify regulators of LRRK2 activity and identified Rab12 as a novel modulator of LRRK2-dependent phosphorylation of one of its substrates, Rab10. Using a combination of imaging and immunopurification methods to isolate lysosomes, we demonstrated that Rab12 is actively recruited to damaged lysosomes and leads to a local and LRRK2-dependent increase in Rab10 phosphorylation. PD-linked variants, including LRRK2 R1441G and VPS35 D620N, lead to increased recruitment of LRRK2 to the lysosome and a local elevation in lysosomal levels of pT73 Rab10. Together, these data suggest a conserved mechanism by which Rab12, in response to damage or expression of PD-associated variants, facilitates the recruitment of LRRK2 and phosphorylation of its Rab substrate(s) at the lysosome.
Article activity feed
-

-

Author response
Reviewer #1 (Public Review):
This careful study reports the importance of Rab12 for Parkinson's disease associated LRRK2 kinase activity in cells. The authors carried out a targeted siRNA screen of Rab substrates and found lower pRab10 levels in cells depleted of Rab12. It has previously been reported that LLOMe treatment of cells breaks lysosomes and with time, leads to major activation of LRRK2 kinase. Here they show that LLOMe-induced kinase activation requires Rab12 and does not require Rab12 phosphorylation to show the effect.
We thank the reviewer for their comments regarding the carefulness and importance of our work and for their specific feedback which has substantially improved our revised manuscript.
- Throughout the text, the authors claim that "Rab12 is required for LRRK2 dependent phosphorylation" (Page …
Author response
Reviewer #1 (Public Review):
This careful study reports the importance of Rab12 for Parkinson's disease associated LRRK2 kinase activity in cells. The authors carried out a targeted siRNA screen of Rab substrates and found lower pRab10 levels in cells depleted of Rab12. It has previously been reported that LLOMe treatment of cells breaks lysosomes and with time, leads to major activation of LRRK2 kinase. Here they show that LLOMe-induced kinase activation requires Rab12 and does not require Rab12 phosphorylation to show the effect.
We thank the reviewer for their comments regarding the carefulness and importance of our work and for their specific feedback which has substantially improved our revised manuscript.
- Throughout the text, the authors claim that "Rab12 is required for LRRK2 dependent phosphorylation" (Page 4 line 78; Page 9 line 153; Page 22 line 421). This is not correct according to Figure 1 Figure Supp 1B - there is still pRab10. It is correct only in relation to the LLOMe activation. Please correct this error.
We appreciate the reviewer’s comment around the requirement of Rab12 for LRRK2-dependent phosphorylation of Rab10 and question regarding whether this is relevant under baseline conditions or only in relation to LLOMe activation. Using our MSD-based assay to quantify pT73 Rab10 levels under basal conditions, we observed a similar reduction in Rab10 phosphorylation when we knockdown Rab12 as we also observed with LRRK2 knockdown (Figure 1A). Further, we see comparable reduction in Rab10 phosphorylation in RAB12 KO cells as that observed in LRRK2 KO cells using our MSD-based assay (Figure 2A and B). Based on this data, we believe Rab12 is a key regulator of LRRK2 activation under basal conditions without additional lysosomal damage. However, as the reviewer noted, we do observe some residual Rab10 phosphorylation upon Rab12 knockdown when assessed by western blot analysis (Figure 1D and Figure 1- figure supplement 1). A similar signal is observed upon LRRK2 knockdown, which may suggest that some small amount of Rab10 phosphorylation may be mediated by another kinase in this cell model. Nevertheless, we appreciate this reviewer’s point and have therefore modified the text to remove any reference to Rab12 being required for LRRK2-dependent Rab phosphorylation and now instead refer to Rab12 as a regulator of LRRK2 activity.
As noted by the reviewer, our data does suggest that Rab12 is required for the increase in Rab10 phosphorylation observed following LLOMe treatment to elicit lysosomal damage, and we now refer to this appropriately throughout the text.
- The authors conclude that Rab12 recruitment precedes that of LRRK2 but the rate of recruitment (slopes of curves in 3F and G) is actually faster for LRRK2 than for Rab12 with no proof that Rab12 is faster-please modify the text-it looks more like coordinated recruitment.
