PKR activation-induced mitochondrial dysfunction in HIV-transgenic mice with nephropathy
Curation statements for this article:-
Curated by eLife

eLife assessment
This study presents valuable new insights into a HIV-associated nephropathy (HIVAN) kidney phenotype in the Tg26 transgenic mouse model, and delineates the kidney cell types that express HIV genes and are injured in these HIV-transgenic mice. A series of compelling experiments demonstrated that PKR inhibition can ameliorate HIVAN with reversal of mitochondrial dysfunction (mainly confined to endothelial cells), a prominent feature shared in other kidney diseases. The data support that inhibition of PKR and mitochondrial dysfunction has potential clinical significance for HIVAN.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
HIV disease remains prevalent in the USA and chronic kidney disease remains a major cause of morbidity in HIV-1-positive patients. Host double-stranded RNA (dsRNA)-activated protein kinase (PKR) is a sensor for viral dsRNA, including HIV-1. We show that PKR inhibition by compound C16 ameliorates the HIV-associated nephropathy (HIVAN) kidney phenotype in the Tg26 transgenic mouse model, with reversal of mitochondrial dysfunction. Combined analysis of single-nucleus RNA-seq and bulk RNA-seq data revealed that oxidative phosphorylation was one of the most downregulated pathways and identified signal transducer and activator of transcription (STAT3) as a potential mediating factor. We identified in Tg26 mice a novel proximal tubular cell cluster enriched in mitochondrial transcripts. Podocytes showed high levels of HIV-1 gene expression and dysregulation of cytoskeleton-related genes, and these cells dedifferentiated. In injured proximal tubules, cell-cell interaction analysis indicated activation of the pro-fibrogenic PKR-STAT3-platelet-derived growth factor (PDGF)-D pathway. These findings suggest that PKR inhibition and mitochondrial rescue are potential novel therapeutic approaches for HIVAN.
Article activity feed
-

-
-
-

eLife assessment
This study presents valuable new insights into a HIV-associated nephropathy (HIVAN) kidney phenotype in the Tg26 transgenic mouse model, and delineates the kidney cell types that express HIV genes and are injured in these HIV-transgenic mice. A series of compelling experiments demonstrated that PKR inhibition can ameliorate HIVAN with reversal of mitochondrial dysfunction (mainly confined to endothelial cells), a prominent feature shared in other kidney diseases. The data support that inhibition of PKR and mitochondrial dysfunction has potential clinical significance for HIVAN.
-
Reviewer #1 (Public Review):
Summary:
HIV associated nephropathy (HIVAN) is a rapidly progressing form of kidney disease that manifests secondary to untreated HIV infection and is predominantly seen in individuals of African descent. Tg26 mice carrying an HIV transgene lacking gag and pol exhibit high levels of albuminuria and rapid decline in renal function that recapitulates many features of HIVAN in humans. HIVAN is seen predominantly in individuals carrying two copies of missense variants in the APOL1 gene, and the authors have previously shown that APOL1 risk variant mRNA induces activity of the double strand RNA sensor kinase PKR. Because of the tight association between the APOL1 risk genotype and HIVAN, the authors hypothesized that PKR activation may mediate the renal injury in Tg26 mice, and tested this hypothesis by treating …
Reviewer #1 (Public Review):
Summary:
HIV associated nephropathy (HIVAN) is a rapidly progressing form of kidney disease that manifests secondary to untreated HIV infection and is predominantly seen in individuals of African descent. Tg26 mice carrying an HIV transgene lacking gag and pol exhibit high levels of albuminuria and rapid decline in renal function that recapitulates many features of HIVAN in humans. HIVAN is seen predominantly in individuals carrying two copies of missense variants in the APOL1 gene, and the authors have previously shown that APOL1 risk variant mRNA induces activity of the double strand RNA sensor kinase PKR. Because of the tight association between the APOL1 risk genotype and HIVAN, the authors hypothesized that PKR activation may mediate the renal injury in Tg26 mice, and tested this hypothesis by treating mice with a commonly used PKR inhibitory compound called C16. Treatment with C16 substantially attenuated renal damage in the Tg26 model as measured by urinary albumin/creatinine ratio, urinary NGAL/creatinine ratio and improvement in histology. The authors then performed bulk and single-nucleus RNAseq on kidneys from mice from different treatment groups to identify pathways and patterns of cell injury associated with HIV transgene expression as well as to determine the mechanistic basis for the effect of C16 treatment. They show that proximal tubule nuclei from Tg26 mice appear to have more mitochondrial transcripts which was reversed by C16 treatment and suggest that this may provide evidence of mitochondrial dysfunction in this model. They explore this hypothesis by showing there is a decrease in the expression of nuclear encoded genes and proteins involved in oxidative phosphorylation as well as a decrease in respiratory capacity via functional assessment of respiration in tubule and glomerular preparations from these mouse kidneys. All of these changes were reversed by C16 treatment. The authors propose the existence of a novel injured proximal tubule cell-type characterized by the leak of mitochondrial transcripts into the nucleus (PT-Mito). Analysis of HIV transgene expression showed high level expression in podocytes, consistent with the pronounced albuminuria that characterizes this model and HIVAN, but transcripts were also detected in tubular and endothelial cells. Because of the absence of mitochondrial transcripts in the podocytes, the authors speculate that glomerular mitochondrial dysfunction in this model is driven by damage to glomerular endothelial cells.
Strengths:
The strengths of this study include the comprehensive transcriptional analysis of the Tg26 model, including an evaluation of HIV transgene expression, which has not been previously reported. This data highlights that HIV transcripts are expressed in a subset of podocytes, consistent with the highly proteinuric disease seen in mouse and humans. However, transcripts were also seen in other tubular cells, notably intercalated cells, principal cells and injured proximal tubule cells. Though the podocyte expression makes sense, the relevance of the tubular expression to human disease is still an open question.
The data in support of mitochondrial dysfunction are also robust and rely on combined evidence from downregulation of transcripts involved in oxidative phosphorylation, decreases in complex I and II as determined by immunoblot, and assessments of respiratory capacity in tubular and glomerular preparations. These data are largely consistent with other preclinical renal injury model reported in the literature as well as previous, less thorough assessments in the Tg26 model.
Comments on latest version:
The authors have revised the manuscript to acknowledge the potential limitations of the C16 tool compound used and have performed some additional analyses that suggest the PT-Mito population can be identified in samples from KPMP. The authors added some control images for the in situ hybridizations, which are helpful, though they don't get to the core issue of limited resolution to determine whether mitochondrial RNA is present in the nuclei of injured PT cells. Some additional work has been done to show that C16 treatment results in a decrease in phospho-PKR, a readout of PKR inhibition. These changes strengthen the manuscript by providing some evidence for the translatability of the PT-mito cluster to humans and some evidence for on-target activity for C16. It would be helpful if the authors could quantify the numbers of cells in IHC with nuclear transcripts as well as pointing out some specific examples in the images provided, as comparator data for the snRNAseq studies in which 3-6% of cortex cells had evidence of nuclear mitochondrial transcripts.
