Proteomic characteristics reveal the signatures and the risks of T1 colorectal cancer metastasis to lymph nodes
Curation statements for this article:-
Curated by eLife

eLife assessment
This is an important contribution to colorectal cancer research to understand how we can predict high-risk patients for recurrence. The strength of the evidence is solid and looks at a novel approach but an approach that still has opportunities to improve.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The presence of lymph node metastasis (LNM) affects treatment strategy decisions in T1NxM0 colorectal cancer (CRC), but the currently used clinicopathological-based risk stratification cannot predict LNM accurately. In this study, we detected proteins in formalin-fixed paraffin-embedded (FFPE) tumor samples from 143 LNM-negative and 78 LNM-positive patients with T1 CRC and revealed changes in molecular and biological pathways by label-free liquid chromatography tandem mass spectrometry (LC-MS/MS) and established classifiers for predicting LNM in T1 CRC. An effective 55-proteins prediction model was built by machine learning and validated in a training cohort (N=132) and two validation cohorts (VC1, N=42; VC2, N=47), achieved an impressive AUC of 1.00 in the training cohort, 0.96 in VC1 and 0.93 in VC2, respectively. We further built a simplified classifier with nine proteins, and achieved an AUC of 0.824. The simplified classifier was performed excellently in two external validation cohorts. The expression patterns of 13 proteins were confirmed by immunohistochemistry, and the IHC score of five proteins was used to build an IHC predict model with an AUC of 0.825. RHOT2 silence significantly enhanced migration and invasion of colon cancer cells. Our study explored the mechanism of metastasis in T1 CRC and can be used to facilitate the individualized prediction of LNM in patients with T1 CRC, which may provide a guidance for clinical practice in T1 CRC.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
This paper by Zhuang and colleagues seeks to answer an important clinical question by trying to come up with novel predictive biomarkers to predict high-risk T1 colorectal cancers that are at risk for nodal involvement. The current clinical features may both miss patients who underwent local therapy and who should have gone on to have surgery and patients for whom surgery was done based on risk features but perhaps unnecessarily. Using a training and validation set, they developed a protein-based classifier with an AUC of 0.825 based on mass spec analyses and proteomic analyses of patients with and without LN importantly linking biological rationale to the proteomic discoveries.
In the training cohort, they took 105 candidate proteins reduced to 55, and did a validation in the training …
Author Response
Reviewer #1 (Public Review):
This paper by Zhuang and colleagues seeks to answer an important clinical question by trying to come up with novel predictive biomarkers to predict high-risk T1 colorectal cancers that are at risk for nodal involvement. The current clinical features may both miss patients who underwent local therapy and who should have gone on to have surgery and patients for whom surgery was done based on risk features but perhaps unnecessarily. Using a training and validation set, they developed a protein-based classifier with an AUC of 0.825 based on mass spec analyses and proteomic analyses of patients with and without LN importantly linking biological rationale to the proteomic discoveries.
In the training cohort, they took 105 candidate proteins reduced to 55, and did a validation in the training cohort first and then in two validation cohorts (one of which was prospective). They also looked at a 9-protein classifier which also performed well and furthermore looked at IHC for clinical ease.
We appreciate the reviewers for the positive review and valuable comments. We have revised the manuscript according to the comments.
Reviewer #2 (Public Review):
The authors utilized a label-free LC-MS/MS analysis in formalin-fixed paraffin-embedded (FFPE) tumors from 143 LNM-negative and 78 LNM-positive patients with T1 CRC to identify protein biomarkers to determine LNM in T1 CRC.
The authors used a fair number of clinical samples for the proteomics investigation. The experimental design is reasonable, and the statistical methods used in this manuscript are solid.
The authors largely achieved their aims and the results supported their conclusion. The method used in this proteomic study can also be used for the proteomics analysis of other cancer types to identify diagnostic and prognostic biomarkers. In addition, the 9-marker panel has a potential clinical diagnosis practice in determining LNM in T1 CRC.