The reviewer raises an excellent point regarding our ability to delineate whether Rab12 recruitment precedes that of LRRK2 on lysosomes following LLOMe treatment. As noted by the reviewer, we do see both the recruitment of Rab12 and LRRK2 to lysosomes increase on a similar timescale, so we cannot truly resolve whether Rab12 recruitment precedes LRRK2 recruitment in our studies. Based on this, we have modified the text to emphasize that this data supports coordinated recruitment, as suggested, and we have further removed any mention of Rab12 preceding LRRK2. The specific change is as follows “Rab12 colocalization with LRRK2 increased over time following LLOMe treatment, supporting potential coordinated recruitment of these proteins to lysosomes upon damage (Figure 3I). Together, these data demonstrate that Rab12 and LRRK2 both associate with lysosomes following membrane rupture.” and can be found on lines 460-463 of the updated manuscript.
- The title is misleading because the authors do not show that Rab12 promotes LRRK2 membrane association. This would require Rab12 to be sufficient to localize LRRK2 to a mislocalized Rab12. The authors DO show that Rab12 is needed for the massive LLOME activation at lysosomes. Please re-word the title.
To address the reviewer’s concern regarding the title of our manuscript, we have modified the title from “Rab12 regulates LRRK2 activity by promoting its localization to lysosomes” to “Rab12 regulates LRRK2 activity by facilitating its localization to lysosomes” to soften the language around the sufficiency of Rab12 in regulating the localization of LRRK2 to lysosomes. We show that Rab12 deletion significantly reduces LRRK2 activity (as assessed by Rab10 phosphorylation on lysosomes) and significantly increases the localization of LRRK2 to lysosomes upon lysosomal damage. The updated title better reflects the regulatory role of Rab12 in modulating LRRK2 activity, and we thank the reviewer for their suggestion to modify this accordingly.
Reviewer #2 (Public Review):
This study shows that rab12 has a role in the phosphorylation of rab10 by LRRK2. Many publications have previously focused on the phosphorylation targets of LRRK2 and the significance of many remains unclear, but the study of LRRK2 activation has mostly focused on the role of disease-associated mutations (in LRRK2 and VPS35) and rab29. The work is performed entirely in an alveolar lung cell line, limiting relevance for the nervous system. Nonetheless, the authors take advantage of this simplified system to explore the mechanism by which rab12 activates LRRK2. In general, the work is performed very carefully with appropriate controls, excluding trivial explanations for the results, but there are several serious problems with the experiments and in particular the interpretation.
We appreciate the reviewer’s comments regarding the rigor of our work and the potential impact of our studies to address a key unanswered question in the field regarding the mechanisms by which LRRK2 activation is mediated. Our studies focused on the A549 cell model given its high endogenous expression of LRRK2 and Rab10, and this cell line provided a simple system to investigate the mechanism and impact of Rab12-dependent regulation of LRRK2 activity. We agree with the reviewer that future studies are warranted to understand whether similar Rab12-dependent regulation of LRRK2 occurs in relevant CNS cell types.
First, the authors note that rab29 appears to have a smaller or no effect when knocked down in these cells. However, the quantitation (Fig1-S1A) shows a much less significant knockdown of rab29 than rab12, so it would be important to repeat this with better knockdown or preferably a KO (by CRISPR) before making this conclusion. And the relationship to rab29 is important, so if a better KD or KO shows an effect, it would be important to assess by knocking down rab12 in the rab29 KO background.
The reviewer raises a good point regarding the importance of confirming that loss of Rab29 has no effect on Rab10 phosphorylation. To address potential concerns about insufficient Rab29 knockdown, we measured the levels of pT73 Rab10 in RAB29 KO A549 cells by MSD-based analysis. RAB29 deletion had no effect on Rab10 phosphorylation, confirming findings from our RAB siRNA screen and the observations of Dario Alessi’s group reported previously (Kalogeropulou et al Biochem J 2020; PMID: 33135724). We have included this new data into our updated manuscript in Figure 1- figure supplement 1 and comment on it on page 6 in the updated Results section.