-
Reviewer #2 (Public Review):
Summary:
Numerous studies by the authors and other groups have demonstrated an important role for HIV gene expression kidney cells in promoting progressive chronic kidney disease, especially HIV associated nephropathy. The authors had previously demonstrated a role for protein kinase R (PKR) in a non-HIV transgenic model of kidney disease (Okamoto, Commun Bio, 2021). In this study, the authors used innovative techniques including bulk and single nuclear RNAseq to demonstrate that mice expressing a replication-incompetent HIV transgene have prominent dysregulation of mitochondrial gene expression and activation of PKR and that treatment of these mice with a small molecule PKR inhibitor ameliorated the kidney disease phenotype in HIV-transgenic mice. They also identified STAT3 as a key upstream regulator of …
Reviewer #2 (Public Review):
Summary:
Numerous studies by the authors and other groups have demonstrated an important role for HIV gene expression kidney cells in promoting progressive chronic kidney disease, especially HIV associated nephropathy. The authors had previously demonstrated a role for protein kinase R (PKR) in a non-HIV transgenic model of kidney disease (Okamoto, Commun Bio, 2021). In this study, the authors used innovative techniques including bulk and single nuclear RNAseq to demonstrate that mice expressing a replication-incompetent HIV transgene have prominent dysregulation of mitochondrial gene expression and activation of PKR and that treatment of these mice with a small molecule PKR inhibitor ameliorated the kidney disease phenotype in HIV-transgenic mice. They also identified STAT3 as a key upstream regulator of kidney injury in this model, which is consistent with previously published studies. Other important advances include identifying the kidney cell types that express the HIV transgene and have dysregulation of cellular pathways.
Strengths:
Major strengths of the study include the use of a wide variety of state-of-the-art molecular techniques to generate important new data on the pathogenesis of kidney injury in this commonly used model of kidney disease and the identification of PKR as a potential druggable target for the treatment of HIV-induced kidney disease. The authors also identify a potential novel cell type within the kidney characterized by high expression of mitochondrial genes.
Weaknesses:
Though the HIV-transgenic model used in these studies results in a phenotype that is very similar to HIV-associated nephropathy in humans, the model has several limitations that may prevent direct translation to human disease, including the fact that mice lack several genetic factors that are important contributors to HIV and kidney pathogenesis in humans. Additional studies are therefore needed to confirm these findings in human kidney disease.
-
Author response:
The following is the authors’ response to the previous reviews.
Responses to recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
The manuscript would be strengthened with the following key revisions mostly having to do with image quality:
(1) It is very difficult in Figure 4B to see which nuclei actually have evidence of mitochondrial transcripts. It might be helpful to provide arrows to specific cells and also to provide some estimate of the percentage of cells with nuclear mt-transcripts as measured by ISH compared to the 3-6% of cortex cell estimate seen in the snRNAseq analysis.
As suggested, now we have added arrows to help readers to see the signals in nuclei. The detection threshold of ISH and single-nucleus RNA-seq should be different, and therefore, measuring estimates of PT-Mito …
Author response:
The following is the authors’ response to the previous reviews.
Responses to recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
The manuscript would be strengthened with the following key revisions mostly having to do with image quality:
(1) It is very difficult in Figure 4B to see which nuclei actually have evidence of mitochondrial transcripts. It might be helpful to provide arrows to specific cells and also to provide some estimate of the percentage of cells with nuclear mt-transcripts as measured by ISH compared to the 3-6% of cortex cell estimate seen in the snRNAseq analysis.
As suggested, now we have added arrows to help readers to see the signals in nuclei. The detection threshold of ISH and single-nucleus RNA-seq should be different, and therefore, measuring estimates of PT-Mito by ISH would not be reliable.
(2) The phospho-PKR images provided as evidence of C16 activity (Supplemental Figure 1) are too dim to be very useful. Could brighter images be provided?
We have now adjusted the LUTs of images in Supplemental Figure 1.
-
-
Author response:
The following is the authors’ response to the original reviews.
eLife assessment
This study presents valuable new insights into HIV-associated nephropathy (HIVAN) kidney phenotype in the Tg26 transgenic mouse model and delineates the kidney cell types that express HIV genes and are injured in these HIV-transgenic mice. A series of compelling experiments demonstrated that PKR inhibition can ameliorate HIVAN with reversal of mitochondrial dysfunction (mainly confined to endothelial cells), a prominent feature shared in other kidney diseases. Although there are concerns regarding the specificity of C16 to PKR inhibition, as well as with the in situ hybridization studies, the data suggests that inhibition of PKR and mitochondrial dysfunction has potential clinical significance for HIVAN.
Public Reviews:
Reviewer #1 …
Author response:
The following is the authors’ response to the original reviews.
eLife assessment
This study presents valuable new insights into HIV-associated nephropathy (HIVAN) kidney phenotype in the Tg26 transgenic mouse model and delineates the kidney cell types that express HIV genes and are injured in these HIV-transgenic mice. A series of compelling experiments demonstrated that PKR inhibition can ameliorate HIVAN with reversal of mitochondrial dysfunction (mainly confined to endothelial cells), a prominent feature shared in other kidney diseases. Although there are concerns regarding the specificity of C16 to PKR inhibition, as well as with the in situ hybridization studies, the data suggests that inhibition of PKR and mitochondrial dysfunction has potential clinical significance for HIVAN.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
HIV-associated nephropathy (HIVAN) is a rapidly progressing form of kidney disease that manifests secondary to untreated HIV infection, and is predominantly seen in individuals of African descent. Tg26 mice carrying an HIV transgene lacking gag and pol exhibit high levels of albuminuria and rapid decline in renal function that recapitulates many features of HIVAN in humans. HIVAN is seen predominantly in individuals carrying two copies of missense variants in the APOL1 gene, and the authors have previously shown that APOL1 risk variant mRNA induces activity of the double-strand RNA sensor kinase PKR. Because of the tight association between the APOL1 risk genotype and HIVAN, the authors hypothesized that PKR activation may mediate renal injury in Tg26 mice and tested this hypothesis by treating mice with a commonly used PKR inhibitory compound called C16. Treatment with C16 substantially attenuated renal damage in the Tg26 model as measured by urinary albumin/creatinine ratio, urinary NGAL/creatinine ratio, and improvement in histology. The authors then performed bulk and single-nucleus RNAseq on kidneys from mice from different treatment groups to identify pathways and patterns of cell injury associated with HIV transgene expression as well as to determine the mechanistic basis for the effect of C16 treatment. They show that proximal tubule nuclei from Tg26 mice appear to have more mitochondrial transcripts which was reversed by C16 treatment and suggest that this may provide evidence of mitochondrial dysfunction in this model. They explore this hypothesis by showing there is a decrease in the expression of nuclear-encoded genes and proteins involved in oxidative phosphorylation as well as a decrease in respiratory capacity via functional assessment of respiration in tubule and glomerular preparations from these mouse kidneys. All of these changes were reversed by C16 treatment. The authors propose the existence of a novel injured proximal tubule cell-type characterized by the leak of mitochondrial transcripts into the nucleus (PT-Mito). Analysis of HIV transgene expression showed high level expression in podocytes, consistent with the pronounced albuminuria that characterizes this model and HIVAN, but transcripts were also detected in tubular and endothelial cells. Because of the absence of mitochondrial transcripts in the podocytes, the authors speculate that glomerular mitochondrial dysfunction in this model is driven by damage to glomerular endothelial cells.