Nevertheless, the authors need to justify their standards in selecting the biomarkers. For example, a p-value cut-off of 0.1 is not a usual criterion in similar proteomic studies. In addition, an identification frequency of 30% in patients seems not preferable for biomarker identification. The authors also need to justify the definition of fold change in the three subtypes with Kruskal-Walli's test. The authors need to describe more details on how they identified the 13 proteins from a 55-protein database. In addition, what is the connection between the final 9 proteins and the 19 proteins? What is the criterion to select 5 proteins for IHC validation from the 9 proteins?
We appreciate the reviewers for the positive review and valuable comments. We have revised the manuscript according to the comments.
The criteria and details of our standards in selecting are as follows.
- About p-value cut-off of 0.1:
The purpose of this step is to screen appropriate variables for subsequent machine learning, rather than comparing differences between groups. The p-value cut-off of 0.1 is also a reliable strategy for variable selection in proteomics research. For example, it has been used in studies to predict the response to tumor necrosis factor-α inhibitors in rheumatoid arthritis (PMID: 28650254); the research about circadian clock in mouse liver (PMID: 29674717); the proteomic biomarker discovery in atherosclerosis (PMID: 15496433); and the proteomics and transcriptomics analysis in bacillus subtilis (PMID: 19948795).
Based on reviewer’s suggestion, we used a cutoff of p-value 0.05 to screen for variables. In a training set of 70 lymph node-negative and 62 lymph node-positive cases, we identified 355 protein markers. We further incorporated these proteins into a lasso regression analysis and ultimately developed a lymph node metastasis prediction model consisting of 52 protein markers. We validated the model in VC1 and VC2, with AUC values of 1.000, 0.824, and 0.918 for the training set, VC1, and VC2, respectively, the predictive performance was slightly inferior to that of the model developed in this study (Figure 3- figure supplement 1C).
- About identification frequency of 30%:
The analysis focusing on the proteins identified in > 30% of the samples has been applied in the previous published studies. For instance, the study of using proteomic biomarkers to build diagnostic model in lung cancer (PMID: 29576497), proteins identified in > 30% cohort samples were used for downstream analysis. In the study on the impact of Reptin on protein-protein interaction (PMID: 30862565) have demonstrated that proteins were required to have at least in > 30% of samples in order to be included in the proteome dataset.
We compared our cohort with Jun Qin et al. and Bing Zhang et al., study published in Nature (PMID: 25043054), according to the number of the proteins detected in more than 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%, 90%, 100% of samples, respectively (Figure 2- figure supplement 1). The proportion proteins detected at different cutoff of the samples in the three cohort were, 10% (0.60, 0.94, 0.48), 20% (0.52, 0.83, 0.38), 30% (0.46,0.75, 0.31), 40% (0.41, 0.69, 0.26), 50% (0.37, 0.63, 0.23), 60% (0.33, 0.57, 0.18), 70% (0.29, 0.52, 0.15), 80% (0.25, 0.45, 0.11), 90% (0.19, 0.37, 0.11), 100% (0.07, 0.23, 0.10), respectively. The results showed that our cohort was reliable.
To investigate the impacts of protein identification frequency cutoff in our study, we performed comparative pathway enrichment analysis of the differential expressed proteins (LNM+ vs. LNM-: p-value < 0.05, Wilcoxon rank-sum tests) under different observation percentiles, which were detected in more than 10%, more than 30% and more than 50% of samples, respectively. The results revealed that proteins from three thresholds (10%, 30% and 50%) represented similar pathway enrichment, such as mTOR signaling pathway and amino acid metabolism pathways were dominant in LNM-negative patients, coagulation cascades and Lipid metabolism pathways were overrepresented in the LNM-positive patients (Figure 2- figure supplement 1)
Based on reviewer’s suggestion, we used a cutoff of 50% as identification frequency for variables. The lasso regression was carried out in training cohort (70 LNM-negative and 62 LNM-positive), with AUC of 0.999. The model was validated in VC1 and VC2, with AUC of 0.812 and 0.886, respectively. (Figure 2- figure supplement 1).