Secondly, the knockdown of rab12 generally has a strong effect on the phosphorylation of the LRRK2 substrate rab10 but I could not find an experiment that shows whether rab12 has any effect on the residual phosphorylation of rab10 in the LRRK2 KO. There is not much phosphorylation left in the absence of LRRK2 but maybe this depends on rab12 just as much as in cells with LRRK2 and rab12 is operating independently of LRRK2, either through a different kinase or simply by making rab10 more available for phosphorylation. The epistasis experiment is crucial to address this possibility. To establish the connection to LRRK2, it would also help to compare the effect of rab12 KD on the phosphorylation of selected rabs that do or do not depend on LRRK2.
The reviewer raises an interesting question regarding whether Rab12 can further reduce Rab10 phosphorylation independently of LRRK2. Using our quantitative MSD-based assay, we observe that pRab10 levels are at the lower limits of detection of the assay in LRRK2 KO A549 cells. Unfortunately, this means that we are unable to detect whether there might be any additional minor reduction in Rab10 phosphorylation with Rab12 knockdown in LRRK2 KO cells. We cannot rule out that Rab12 may play a LRRK2-independent role in regulating Rab10 phosphorylation in other cell lines, and future studies are warranted to explore whether Rab12 knockdown can further reduce Rab10 phosphorylation in other systems, including in CNS cells.
Regarding exploring the effects of RAB12 knockdown on the phosphorylation of other Rabs, we also assessed the impact of RAB12 KO on phosphorylation of another LRRK2-Rab substrate, Rab8a. We observed a strong reduction in pT72 Rab8a levels in RAB12 KO cells compared to wildtype cells, suggesting the impact of RAB12 deletion extends beyond Rab10 (see representative western blot in Author response image 1). Due to potential concerns with the selectivity of the pT72 Rab8a antibody (potentially detecting the phosphorylation of other LRRK2-Rabs), we cannot definitively demonstrate that Rab12 mediates the phosphorylation of other Rabs. This question should be revisited when additional phospho-Rab antibodies become available that enable us to selectively detect LRRK2-dependent phosphorylation of additional Rab substrates under endogenous expression conditions.
Author response image 1.
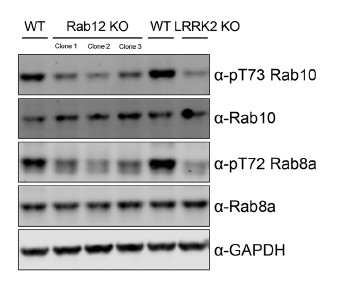
A strength of the work is the demonstration of p-rab10 recruitment to lysosomes by biochemistry and imaging. The demonstration that LRRK2 is required for this by biochemistry (Fig 4A) is very important but it would also be good to determine whether the requirement for LRRK2 extends to imaging. In support of a causal relationship, the authors also state that lysosomal accumulation of rab12 precedes LRRK2 but the data do not show this. Imaging with and without LRRK2 would provide more compelling evidence for a causative role.
We thank the reviewer for their suggestion to assess Rab12 recruitment to damaged lysosomes with and without LRRK2 using imaging-based analyses to add confidence to our findings from biochemical approaches. To address this comment, we have imaged the recruitment of mCherry-tagged Rab12 to lysosomes (as assessed using an antibody against endogenous LAMP1) and observed a significant increase in Rab12 levels on lysosomes following LLOMe treatment. This occurs to a similar extent in LRRK2 KO A549 cells, suggesting that Rab12 is an upstream regulator of LRRK2 activity. This new data has been incorporated into the revised manuscript (Figure 3E) and is presented on page 20 of the updated manuscript.
Our conclusions on this are further strengthened by new data assessing Rab12 recruitment to lysosomes using orthogonal analysis of isolated lysosomes biochemically. Using the Lyso-IP method, we observed a strong increase in the levels of Rab12 on lysosomes following LLOMe treatment that was maintained in LRRK2 KO cells. These data have been added to the updated manuscript (new data added to Figure 3- figure supplement 1).