Strengths:
The strengths of this study include the comprehensive transcriptional analysis of the Tg26 model, including an evaluation of HIV transgene expression, which has not been previously reported. This data highlights that HIV transcripts are expressed in a subset of podocytes, consistent with the highly proteinuric disease seen in mice and humans. However, transcripts were also seen in other tubular cells, notably intercalated cells, principal cells and injured proximal tubule cells. Though the podocyte expression makes sense, the relevance of the tubular expression to human disease is still an open question.
The data in support of mitochondrial dysfunction are also robust and rely on combined evidence from downregulation of transcripts involved in oxidative phosphorylation, decreases in complex I and II as determined by immunoblot, and assessments of respiratory capacity in tubular and glomerular preparations. These data are largely consistent with other preclinical renal injury models reported in the literature as well as previous, less thorough assessments in the Tg26 model.
Weaknesses:
The key weakness of the study lies in the use of a PKR inhibitor with questionable specificity. C16 has been reported to inhibit numerous other kinases including cyclin CDKs and GSK3α and -β, and this means that the conclusions of this study with respect to the role of PKR are highly questionable. The rationale for the dose used was not provided (and is lower than used in other publications with C16), and in the absence of drug exposure data and assessment of target engagement, it is difficult to ascertain whether substantial inhibition of PKR was achieved.
A second key weakness lies in the identification of the PT-Mito cell cluster. Though the authors provide some rationale for the identification of this specific cell type, it seems equally plausible the cells merely reflect a high background capture of mitochondria in a subset of droplets. The IHC analysis that was provided is not convincing enough to support the claim and more careful high resolution imaging and in situ hybridization (with appropriate quantitation) will be needed to provide substantive support for the presence of a proximal tubule cell type with mitochondrial transcript that are trafficked to the nucleus.
We appreciate the reviewer’s thoughtful summary.
With regard to non-specificity of C16, we added to the Discussion a description and references that describe non-specificity of C16. as suggested by the reviewer. Of note, the C16 doses that we used were also used previously (Okamoto, CommBiol, 2018). Importantly, newly-added immunofluorescence images using a phospho-PKR specific antibody showed PKR inhibition (Supplemental Figure 1).
Identification of the PT-Mito cluster in tissues was challenging, mainly due to the absence of existence of know marker genes for newly-identified cluster. Finally, We added in situ hybridization images, with a negative control probe, to show specificity of target probes.
Reviewer #2 (Public Review):
Summary:
Numerous studies by the authors and other groups have demonstrated an important role for HIV gene expression kidney cells in promoting progressive chronic kidney disease, especially HIV-associated nephropathy. The authors had previously demonstrated a role for protein kinase R (PKR) in a non-HIV transgenic model of kidney disease (Okamoto, Commun Bio, 2021). In this study, the authors used innovative techniques including bulk and single nuclear RNAseq to demonstrate that mice expressing a replication-incompetent HIV transgene have prominent dysregulation of mitochondrial gene expression and activation of PKR and that treatment of these mice with a small molecule PKR inhibitor ameliorated the kidney disease phenotype in HIV-transgenic mice. They also identified STAT3 as a key upstream regulator of kidney injury in this model, which is consistent with previously published studies. Other important advances include identifying the kidney cell types that express the HIV transgene and have dysregulation of cellular pathways.
Strengths:
Major strengths of the study include the use of a wide variety of state-of-the-art molecular techniques to generate important new data on the pathogenesis of kidney injury in this commonly used model of kidney disease and the identification of PKR as a potential druggable target for the treatment of HIV-induced kidney disease. The authors also identify a potential novel cell type within the kidney characterized by high expression of mitochondrial genes.
Weaknesses:
Though the HIV-transgenic model used in these studies results in a phenotype that is very similar to HIV-associated nephropathy in humans, the model has several limitations that may prevent direct translation to human disease, including the fact that mice lack several genetic factors that are important contributors to HIV and kidney pathogenesis in humans. Additional studies are therefore needed to confirm these findings in human kidney disease.
We appreciate the succinct summary of the present work. We agree that the findings from the HIV Tg26 mouse model warrant additional investigation in human kidney disease samples. Further studies will be needed to confirm whether the mechanisms presented here are operative in human HIVAN or other RNA virus-associated kidney diseases.
Reviewer #1 (Recommendations For The Authors)
The specificity of the C16 tool has been called into question in 3 publications - Chen et al, 2008, PMID: 19046382; Lopez-Grancha et al, 2021, PMID: 34531308; and Cusak et al, 2023, PMID: 36400288. Lopez-Grancha et al have reported a novel, more selective PKR inhibitor with good pharmacological properties that might enable a more robust test of the PKR hypothesis. Regardless, compound exposures and target engagement (i.e. by monitoring phosphorylation of PKR targets such eIF2α) should accompany these studies. Alternatively, it may be easier to probe the role of PKR in Tg26 pathogenicity by crossing the Tg26 line to a PKR knockout mouse.
In response, we have added a description and references about the the possibility of non-specificity of C16 in the Discussion as a limitation as suggested. (Page 21).
“Third, we acknowledge possibility of a non-specific effect of C16 as an inhibitor of PKR.66-68”
Further, we added immunohistochemistry images of pPKR on kidney tissue as shown in Supplemental Figure 1A-D. Images showed PKR activation in Tg26 tubular cells, which was inhibited by C16 treatment.
Author response image 1.
Immunofluorescent images showing pPKR. (A-D) Immunofluorescent images showed PKR activation by detecting pPKR in Tg26 mouse kidney. pPKR was inhibited by C16 treatments.