- About identification of the 13 proteins and the criterion to select 5 proteins for IHC validation from 55-protein database:
The process of reducing the number of proteins from 55 to 13 and finally establishing a 5-molecule classifier based on the IHC score is as shown in Figure 1- figure supplement 2 in the revision. We first selected 19 proteins with [log2FC] > 1 or < -1 and p<0.05 (Wilcoxon rank-sum test) between the LNM-negative and LNM-positive in 221 patients from 55 proteins. Then we started looking for antibodies to these 19 proteins. We finally obtained 13 antibodies for further immunohistochemistry. We did immunohistochemical staining to the FFPE samples with 13 antibodies, and got the IHC score of each protein to build the single molecular prediction model by SPSS on ROC curve. For the principles of MS based proteomic and IHC stain are different, not all identified proteins can be converted into IHC. Finally, 5 IHC makers with p-value of IHC score less than 0.05 (Student’s t-test) were selected to build the IHC classifier using Logistic Regression. We also updated the description in the “Result” section in the revised manuscript (line 718-722, page 34-35 in the revision).
- About the connection between the final 9 proteins and the 19 proteins:
To facilitate the clinical translation of the model, Multiple Logistic Regression was used to obtain 9 core proteins from 19 proteins (Figure 1- figure supplement 2 in the revision). We first performed logistic regression in 19 proteins, and eliminated 10 proteins with insignificant Estimate Std. Error z value (Pr (>|z|) > 0.05, and obtained 9 proteins with Pr(>|z|) < 0.05. After that, we carried out Binary Logistic Regression calculation again with 9 proteins to build the simplified classifier. We also updated the description in the “Materials and methods” section in the revised manuscript (line 1092, page 51 in the revision).
- About the definition of fold change in the three subtypes with Kruskal-Walli's test:
The fold change in the three subtypes is the ratio of the mean of the expressions in each group (well to moderately differentiated adenocarcinoma, poorly differentiated adenocarcinoma and mucinous adenocarcinoma) to the mean of the other two group. Kruskal-Walli's test was performed between three subtypes.
We also updated the description in the “Result” section in the revised manuscript (line 506-517, page 25 in the revision), and “Figure 1- figure supplement 2H in the revision”.
Reviewer #3 (Public Review):
This work provides a proteomic analysis of 132 early-stage (pT1) colorectal cancers (CRC) to attempt to identify proteins (or a signature pattern thereof) that might be used to predict the patient risk of lymph node metastases (LNM) and potentially stratify patients for further treatment or surveillance. The generated dataset is extensive and the methods appear solid. The work identifies a 55-protein signature that is strongly predictive of LNM in the training cohort and two validation cohorts and then generates two simplified classifiers: a 9-protein proteomic and a 5-protein immunohistochemical classifier. These also perform very well in predicting LNM. Loss of the small GTPase RHOT2 is identified as a poor prognostic factor and validated in a migration assay. The findings could allow better prognostication in CRC and, if confirmed and better validated and contextualized, might impact patient care.
Strengths:
A large training cohort of resected early-stage (pT1M0) CRCs was analyzed by rigorous methods including careful quantitative analysis. The data generated are unbiased and potentially useful. A number of proteins are found to be different between CRCs with and without lymph node metastases, which are used to train a machine learning model that performs flawlessly in predicting LNM in the training cohort and very well in predicting LNM in two validation cohorts. The authors then develop two simplified classifiers that might be more readily extended into clinical care: a 9-protein proteomic assay and a 5-protein immunohistochemical assay; both of these also perform well in predicting LNM. Because LNM is a key prognostic factor, and colectomy (which includes removal of lymph nodes needed to assess LNM) carries significant risk and morbidity, particularly in rectal cancer, classifiers like these are potentially interesting. Finally, the authors identify the loss of expression of RHOT2 as a novel prognostic factor.
Weaknesses:
Major points:
The data are limited by a number of assumptions about metastasis, minimal contextualization of the results, and claims that are too strong given the data. Critically, the authors use the presence or absence of LNM as the study's only outcome; while LNM is a key predictor in CRC, it is uncommon in T1 CRC (generally 3-10%, 12% in this study), stochastic, inefficient, and incompletely identified by histologic evaluation. Larger resection (here, colectomy) removes both identified and occult LNM, which is probably best studied in randomized trials of lymphadenectomy in Japanese gastric cancer cohorts and should be better discussed. Critically, patient survival or disease-free survival would be more relevant outcomes. Further, absent longer-term data, many patients without identified LNM might nonetheless be high-risk and skew the cohorts. It is also not clear whether these findings would be generalizable to other early-stage colon cancers.