Together, these data support our hypothesis that Rab12 recruitment to damaged lysosomes is upstream, and independent, of LRRK2.
The authors also touch base with PD mutations, showing that loss of rab12 reduces the phosphorylation of rab10. However, it is interesting that loss of rab12 has the same effect with R1441G LRRK2 and D620N VPS35 as it does in controls. This suggests that the effect of rab12 does not depend on the extent of LRRK2 activation. It is also surprising that R1441G LRRK2 does not increase p-rab10 phosphorylation (Fig 2G) as suggested in the literature and stated in the text.
We agree with the reviewer that it is quite interesting that RAB12 knockdown significantly attenuates Rab10 phosphorylation in the context of PD-linked variants in addition to that observed in wildtype cells basally and after LLOMe treatment. As noted by the reviewer, we did not observe increased levels of phospho-Rab10 in LRRK2 R1441G KI A549 cells at the whole cell level (Figure 2G). However, we observed a significant increase in Rab10 phosphorylation on isolated lysosomes from LRRK2 R1441G KI cells compared to WT cells (Figure 4B). This may suggest that the LRRK2 R1441G variant leads to a more modest increase in LRRK2 activity in this cell model. Previous studies in MEFs from LRRK2 R1441G KI mice or neutrophils from human subjects that carry the LRRK2 R1441G variant showed a 3-4 fold increase in Rab10 phosphorylation (Fan et al Acta Neuropathol 2021 PMID: 34125248 and Karaye et al Mol Cell Proteomics 2020 PMID: 32601174), supporting that this variant does lead to increased Rab10 phosphorylation and that the extent of LRRK2 activation may vary across different cell types.
Most important, the final figure suggests that PD-associated mutations in LRRK2 and VPS35 occlude the effect of lysosomal disruption on lysosomal recruitment of LRRK2 (Fig 4D) but do not impair the phosphorylation of rab10 also triggered by lysosomal disruption (4A-C). Phosphorylation of this target thus appears to be regulated independently of LRRK2 recruitment to the lysosome, suggesting another level of control (perhaps of kinase activity rather than localization) that has not been considered.
The reviewer suggests an interesting hypothesis around the existence of additional levels of control beyond the lysosomal levels of LRRK2 to lead to increased Rab10 phosphorylation of lysosomes. Given the variability we have observed in measuring endogenous LRRK2 levels on lysosomes, we performed two additional replicates to assess lysosomal LRRK2 levels in LRRK2 R1441G KI and VPS35 D620N KI cells at baseline and after treatment with LLOMe. We observed a significant increase in LRRK2 levels on lysosomes in cells expressing either PD-linked variant and a trend toward a further increase in the levels of LRRK2 on lysosomes after LLOMe treatment in these cells (Figure 4D in the updated manuscript). We have updated the text on page 24 to reflect this change, suggesting that the PD-linked variants do not fully occlude the effect of lysosomal disruption on the lysosomal recruitment of LRRK2.
LLOMe treatment leads to a stronger increase in Rab10 phosphorylation on lysosomes from LRRK2 R1441G and VPS35 D620N cells compared to the modest increase in LRRK2 levels observed. This could suggest that, as the reviewer noted, additional mechanisms beyond increased lysosomal localization of LRRK2 may be driving the robust increase in Rab10 phosphorylation observed. We have modified the results section on lines 548-551 to highlight this possibility: “Rab10 phosphorylation showed a more significant increase in response to LLOMe treatment than LRRK2 on lysosomes from LRRK2 R1441G and VPS35 D620N KI cells, suggesting that there may be more regulation beyond the enhanced proximity between LRRK2 and Rab that contribute to LRRK2 activation in response to lysosomal damage.”