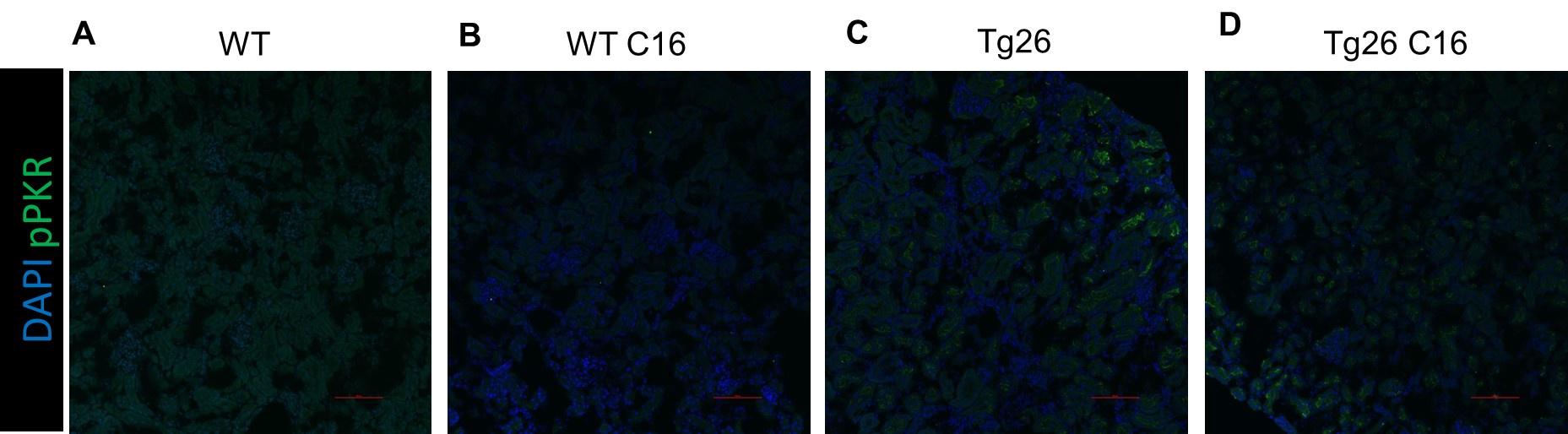
The suggested PKR knockout mice experiment is an excellent idea for future work but we believe Is outside the scope of the current manuscript.
To enhance the evidentiary base for the PT-Mito cell type, it would be interesting to know whether these cells can also be found in human datasets like KPMP, though this might require reprocessing the original snRNAseq data. Further in situ hybridization in both mouse and human samples using fluorescent rather than colorimetric approaches should yield a more compelling dataset to provide evidence for this cell type. These approaches would also allow for more precise quantification of the PT-Mito cells compared to the population of proximal tubule cells. Again, the default assumption here should be that the mitochondrial transcripts represent a contamination, and the purpose of these additional experiments is to definitively rule out that explanation.
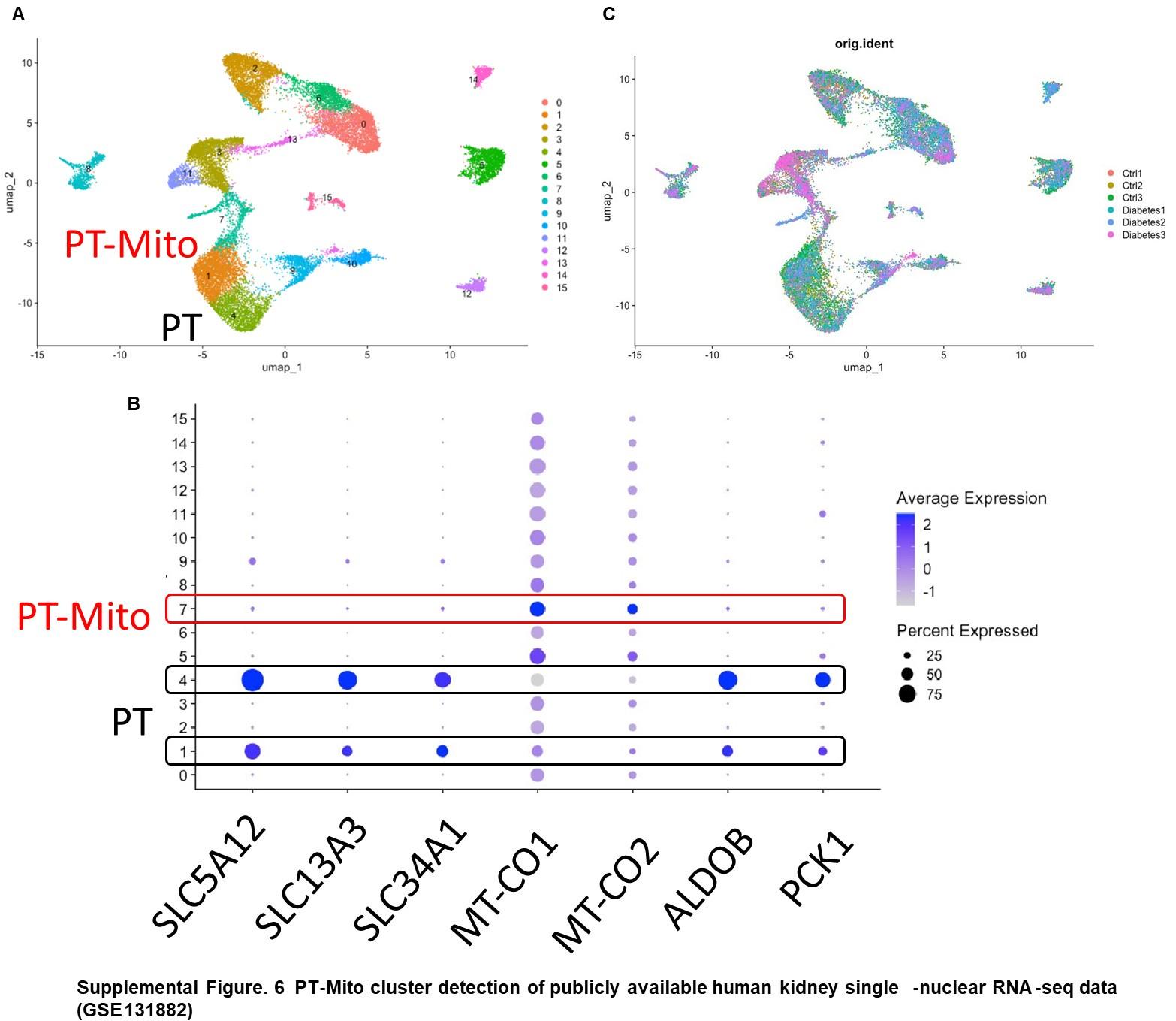
Authors: First, as suggested, we carried out additional analyses. We examined a publiclyavailable human kidney snRNA-seq dataset (GSE131882) and found in it the same PT-Mito cluster as shown in Supplemental Figure 6. The PT-Mito cluster was located in close proximity to the PT cluster in a UMAP plot. We added this finding in the Results as follows (Page 12):
“We also confirmed the existence of similar PT-Mito cluster in published human kidney single-nuclear RNA-seq data47 by the re-analysis of the original data. (Supplemental Figure 6A-C).”