The data are also not correlated with the genetics of the cases, which were not discussed.
The results would benefit from the inclusion of standard-of-care MSI status. The classifiers would also be much more impactful if they were generalizable beyond T1 CRCs; this could be readily tested in public datasets.
The authors explain the data as mechanistic, but, aside from one experiment modulating RHOT2 levels, they are fundamentally correlative and should be described as such.
Although they focused on areas containing >80% tumor as judged by the reading pathologist, it is unclear whether the identified proteomic changes originate from the tumor or the microenvironment.
The authors fail to properly contextualize the results or overstate the novelty of their study. A number of examples - the study is claimed as "the first proteomic study of T1 CRC" and "the first comprehensive proteomics study to focus on LNM in patients with submucosal T1 CRCs"; neither of these appears to be true, for example, Steffen et al. (Journal of Proteome Research, 2021, reference 18) may satisfy both of these, although the numbers are smaller. Many other results are reported without context, for example, proteomic characterization of mucinous carcinomas has been performed previously, a modest correlation in mucinous carcinoma is ascribed a large mechanistic role, and PDPN is discussed but is not contextualized as a protein that has been well-studied in the context of metastasis.
The data on RHOT2 are promising but very preliminary. RHOT2 is described as ubiquitous in colorectal cancer cell lines; a brief search in Human Protein Atlas shows RHOT2 RNA and proteins are ubiquitously expressed throughout the body. While its loss appears potentially prognostic, it is unclear whether this is simply a surrogate for other features, such as loss of differentiation state, and whether this is unique to CRC; multivariate analysis would be important.
We appreciate the reviewer for the constructive and insightful comments, which help to improve the quality of this manuscript. Here, we summarized the reviewer’s comments as following: (1) Lack of longer-term data and micrometastasis; (2) test the classifier in public datasets; (3) inclusion of standard genetics and gene alterations; (4) about the tumor purity of all tumor samples and whether the results were influenced by the tumor microenvironment; (5) contextualize the results; (6) multivariate analysis of RHOT2.
- Lack of longer-term data and micrometastasis:
Thank the reviewer for the comments. We fully acknowledge the limitations of our study, including the uncertainty associated with the detection of lymph node micrometastasis and the lack of long-term survival data, which can impact the strength of our conclusions. We agree that LNM is a key predictor in CRC and that it is uncommon in T1 CRC, with a reported incidence of 3-10%. We acknowledge that larger resections, such as colectomy, are generally recommended for patients with T1 CRC with LNM due to the potential risk of metastasis. However, our study aimed to establish a predictive model for LNM in T1 CRC, which could potentially help guide clinical decision-making on whether additional surgery is needed after endoscopic resection, according to the current NCCN guidelines.
We have taken following methods to address these limitations:
We matched propensity-score of patients to reduce confounding biases in our training cohort, and patients were prospectively enrolled in our validation cohort, which was designed as a single-blinded prospective study to enhance the rigor and reliability of our findings.
For the influence of micrometastases in our study. According to reviewer's suggestion, we discussed the reports related to lymph nodes micrometastases in Japanese gastric cancer cohorts (PMID: 17377930, 9070482), and at the same time, we consulted the articles about micrometastases in T1 CRC (PMID: 17661146, 16412600). There were about 5% pT1N0 gastric cancer patients have ITCs in LN, and 10% in pT1Nx CRC. The effect of MMs on prognosis in pT1N0 CRC is still unclear. The present of ITCs/MMs in LN may explain why there are nearly 13% (29 of 221) LNM-negative patients were classified into high-risk group by the prediction model in our study.