Reviewer #3 (Public Review):
Increased LRRK2 kinase activity is known to confer Parkinson's disease risk. While much is known about disease-causing LRRK2 mutations that increase LRRK2 kinase activity, the normal cellular mechanisms of LRRK2 activation are less well understood. Rab GTPases are known to play a role in LRRK2 activation and to be substrates for the kinase activity of LRRK2. However, much of the data on Rabs in LRRK2 activation comes from over-expression studies and the contributions of endogenously expressed Rabs to LRRK2 activation are less clear. To address this problem, Bondar and colleagues tested the impact of systematically depleting candidate Rab GTPases on LRRK2 activity as measured by its ability to phosphorylate Rab10 in the human A549 type 2 pneumocyte cell line. This resulted in the identification of a major role for Rab12 in controlling LRRK2 activity towards Rab10 in this model system. Follow-up studies show that this role for Rab12 is of particular importance for the phosphorylation of Rab10 by LRRK2 at damaged lysosomes. Increases in LRRK2 activity in cells harboring disease-causing mutants of LRRK2 and VPS35 also depend (at least partially) on Rab12. Confidence in the role of Rab12 in supporting LRRK2 activity is strengthened by parallel experiments showing that either siRNA-mediated depletion of Rab12 or CRISPR-mediated Rab12 KO both have similar effects on LRRK2 activity. Collectively, these results demonstrate a novel role for Rab12 in supporting LRRK2 activation in A549 cells. It is likely that this effect is generalizable to other cell types. However, this remains to be established. It is also likely that lysosomes are the subcellular site where Rab12-dependent activation of LRRK2 occurs. Independent validation of these conclusions with additional experiments would strengthen this conclusion and help to address some concerns that much of the data supporting a lysosome localization for Rab12-dependent activation of LRRK2 comes from a single method (LysoIP). Furthermore, there is a discrepancy between panel 4A versus 4D in the effect of LLoMe-induced lysosome damage on LRRK2 recruitment to lysosomes that will need to be addressed to strengthen confidence in conclusions about lysosomes as sites of LRRK2 activation by Rab12.
We thank the reviewer for their comments regarding our work that identifies Rab12 as a novel regulator of LRRK2 activation and the appreciation of the parallel approaches we employed to add confidence in this effect.
As suggested by the reviewer, we have updated our manuscript to now include independent validation of our conclusions using imaging-based analyses to complement our data from biochemical analyses using the Lyso-IP method. Specifically, we have included new imaging data that confirms that Rab12 levels are increased on lysosomes following membrane permeabilization with LLOMe treatment and demonstrates that this occurs independent of LRRK2, providing additional support that Rab12 is an upstream regulator of LRRK2 activity (Figure 3E in the updated manuscript).
Regarding the reviewer’s comment on a discrepancy between our findings in Figure 4A and Figure 4D, we have performed additional independent replicates in Figure 4D to assess the impact of lysosomal damage on the lysosomal levels of LRRK2 at baseline or upon the expression of genetic variants. We observed a significant increase in LRRK2 levels on lysosomes following LLOMe treatment in our set of experiments included in Figure 4A and a non-significant trend toward an increase in LRRK2 levels on isolates lysosomes in Figure 4D. As described in more detail below (in response to the second point raised by this reviewer), we think this variability arises because of a combination of low levels of LRRK2 on lysosomes with endogenous expression and variability across experiments in the efficiency of lysosomal isolation. Our observations of increased recruitment of LRRK2 to lysosomes upon damage are further supported by parallel imaging-based studies (Figure 3F-I) and are consistent with previous studies using overexpression systems.
We thank the reviewer for all of the suggestions which have added further confidence to our conclusions and substantially improved the manuscript.
-
eLife assessment
This valuable study shows that Rab12 is required for LRRK2 activation. However, while some of the data are compelling, some claims, especially the ones related to LRRK2's membrane association are not supported. Addressing discrepancies between figures (pointed out by reviewers) and re-writing certain sections will greatly improve this manuscript.
-
Reviewer #1 (Public Review):
This careful study reports the importance of Rab12 for Parkinson's disease associated LRRK2 kinase activity in cells. The authors carried out a targeted siRNA screen of Rab substrates and found lower pRab10 levels in cells depleted of Rab12. It has previously been reported that LLOME treatment of cells breaks lysosomes and with time, leads to major activation of LRRK2 kinase. Here they show that LLOME-induced kinase activation requires Rab12 and does not require Rab12 phosphorylation to show the effect.