Author response image 2.
PT-Mito cluster detection of publicly available human kidney single-nuclear RNA-seq data (GSE131882) (A) UMAP plot of human kidney single-nuclear RNA-seq data shows 16 clusters. Cluster 1, 4 are proximal tubule (PT) clusters, and cluster 7 is PT-Mito cluster. (B) Dot plot shows expression of PT marker genes and PT-Mito marker genes obtained from current manuscript data. PTMito markers including MT-CO1 and MT-CO2 had high expression in cluster 7. (C) UMAP plot shows all six samples are contributing to all cell clusters.
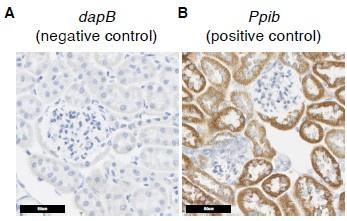
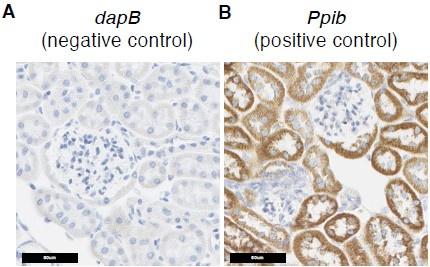
Second, as suggested, we also included negative control data from in situ hybridization studies (Supplementary Figure 5A, 5B), which shows that the signals in Figure 4B, 4C are true signals.
Author response image 3.
Additional in situ hybridization images. (A) In situ hybridization images probing dapB (negative control probe) showed no signals. (B) In situ hybridization images probing Ppib (positive control probe) showed strong signals.
Reviewer #2 (Recommendations For The Authors)
(1) The supplementary data file seems to have been uploaded twice but the supplementary methods were not available which would have been helpful when assessing some methods such as using PodoCount to count podocytes.
We acknowledge that we inadvertently failed to upload the Supplementary Methods section-thank you for pointing this out. The supplementary methods are now provided in the revised submission, including detailed methods about PodoCount. Corresponding descriptions are as follows:
“Estimation of glomerular podocyte count
PodoCount5, a computational tool for whole slide podocyte estimation from digitized histologic sections, was used to detect, enumerate, and characterize podocyte nuclear profiles in the glomeruli of immunohistochemically labeled (IHC-labeled) murine kidney sections. Formalin-fixed, paraffin embedded tissues (2 µm thickness) were IHC-labeled for p57kip2, a marker of podocyte terminal differentiation (ab75974, Abcam, Cambridge, UK), and detected with horse radish peroxidase (RU-HRP1000, Diagnostic BioSystems, Pleasanton, CA) and diaminobenzidine chromogen substrate (BSB0018A, Bio SB, Santa Barbara, CA). A periodic acid-Schiff post-stain was applied without hematoxylin counterstain. The tool uses a combination of stain deconvolution, digital image processing, and feature engineering to compute histologic podometrics6 with correction for section thickness7. In this study, PodoCount was used to assess mean glomerular podocyte count per mouse.“
(2) In the abstract, the authors give the impression that they know definitively the sequence of HIV gene expression, cytoskeletal dysregulation, dedifferentiation, then loss from glomeruli. Since they could only examine cells that were present in glomeruli, they can't definitively say much about the cells that were lost from glomeruli.
As suggested, deleted the following text: “and were lost from glomeruli tuft”
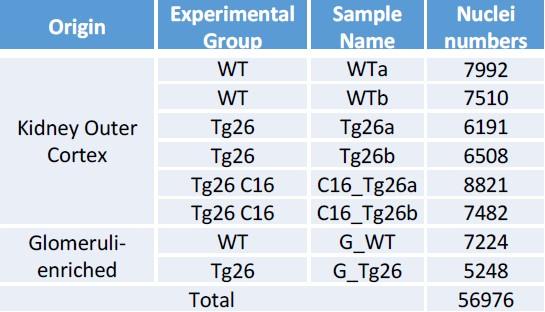
(3) The authors state that 56,976 cells were used for snRNAseq studies. Was the number of cells similar for each of the 8 mice (from 4 different groups)?
In response, we have created a new table summarizing numbers of nuclei from each sample (i.e. each mouse) added to the Supplemental Figure 2D as follows:
Author response table 1.
Pre-processing of single-nuclear RNA-seq data, Breakdown of nuclei numbers from each sample showed comparable numbers of nuclei analyzed.
(4) Please provide information on the assay that was used to measure creatinine since some methods can be unreliable in mice
This is now provided in the revised submission, including creatinine measurement methods (LC-MS/MS) on page 3 of Supplementary Material:
“Mouse chemistry measurements
Plasma creatinine was measured by isotope dilution LC-MS/MS at The University of Alabama at Birmingham O’Brien Center Core C (Birmingham, AL).”
(5) The authors state that expression of PKR (Eif2ak2) was expressed in all nephron segments. However, it appears on visual inspection of the UMAP in Fig S2B that the percentage of cells expressing Eif2ak2 was low. What percent of cells expressed Eif2ak2 and if it was a low percentage, what is the authors hypothesis for how expression in a small percentage of cells led to the kidney phenotype?
Supplemental Figure 2B (now 3B) does show modest expression of Eif2ak2, approximately 10%. The technique may lack sensitivity to detect low gene expression and even low gene expression may be sufficient to cause phenotypic change.
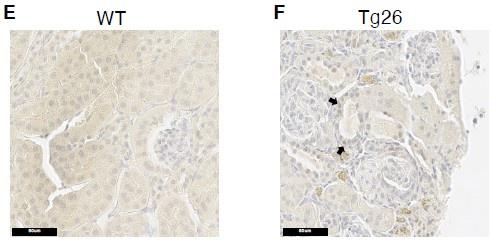
(6a) In figure 4B and C, it is not clear what genotype/treatment group is shown.
The legend for figure 4B, 4C has been modified to state that the group was wildtype mice
(B, C) In situ hybridization of mt-Co1 and mt-Atp6 genes showed signals inside nuclei of WT mice
(6b) Also, if these ISH images are from Tg26 mice, it would be helpful to do ISH in mice with/without C16 treatment.
These images of ISH for these two genes are from wild-type mice, as now stated in the revised legend. Our purpose was to show that these mitochondrial-encoded gene transcripts (mt-Co1 and mt-Atp6) are transported to nuclei from the cytoplasm. We believe it is not necessary to do ISH in Tg26 mice because these genes are not disease-specific.