We have also added a section to the “Discussion” in the revised manuscript to discuss the potential impact of these limitations on the interpretation of our findings (line 856-873, page 41) in the revision, as follow:
“In this study, to ensure the accuracy of LN status of the enrolled patients, the dissected number of LN in all patients including both surgical resection and ESD was more than 12. However, the longer-term follow-up data, including DFS, PFS, etc., are not available, due to limitations in sample collection time and the prognosis of such patients needs to be tracked over long periods of time, and may impact the strength of our conclusions. To address this limitation, we used propensity-score matching to reduce confounding biases in our training cohort. Patients were prospectively enrolled in our validation cohort (VC2), which was designed as a single-blinded prospective study to enhance the rigor and reliability of our findings. Furthermore, the presence of isolated tumor cells (ITCs) or micrometastases (MMs) within regional LN are not considered, due to conventional histopathologic examination cannot detected them. According to previous studies, there were about 5% pT1N0 gastric cancer patients have ITCs in LN, and 10% in pT1Nx CRC. The effect of MMs on prognosis in pT1N0 CRC is still unclear. The present of ITCs/MMs in LN may explain why there are nearly 13% (29 of 221) LNM-negative patients were classified into high-risk group by the prediction model in our study. Our study would provide a valuable database and could help for clinical decision-making in the context of T1 CRC. We will continuously follow the prognosis of the patients, and the ITCs/MMs in LN also need to be further validated in the future studies.”
In conclusion, we appreciate reviewer’s comments and acknowledge the limitations of our study. We believe that our study provides valuable insights into the development of a predictive model for LNM in T1 CRC, which could potentially aid in clinical decision-making according to the current NCCN guidelines.
- Test the classifier in public datasets:
According to reviewer’s suggestions, we tested our classifier in two different public datasets, including the colon and rectal cancer study from CPTAC published in Nature (PMID: 25043054), and the metastatic colorectal cancer study published in Cancer Cell (PMID: 32888432). The detail was further discussed in “point-to-point responses R3 Q2.”.
- Standard genetics and gene alterations:
According to reviewer’s suggestions, we assessed MSI status and CRC-associated gene mutations (RAS, BRAF and PIK3CA) in our cohort. The detail was further discussed in “point-to-point responses R3 Q1.”
- The influence of microenvironment:
We apologized for not explaining it clearly. To the question of whether the differences between two groups (LNM+ and LNM-) are caused by tumor microenvironment or the tumor tissues, we firstly, used xCell (PMID: 29141660) to study the composition of the tumor microenvironment (Figure2-source data 4 in the revision). The results showed that there was no difference in the tumor microenvironment between the LNM-positive and negative groups (P > 0.05, Wilcoxon rank-sum test) (Figure RL1A). However, when we compared the xCell algorism-based cell deconvolution results between the LNM-positive and -negative groups, we found 8 microenvironment associated cell features differed in two groups (p<0.05) (Figure RL1B). LNM-positive patients were featured with Chondrocytes and Th1 cells. And the remaining 6 features are all high in LNM-negative patients, including, B cells, cDC, Myocytes, etc. Correspondingly, 7 immune cell markers were also observed to be significantly different between the two groups (Log2FC>1 or <-1, P > 0.05, Wilcoxon rank-sum test) (Figure RL1C).
Secondly, we checked the expression profile of the signature proteins detected in our study by The Human Protein Atlas (HPA). Among 9404 identified proteins, 7852 (83.4%) have HPA’s CRC IHC staining data, and 6249 (79.6%) showed medium to high tumor-specific staining in CRC samples (Figure RL1D). Of the signature proteins up-regulated in LNM-positive patients (LNM+ vs. LNM-: log2FC > 1 and p<0.05, Wilcoxon rank-sum test), 76 of 84 (90.5%) have IHC staining data in HPA, and 63 (82.9%) showed medium to high tumor-specific staining in CRC samples (Figure RL1E). For specific proteins of LNM-negative patients (LNM+ vs. LNM-: log2FC <-1 and p<0.05, Wilcoxon rank-sum test), 72 of 82 (87.8%) have IHC staining data in HPA, and 60 (83.3%) showed medium to high tumor-specific staining in CRC samples (Figure RL1F).