1. Throughout the text, the authors claim that "Rab12 is required for LRRK2 dependent phosphorylation" (Page 4 line 78; Page 9 line 153; Page 22 line 421). This is not correct according to Figure 1 Figure Supp 1B - there is still pRab10. It is correct only in relation to the LLOME activation. Please correct …
Reviewer #1 (Public Review):
This careful study reports the importance of Rab12 for Parkinson's disease associated LRRK2 kinase activity in cells. The authors carried out a targeted siRNA screen of Rab substrates and found lower pRab10 levels in cells depleted of Rab12. It has previously been reported that LLOME treatment of cells breaks lysosomes and with time, leads to major activation of LRRK2 kinase. Here they show that LLOME-induced kinase activation requires Rab12 and does not require Rab12 phosphorylation to show the effect.
1. Throughout the text, the authors claim that "Rab12 is required for LRRK2 dependent phosphorylation" (Page 4 line 78; Page 9 line 153; Page 22 line 421). This is not correct according to Figure 1 Figure Supp 1B - there is still pRab10. It is correct only in relation to the LLOME activation. Please correct this error.
2. The authors conclude that Rab12 recruitment precedes that of LRRK2 but the rate of recruitment (slopes of curves in 3F and G) is actually faster for LRRK2 than for Rab12 with no proof that Rab12 is faster-please modify the text-it looks more like coordinated recruitment.
3. The title is misleading because the authors do not show that Rab12 promotes LRRK2 membrane association. This would require Rab12 to be sufficient to localize LRRK2 to a mislocalized Rab12. The authors DO show that Rab12 is needed for the massive LLOME activation at lysosomes. Please re-word the title.
-
Reviewer #2 (Public Review):
This study shows that rab12 has a role in the phosphorylation of rab10 by LRRK2. Many publications have previously focused on the phosphorylation targets of LRRK2 and the significance of many remains unclear, but the study of LRRK2 activation has mostly focused on the role of disease-associated mutations (in LRRK2 and VPS35) and rab29. The work is performed entirely in an alveolar lung cell line, limiting relevance for the nervous system. Nonetheless, the authors take advantage of this simplified system to explore the mechanism by which rab12 activates LRRK2. In general, the work is performed very carefully with appropriate controls, excluding trivial explanations for the results, but there are several serious problems with the experiments and in particular the interpretation.
First, the authors note that …
Reviewer #2 (Public Review):
This study shows that rab12 has a role in the phosphorylation of rab10 by LRRK2. Many publications have previously focused on the phosphorylation targets of LRRK2 and the significance of many remains unclear, but the study of LRRK2 activation has mostly focused on the role of disease-associated mutations (in LRRK2 and VPS35) and rab29. The work is performed entirely in an alveolar lung cell line, limiting relevance for the nervous system. Nonetheless, the authors take advantage of this simplified system to explore the mechanism by which rab12 activates LRRK2. In general, the work is performed very carefully with appropriate controls, excluding trivial explanations for the results, but there are several serious problems with the experiments and in particular the interpretation.
First, the authors note that rab29 appears to have a smaller or no effect when knocked down in these cells. However, the quantitation (Fig1-S1A) shows a much less significant knockdown of rab29 than rab12, so it would be important to repeat this with better knockdown or preferably a KO (by CRISPR) before making this conclusion. And the relationship to rab29 is important, so if a better KD or KO shows an effect, it would be important to assess by knocking down rab12 in the rab29 KO background.
Secondly, the knockdown of rab12 generally has a strong effect on the phosphorylation of the LRRK2 substrate rab10 but I could not find an experiment that shows whether rab12 has any effect on the residual phosphorylation of rab10 in the LRRK2 KO. There is not much phosphorylation left in the absence of LRRK2 but maybe this depends on rab12 just as much as in cells with LRRK2 and rab12 is operating independently of LRRK2, either through a different kinase or simply by making rab10 more available for phosphorylation. The epistasis experiment is crucial to address this possibility. To establish the connection to LRRK2, it would also help to compare the effect of rab12 KD on the phosphorylation of selected rabs that do or do not depend on LRRK2.