(6c) Also, only 3-6% of cells express these "PT-mito" markers by snRNAseq, but it appears that far more are expressed by ISH, raising concerns for nonspecific binding of the ISH probe.
(6d) Also, nonsense controls should be included to demonstrate the specificity of the ISH data.
First (comment 6c), the PT-mito cluster does not have specific markers, to our knowledge. Second (comment 6d) , to address the concern for non-specific binding of the ISH probes, we have now added additional ISH images, together with a negative control probe (C. elegans gene dapB) and a positive control probe (mouse Ppib), as shown in Supplementary Figure 5A and 5B, respectively.
Author response image 4.
Additional in situ hybridization images. (A) In situ hybridization images probing dapB (negative control probe) showed no signals. (B) In situ hybridization images probing Ppib (positive control probe) showed strong signals.
(7) The authors state that "mitochondrial dysfunction was most pronounced in the PT-Mito cluster" but in Figure 4D, the oxidative phosphorylation activation Z score was most down in the PT-inj (injured PT cells) and the PT-Mito cells were the 4-most downregulated cell type.
We appreciate the careful reading and agree with reviewer’s comment. In the revision, we have deleted “most” from this description.
(8) In Fig 4F, please state what "Cp expression" means.
We have spelled out ceruloplasmin (Cp).
(9) It is not clear in immunohistochemistry images in Fig 5F where the p-stat3 was detected due to the hematoxylin counterstain which may have obscured subtle nuclear staining. Also, some of the strongest staining appears to be in peritubular capillaries, instead of tubular and glomerular epithelial cells.
We have added arrows to help readers see where we show that p-Stat3 was detected as faintly-brown and distinct cytoplasmic granules in injured tubular cells in Tg26 mice (panel F), as opposed to diffuse in tubular cytoplasmic color in wild-type mice (panel E).
Author response image 5.
(10) For the studies of mitochondrial oxygen consumption (Fig 6), it would be helpful to also provide data on the effect of C16 in wild-type kidneys, in case C16 somehow causes a primary increase in mitochondrial oxygen consumption rather than preventing HIV-induced loss in kidney cells from HIV-transgenic mice.
We did not include Seahorse data regarding oxygen consumption from WT mice treated with C16, as C16 did not affect either renal function or transcriptomes in WT mice, in contrast to the Tg26 mice (Figure 1A-G).
(11) The authors emphasize that podocytes had the highest expression of HIV genes (Fig 7). However, it appears that <2% of podocytes expressed HIV genes. How do the authors explain the severe renal phenotype given the relatively small number of cells expressing the HIV transgene? Also, did the same cells express all/most of the HIV transcripts, or did some cells express some HIV transcripts? For instance, since the authors state that vpr and nef have the most important role in kidney injury, were the same cells that expressed nef also expressing Vpr?
We know that snRNA-seq cannot detect the whole transcriptome in each cell, due to the well-known drop-out effect characteristic of the method. Several factors may contribute to this drop-out effect, including stochastic patterns of gene expression, low RNA amounts and inefficient mRNA capture (Qiu, Nature Comm, 2020; Ran, Bioinformatics, 2020).
Our interpretation is that HIV gene expressing-podocytes had higher expression of HIV genes, but it does not mean that other kidney cells entirely lack HIV gene expression. With regard to co-expression of other HIV transcripts, nef and vpr were more often coexpressed as shown in Figure 7J. Vpr was expressed in nef-positive podocytes and not detected in nef-negative podocytes.
(12) In figure 8, the authors emphasize the dysregulation of genes involved in cell-cell interaction, particularly PDGF-D. They show some data for the effect of C16 in this system in Fig 8 but it would be helpful if they can state the effect in the text of the Results section.
We have added text in the Results describing activating interactions in Tg26 mice, that were reduced by C16 exposure, as follows: (page 18)
“For example, platelet derived growth factor D (PDGF-D) was upregulated in PT-Inj in Tg26 mice and was downregulated by C16 treatment (Figure 8D). Further, PDGF-D may interact with PDGFR-B in fibroblasts.”
-
eLife assessment
This study presents valuable new insights into a HIV-associated nephropathy (HIVAN) kidney phenotype in the Tg26 transgenic mouse model, and delineates the kidney cell types that express HIV genes and are injured in these HIV-transgenic mice. A series of compelling experiments demonstrated that PKR inhibition can ameliorate HIVAN with reversal of mitochondrial dysfunction (mainly confined to endothelial cells), a prominent feature shared in other kidney diseases. The data support that inhibition of PKR and mitochondrial dysfunction has potential clinical significance for HIVAN.
-
Reviewer #1 (Public Review):
Summary:
HIV associated nephropathy (HIVAN) is a rapidly progressing form of kidney disease that manifests secondary to untreated HIV infection, and is predominantly seen in individuals of African descent. Tg26 mice carrying an HIV transgene lacking gag and pol exhibit high levels of albuminuria and rapid decline in renal function that recapitulates many features of HIVAN in humans. HIVAN is seen predominantly in individuals carrying two copies of missense variants in the APOL1 gene, and the authors have previously shown that APOL1 risk variant mRNA induces activity of the double strand RNA sensor kinase PKR. Because of the tight association between the APOL1 risk genotype and HIVAN, the authors hypothesized that PKR activation may mediate the renal injury in Tg26 mice, and tested this hypothesis by treating …
Reviewer #1 (Public Review):
Summary:
HIV associated nephropathy (HIVAN) is a rapidly progressing form of kidney disease that manifests secondary to untreated HIV infection, and is predominantly seen in individuals of African descent. Tg26 mice carrying an HIV transgene lacking gag and pol exhibit high levels of albuminuria and rapid decline in renal function that recapitulates many features of HIVAN in humans. HIVAN is seen predominantly in individuals carrying two copies of missense variants in the APOL1 gene, and the authors have previously shown that APOL1 risk variant mRNA induces activity of the double strand RNA sensor kinase PKR. Because of the tight association between the APOL1 risk genotype and HIVAN, the authors hypothesized that PKR activation may mediate the renal injury in Tg26 mice, and tested this hypothesis by treating mice with a commonly used PKR inhibitory compound called C16. Treatment with C16 substantially attenuated renal damage in the Tg26 model as measured by urinary albumin/creatinine ratio, urinary NGAL/creatinine ratio and improvement in histology. The authors then performed bulk and single-nucleus RNAseq on kidneys from mice from different treatment groups to identify pathways and patterns of cell injury associated with HIV transgene expression as well as to determine the mechanistic basis for the effect of C16 treatment. They show that proximal tubule nuclei from Tg26 mice appear to have more mitochondrial transcripts which was reversed by C16 treatment and suggest that this may provide evidence of mitochondrial dysfunction in this model. They explore this hypothesis by showing there is a decrease in the expression of nuclear encoded genes and proteins involved in oxidative phosphorylation as well as a decrease in respiratory capacity via functional assessment of respiration in tubule and glomerular preparations from these mouse kidneys. All of these changes were reversed by C16 treatment. The authors propose the existence of a novel injured proximal tubule cell-type characterized by the leak of mitochondrial transcripts into the nucleus (PT-Mito). Analysis of HIV transgene expression showed high level expression in podocytes, consistent with the pronounced albuminuria that characterizes this model and HIVAN, but transcripts were also detected in tubular and endothelial cells. Because of the absence of mitochondrial transcripts in the podocytes, the authors speculate that glomerular mitochondrial dysfunction in this model is driven by damage to glomerular endothelial cells.