Finally, we reviewed again all H&E-stained slides of tumor tissues of patients involved in the study, and supplemented tumor purity values of tumor samples of all the patients in Figure1-source data 1. We compared the tumor purity between the LNM-positive (with average 87.75%) and negative patients (with average 88.27%). The result showed there was no difference between the two groups (P = 0.46, Student’s t-test), demonstrating the high purity and quality of the tumor tissues. (Figure1-supplementary figure 1J in the revision).
These results indicate that, in our study the differences between LNM-positive and LNM-negative groups are mainly caused by tumor tissues. However, the tumor microenvironment may also play a critical but not direct role in T1 CRC development and progression.
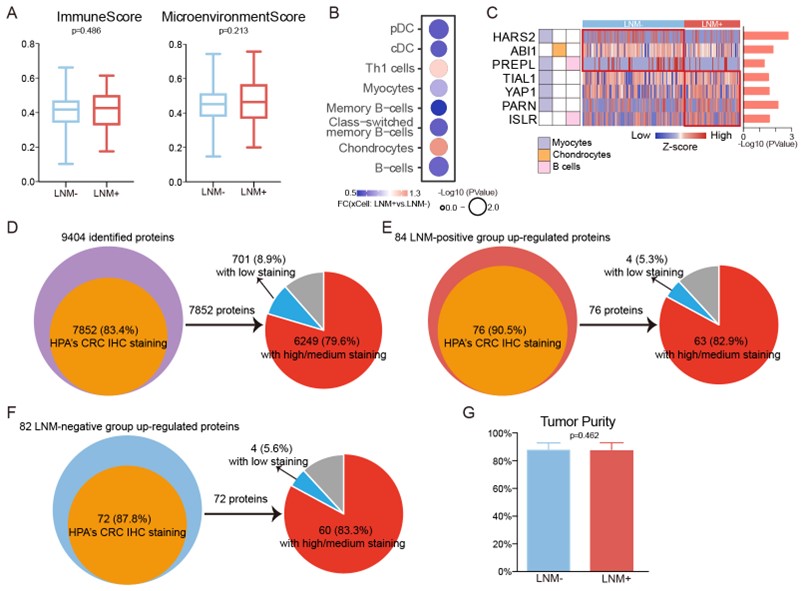
Figure RL1. A. Comparison of xCell scores of immune and microenvironment between the LNM-negative group (n= 143) and LNM-positive group (n= 78). B&C. Immune/stromal signatures identified from xCell, together with derived relative abundance of immune and stromal cell types. D, E, F. Identified signature proteins (D), LNM-positive group up-regulated proteins (E) and LNM-negative group up-regulated proteins (F) were mostly validated by HPA IHC Staining Data. G. Barplot for tumor purity between LNM-negative and -positive patients.
- Contextualize the results:
According to the reviewer’s advice, we have made corresponding adjustments in the revised manuscript, for example:
- “We have made a comprehensive proteomic study of T1 CRC and provides a reliable data source for future research. “(line 342, page 17 in the revision)
-“Here, we present a comprehensive proteomic study to focus on LNM in patients with submucosal T1 CRCs.” (line 788, page 37 in the revision)
With regard to the problem of results are reported without context, we have provided supplementary descriptions of the context of the results in the “Result” section of the revised manuscript, for example:
“Mucinous adenocarcinoma was considered to be a significant risk factor of LNM in T1 CRC (PMID: 31620912).” (line 498, page 24 in the revision)
“Mucinous adenocarcinoma of the colorectal is a lethal cancer with unknown molecular etiology and a high propensity to lymph node metastasis. Previous proteomic studies on mucinous adenocarcinoma have found the proteins associated with treatment response in rectal mucinous adenocarcinoma and mechanisms of metastases in mucinous salivary adenocarcinoma.” (PMID: 34990823, 28249646) (line 534-538, page 26 in the revision)
“Previous studies have shown that PDPN expression correlated with LNM in numerous cancers, especially in early oral squamous cell carcinomas.” (PMID: 21105028).” (line 570, page 27 in the revision)
- Multivariate analysis of RHOT2:
RHOT2 and its paralog RHOT1 plays an important role in mitochondrial trafficking (PMID: 16630562). Although the function of RHOT2 in cancer is still unknown, the expression of RHOT1 affects metastasis in a variety of tumors, including pancreatic cancer (PMID: 26101710), gastric cancer (PMID: 35170374), small cell lung cancer (PMID: 33515563), etc. In addition, previous studies have found that Myc regulation of mitochondrial trafficking through RHOT1 and RHOT2 enables tumor cell motility and metastasis (PMID: 31061095).