A strength of the work is the demonstration of p-rab10 recruitment to lysosomes by biochemistry and imaging. The demonstration that LRRK2 is required for this by biochemistry (Fig 4A) is very important but it would also be good to determine whether the requirement for LRRK2 extends to imaging. In support of a causal relationship, the authors also state that lysosomal accumulation of rab12 precedes LRRK2 but the data do not show this. Imaging with and without LRRK2 would provide more compelling evidence for a causative role.
The authors also touch base with PD mutations, showing that loss of rab12 reduces the phosphorylation of rab10. However, it is interesting that loss of rab12 has the same effect with R1441G LRRK2 and D620N VPS35 as it does in controls. This suggests that the effect of rab12 does not depend on the extent of LRRK2 activation. It is also surprising that R1441G LRRK2 does not increase p-rab10 phosphorylation (Fig 2G) as suggested in the literature and stated in the text.
Most important, the final figure suggests that PD-associated mutations in LRRK2 and VPS35 occlude the effect of lysosomal disruption on lysosomal recruitment of LRRK2 (Fig 4D) but do not impair the phosphorylation of rab10 also triggered by lysosomal disruption (4A-C). Phosphorylation of this target thus appears to be regulated independently of LRRK2 recruitment to the lysosome, suggesting another level of control (perhaps of kinase activity rather than localization) that has not been considered.
-
Reviewer #3 (Public Review):
Increased LRRK2 kinase activity is known to confer Parkinson's disease risk. While much is known about disease-causing LRRK2 mutations that increase LRRK2 kinase activity, the normal cellular mechanisms of LRRK2 activation are less well understood. Rab GTPases are known to play a role in LRRK2 activation and to be substrates for the kinase activity of LRRK2. However, much of the data on Rabs in LRRK2 activation comes from over-expression studies and the contributions of endogenously expressed Rabs to LRRK2 activation are less clear. To address this problem, Bondar and colleagues tested the impact of systematically depleting candidate Rab GTPases on LRRK2 activity as measured by its ability to phosphorylate Rab10 in the human A549 type 2 pneumocyte cell line. This resulted in the identification of a major …
Reviewer #3 (Public Review):
Increased LRRK2 kinase activity is known to confer Parkinson's disease risk. While much is known about disease-causing LRRK2 mutations that increase LRRK2 kinase activity, the normal cellular mechanisms of LRRK2 activation are less well understood. Rab GTPases are known to play a role in LRRK2 activation and to be substrates for the kinase activity of LRRK2. However, much of the data on Rabs in LRRK2 activation comes from over-expression studies and the contributions of endogenously expressed Rabs to LRRK2 activation are less clear. To address this problem, Bondar and colleagues tested the impact of systematically depleting candidate Rab GTPases on LRRK2 activity as measured by its ability to phosphorylate Rab10 in the human A549 type 2 pneumocyte cell line. This resulted in the identification of a major role for Rab12 in controlling LRRK2 activity towards Rab10 in this model system. Follow-up studies show that this role for Rab12 is of particular importance for the phosphorylation of Rab10 by LRRK2 at damaged lysosomes. Increases in LRRK2 activity in cells harboring disease-causing mutants of LRRK2 and VPS35 also depend (at least partially) on Rab12. Confidence in the role of Rab12 in supporting LRRK2 activity is strengthened by parallel experiments showing that either siRNA-mediated depletion of Rab12 or CRISPR-mediated Rab12 KO both have similar effects on LRRK2 activity. Collectively, these results demonstrate a novel role for Rab12 in supporting LRRK2 activation in A549 cells. It is likely that this effect is generalizable to other cell types. However, this remains to be established. It is also likely that lysosomes are the subcellular site where Rab12-dependent activation of LRRK2 occurs. Independent validation of these conclusions with additional experiments would strengthen this conclusion and help to address some concerns that much of the data supporting a lysosome localization for Rab12-dependent activation of LRRK2 comes from a single method (LysoIP). Furthermore, there is a discrepancy between panel 4A versus 4D in the effect of LLoMe-induced lysosome damage on LRRK2 recruitment to lysosomes that will need to be addressed to strengthen confidence in conclusions about lysosomes as sites of LRRK2 activation by Rab12.
-