Strengths:
The strengths of this study include the comprehensive transcriptional analysis of the Tg26 model, including an evaluation of HIV transgene expression, which has not been previously reported. This data highlights that HIV transcripts are expressed in a subset of podocytes, consistent with the highly proteinuric disease seen in mouse and humans. However, transcripts were also seen in other tubular cells, notably intercalated cells, principal cells and injured proximal tubule cells. Though the podocyte expression makes sense, the relevance of the tubular expression to human disease is still an open question.
The data in support of mitochondrial dysfunction are also robust and rely on combined evidence from downregulation of transcripts involved in oxidative phosphorylation, decreases in complex I and II as determined by immunoblot, and assessments of respiratory capacity in tubular and glomerular preparations. These data are largely consistent with other preclinical renal injury model reported in the literature as well as previous, less thorough assessments in the Tg26 model.
Weaknesses:
The key weakness of the study lies in the use of a PKR inhibitor with questionable specificity. C16 has been reported to inhibit numerous other kinases including cyclin CDKs and GSK3α and -β, and this means that the conclusions of this study with respect to the role of PKR are highly questionable. The rationale for the dose used was not provided (and is lower than used in other publications with C16), and in the absence of drug exposure data and assessment of target engagement, it is difficult to ascertain whether substantial inhibition of PKR was achieved.
A second key weakness lies in the identification of the PT-Mito cell cluster. Though the authors provide some rationale for the identification of this specific cell type, it seems equally plausible the cells merely reflect a high background capture of mitochondria in a subset of droplets. The IHC analysis that was provided is not convincing enough to support the claim and more careful high resolution imaging and in situ hybridization (with appropriate quantitation) will be needed to provide substantive support for the presence of a proximal tubule cell type with mitochondrial transcript that are trafficked to the nucleus.
Revision summary:
The authors have revised the manuscript to acknowledge the potential limitations of the C16 tool compound used and have performed some additional analyses that suggest the PT-Mito population can be identified in samples from KPMP. The authors added some control images for the in situ hybridizations, which are helpful, though they don't get to the core issue of limited resolution to determine whether mitochondrial RNA is present in the nuclei of injured PT cells. Some additional work has been done to show that C16 treatment results in a decrease in phospho-PKR, a readout of PKR inhibition. These changes strengthen the manuscript by providing some evidence for the translatability of the PT-mito cluster to humans and some evidence for on-target activity for C16. It would be helpful if the authors could quantify the numbers of cells in IHC with nuclear transcripts as well as pointing out some specific examples in the images provided, as comparator data for the snRNAseq studies in which 3-6% of cortex cells had evidence of nuclear mitochondrial transcripts.
-
Reviewer #2 (Public Review):
Summary:
Numerous studies by the authors and other groups have demonstrated an important role for HIV gene expression kidney cells in promoting progressive chronic kidney disease, especially HIV associated nephropathy. The authors had previously demonstrated a role for protein kinase R (PKR) in a non-HIV transgenic model of kidney disease (Okamoto, Commun Bio, 2021). In this study, the authors used innovative techniques including bulk and single nuclear RNAseq to demonstrate that mice expressing a replication-incompetent HIV transgene have prominent dysregulation of mitochondrial gene expression and activation of PKR and that treatment of these mice with a small molecule PKR inhibitor ameliorated the kidney disease phenotype in HIV-transgenic mice. They also identified STAT3 as a key upstream regulator of …
Reviewer #2 (Public Review):
Summary:
Numerous studies by the authors and other groups have demonstrated an important role for HIV gene expression kidney cells in promoting progressive chronic kidney disease, especially HIV associated nephropathy. The authors had previously demonstrated a role for protein kinase R (PKR) in a non-HIV transgenic model of kidney disease (Okamoto, Commun Bio, 2021). In this study, the authors used innovative techniques including bulk and single nuclear RNAseq to demonstrate that mice expressing a replication-incompetent HIV transgene have prominent dysregulation of mitochondrial gene expression and activation of PKR and that treatment of these mice with a small molecule PKR inhibitor ameliorated the kidney disease phenotype in HIV-transgenic mice. They also identified STAT3 as a key upstream regulator of kidney injury in this model, which is consistent with previously published studies. Other important advances include identifying the kidney cell types that express the HIV transgene and have dysregulation of cellular pathways.
Strengths:
Major strengths of the study include the use of a wide variety of state-of-the-art molecular techniques to generate important new data on the pathogenesis of kidney injury in this commonly used model of kidney disease and the identification of PKR as a potential druggable target for the treatment of HIV-induced kidney disease. The authors also identify a potential novel cell type within the kidney characterized by high expression of mitochondrial genes.
Weaknesses:
Though the HIV-transgenic model used in these studies results in a phenotype that is very similar to HIV-associated nephropathy in humans, the model has several limitations that may prevent direct translation to human disease, including the fact that mice lack several genetic factors that are important contributors to HIV and kidney pathogenesis in humans. Additional studies are therefore needed to confirm these findings in human kidney disease.
-
-
eLife assessment
This study presents valuable new insights into HIV-associated nephropathy (HIVAN) kidney phenotype in the Tg26 transgenic mouse model, and delineates the kidney cell types that express HIV genes and are injured in these HIV-transgenic mice. A series of compelling experiments demonstrated that PKR inhibition can ameliorate HIVAN with reversal of mitochondrial dysfunction (mainly confined to endothelial cells), a prominent feature shared in other kidney diseases. Although there are concerns regarding the specificity of C16 to PKR inhibition, as well as with the in situ hybridization studies, the data suggests that inhibition of PKR and mitochondrial dysfunction has potential clinical significance for HIVAN.