As shown in Figure 4, in our analysis of previous version, we found RHOT2 was significant down-regulated (Log2FC=-1.35; p=0.003, Wilcoxon rank-sum test) in LNM-positive patients compared with LNM-negative patients in our T1 CRC cohort and the low level of RHOT2 is related to low overall survival of patients with colon cancer in TCGA cohort. Knockdown of RHOT2 expression could markedly enhance the migration ability of colon cancer cells.
In order to further explore the influence of RHOT2 on T1 CRC LNM, in addition to the previous results, we carried out the following analysis as shown in Figure4 in the revision.
We, firstly, calculated the correlations between the expression of RHOT2 and other proteins in our cohort (Figure 4). 1,508 proteins were correlated significantly (P < 0.05, Spearman) with RHOT2, and 1,354 proteins showed a positive correlation (coefficient >0) with RHOT2, and 154 proteins were negatively correlated with RHOT2 (coefficient <0). However, when we performed GSEA in RHOT2-associated proteins to identify biological signatures impacted by RHOT2, most of the obtained pathways (p<0.01) showed NES less than 0, which means these pathways were mainly enriched in RHOT2-negative-correlated group, only “mitochondrion” (GOCC) had a positive correlation (Figure 4). As we known RHOT2 is an important protein involved in the regulation of mitochondrial dynamics and mitophagy (PMID: 16630562). This result indicates that the involvement of RHOT2 in regulation of mitochondrial function might contribute to the pathogenesis of metastasis in cancer, especially in early-stage CRC. Consistent with the previous results, RHOT2-negative-correlated group was significantly enriched for EMT (HALLMARK) and complement and coagulation cascades pathways. Proteins up-regulated in LNM-positive group (LNM+ vs. LNM-: Log2FC >0; p<0.05, Wilcoxon rank-sum test) were negatively correlated with RHOT2(p < 0.05, coefficient<0, Spearman), including CAP2, COL6A3, COL6A2, TNC, DPYSL3, PCOLCE and BGN in pathway EMT; and GUCY1B3, VWF and F13A1 in pathway complement and coagulation cascades (Figure 2E, L; Figure 4D in the revision). ECM, focal adhesion and Dilated cardiomyopathy (DCM) pathways were also enriched in negative-correlated group. Degradation of RHOT2 has already been reported to be associated with DCM (PMID: 31455181). Overall, combined with the previous results, RHOT2 may play an important role in T1 CRC LNM (Figure 4D in the revision.).
As reviewer mentioned the data on RHOT2 are promising, but the understanding of it is preliminary. More analytical studies and experiments are needed in our future researches to understand the specific role and mechanism of RHOT2 in the process of tumor metastasis. In the revision, we discussed these limitations of our research.
-
eLife assessment
This is an important contribution to colorectal cancer research to understand how we can predict high-risk patients for recurrence. The strength of the evidence is solid and looks at a novel approach but an approach that still has opportunities to improve.
-
Reviewer #1 (Public Review):
This paper by Zhuang and colleagues seeks to answer an important clinical question by trying to come up with novel predictive biomarkers to predict high-risk T1 colorectal cancers that are at risk for nodal involvement. The current clinical features may both miss patients who underwent local therapy and who should have gone on to have surgery and patients for whom surgery was done based on risk features but perhaps unnecessarily. Using a training and validation set, they developed a protein-based classifier with an AUC of 0.825 based on mass spec analyses and proteomic analyses of patients with and without LN importantly linking biological rationale to the proteomic discoveries.