-
Reviewer #1 (Public Review):
Summary:
HIV-associated nephropathy (HIVAN) is a rapidly progressing form of kidney disease that manifests secondary to untreated HIV infection, and is predominantly seen in individuals of African descent. Tg26 mice carrying an HIV transgene lacking gag and pol exhibit high levels of albuminuria and rapid decline in renal function that recapitulates many features of HIVAN in humans. HIVAN is seen predominantly in individuals carrying two copies of missense variants in the APOL1 gene, and the authors have previously shown that APOL1 risk variant mRNA induces activity of the double-strand RNA sensor kinase PKR. Because of the tight association between the APOL1 risk genotype and HIVAN, the authors hypothesized that PKR activation may mediate renal injury in Tg26 mice, and tested this hypothesis by treating …
Reviewer #1 (Public Review):
Summary:
HIV-associated nephropathy (HIVAN) is a rapidly progressing form of kidney disease that manifests secondary to untreated HIV infection, and is predominantly seen in individuals of African descent. Tg26 mice carrying an HIV transgene lacking gag and pol exhibit high levels of albuminuria and rapid decline in renal function that recapitulates many features of HIVAN in humans. HIVAN is seen predominantly in individuals carrying two copies of missense variants in the APOL1 gene, and the authors have previously shown that APOL1 risk variant mRNA induces activity of the double-strand RNA sensor kinase PKR. Because of the tight association between the APOL1 risk genotype and HIVAN, the authors hypothesized that PKR activation may mediate renal injury in Tg26 mice, and tested this hypothesis by treating mice with a commonly used PKR inhibitory compound called C16. Treatment with C16 substantially attenuated renal damage in the Tg26 model as measured by urinary albumin/creatinine ratio, urinary NGAL/creatinine ratio, and improvement in histology. The authors then performed bulk and single-nucleus RNAseq on kidneys from mice from different treatment groups to identify pathways and patterns of cell injury associated with HIV transgene expression as well as to determine the mechanistic basis for the effect of C16 treatment. They show that proximal tubule nuclei from Tg26 mice appear to have more mitochondrial transcripts which was reversed by C16 treatment and suggest that this may provide evidence of mitochondrial dysfunction in this model. They explore this hypothesis by showing there is a decrease in the expression of nuclear-encoded genes and proteins involved in oxidative phosphorylation as well as a decrease in respiratory capacity via functional assessment of respiration in tubule and glomerular preparations from these mouse kidneys. All of these changes were reversed by C16 treatment. The authors propose the existence of a novel injured proximal tubule cell-type characterized by the leak of mitochondrial transcripts into the nucleus (PT-Mito). Analysis of HIV transgene expression showed high level expression in podocytes, consistent with the pronounced albuminuria that characterizes this model and HIVAN, but transcripts were also detected in tubular and endothelial cells. Because of the absence of mitochondrial transcripts in the podocytes, the authors speculate that glomerular mitochondrial dysfunction in this model is driven by damage to glomerular endothelial cells.
Strengths:
The strengths of this study include the comprehensive transcriptional analysis of the Tg26 model, including an evaluation of HIV transgene expression, which has not been previously reported. This data highlights that HIV transcripts are expressed in a subset of podocytes, consistent with the highly proteinuric disease seen in mice and humans. However, transcripts were also seen in other tubular cells, notably intercalated cells, principal cells and injured proximal tubule cells. Though the podocyte expression makes sense, the relevance of the tubular expression to human disease is still an open question.
The data in support of mitochondrial dysfunction are also robust and rely on combined evidence from downregulation of transcripts involved in oxidative phosphorylation, decreases in complex I and II as determined by immunoblot, and assessments of respiratory capacity in tubular and glomerular preparations. These data are largely consistent with other preclinical renal injury models reported in the literature as well as previous, less thorough assessments in the Tg26 model.
Weaknesses:
The key weakness of the study lies in the use of a PKR inhibitor with questionable specificity. C16 has been reported to inhibit numerous other kinases including cyclin CDKs and GSK3α and -β, and this means that the conclusions of this study with respect to the role of PKR are highly questionable. The rationale for the dose used was not provided (and is lower than used in other publications with C16), and in the absence of drug exposure data and assessment of target engagement, it is difficult to ascertain whether substantial inhibition of PKR was achieved.
A second key weakness lies in the identification of the PT-Mito cell cluster. Though the authors provide some rationale for the identification of this specific cell type, it seems equally plausible the cells merely reflect a high background capture of mitochondria in a subset of droplets. The IHC analysis that was provided is not convincing enough to support the claim and more careful high resolution imaging and in situ hybridization (with appropriate quantitation) will be needed to provide substantive support for the presence of a proximal tubule cell type with mitochondrial transcript that are trafficked to the nucleus.
-
Reviewer #2 (Public Review):
Summary:
Numerous studies by the authors and other groups have demonstrated an important role for HIV gene expression kidney cells in promoting progressive chronic kidney disease, especially HIV-associated nephropathy. The authors had previously demonstrated a role for protein kinase R (PKR) in a non-HIV transgenic model of kidney disease (Okamoto, Commun Bio, 2021). In this study, the authors used innovative techniques including bulk and single nuclear RNAseq to demonstrate that mice expressing a replication-incompetent HIV transgene have prominent dysregulation of mitochondrial gene expression and activation of PKR and that treatment of these mice with a small molecule PKR inhibitor ameliorated the kidney disease phenotype in HIV-transgenic mice. They also identified STAT3 as a key upstream regulator of …Reviewer #2 (Public Review):
Summary:
Numerous studies by the authors and other groups have demonstrated an important role for HIV gene expression kidney cells in promoting progressive chronic kidney disease, especially HIV-associated nephropathy. The authors had previously demonstrated a role for protein kinase R (PKR) in a non-HIV transgenic model of kidney disease (Okamoto, Commun Bio, 2021). In this study, the authors used innovative techniques including bulk and single nuclear RNAseq to demonstrate that mice expressing a replication-incompetent HIV transgene have prominent dysregulation of mitochondrial gene expression and activation of PKR and that treatment of these mice with a small molecule PKR inhibitor ameliorated the kidney disease phenotype in HIV-transgenic mice. They also identified STAT3 as a key upstream regulator of kidney injury in this model, which is consistent with previously published studies. Other important advances include identifying the kidney cell types that express the HIV transgene and have dysregulation of cellular pathways.Strengths:
Major strengths of the study include the use of a wide variety of state-of-the-art molecular techniques to generate important new data on the pathogenesis of kidney injury in this commonly used model of kidney disease and the identification of PKR as a potential druggable target for the treatment of HIV-induced kidney disease. The authors also identify a potential novel cell type within the kidney characterized by high expression of mitochondrial genes.Weaknesses:
Though the HIV-transgenic model used in these studies results in a phenotype that is very similar to HIV-associated nephropathy in humans, the model has several limitations that may prevent direct translation to human disease, including the fact that mice lack several genetic factors that are important contributors to HIV and kidney pathogenesis in humans. Additional studies are therefore needed to confirm these findings in human kidney disease. -