In the training cohort, they took 105 candidate proteins reduced to 55, and did a validation in the training cohort first and then …
Reviewer #1 (Public Review):
This paper by Zhuang and colleagues seeks to answer an important clinical question by trying to come up with novel predictive biomarkers to predict high-risk T1 colorectal cancers that are at risk for nodal involvement. The current clinical features may both miss patients who underwent local therapy and who should have gone on to have surgery and patients for whom surgery was done based on risk features but perhaps unnecessarily. Using a training and validation set, they developed a protein-based classifier with an AUC of 0.825 based on mass spec analyses and proteomic analyses of patients with and without LN importantly linking biological rationale to the proteomic discoveries.
In the training cohort, they took 105 candidate proteins reduced to 55, and did a validation in the training cohort first and then in two validation cohorts (one of which was prospective). They also looked at a 9 protein classifier which also performed well and furthermore looked at IHC for clinical ease.
-
Reviewer #2 (Public Review):
The authors utilized a label-free LC-MS/MS analysis in formalin-fixed paraffin-embedded (FFPE) tumors from 143 LNM-negative and 78 LNM-positive patients with T1 CRC to identify protein biomarkers to determine LNM in T1 CRC.
The authors used a fair number of clinical samples for the proteomics investigation. The experimental design is reasonable, and the statistical methods used in this manuscript are solid.
The authors largely achieved their aims and the results supported their conclusion. The method used in this proteomic study can also be used for the proteomics analysis of other cancer types to identify diagnostic and prognostic biomarkers. In addition, the 9 marker panel has a potential clinical diagnosis practice in determining LNM in T1 CRC.
Nevertheless, the authors need to justify their standards in …
Reviewer #2 (Public Review):
The authors utilized a label-free LC-MS/MS analysis in formalin-fixed paraffin-embedded (FFPE) tumors from 143 LNM-negative and 78 LNM-positive patients with T1 CRC to identify protein biomarkers to determine LNM in T1 CRC.
The authors used a fair number of clinical samples for the proteomics investigation. The experimental design is reasonable, and the statistical methods used in this manuscript are solid.
The authors largely achieved their aims and the results supported their conclusion. The method used in this proteomic study can also be used for the proteomics analysis of other cancer types to identify diagnostic and prognostic biomarkers. In addition, the 9 marker panel has a potential clinical diagnosis practice in determining LNM in T1 CRC.
Nevertheless, the authors need to justify their standards in selecting the biomarkers. For example, a p-value cut-off of 0.1 is not a usual criterion in similar proteomic studies. In addition, an identification frequency of 30% in patients seems not preferable for biomarker identification. The authors also need to justify the definition of fold change in the three subtypes with Kruskal-Walli's test. The authors need to describe more details on how they identified the 13 proteins from a 55-protein database. In addition, what is the connection between the final 9 proteins and the 19 proteins? What is the criterion to select 5 proteins for IHC validation from the 9 proteins?
-
Reviewer #3 (Public Review):
This work provides a proteomic analysis of 132 early-stage (pT1) colorectal cancers (CRC) to attempt to identify proteins (or a signature pattern thereof) that might be used to predict the patient risk of lymph node metastases (LNM) and potentially stratify patients for further treatment or surveillance. The generated dataset is extensive and the methods appear solid. The work identifies a 55-protein signature that is strongly predictive of LNM in the training cohort and two validation cohorts and then generates two simplified classifiers: a 9-protein proteomic and a 5-protein immunohistochemical classifier. These also perform very well in predicting LNM. Loss of the small GTPase RHOT2 is identified as a poor prognostic factor and validated in a migration assay. The findings could allow better …
Reviewer #3 (Public Review):
This work provides a proteomic analysis of 132 early-stage (pT1) colorectal cancers (CRC) to attempt to identify proteins (or a signature pattern thereof) that might be used to predict the patient risk of lymph node metastases (LNM) and potentially stratify patients for further treatment or surveillance. The generated dataset is extensive and the methods appear solid. The work identifies a 55-protein signature that is strongly predictive of LNM in the training cohort and two validation cohorts and then generates two simplified classifiers: a 9-protein proteomic and a 5-protein immunohistochemical classifier. These also perform very well in predicting LNM. Loss of the small GTPase RHOT2 is identified as a poor prognostic factor and validated in a migration assay. The findings could allow better prognostication in CRC and, if confirmed and better validated and contextualized, might impact patient care.
Strengths:
A large training cohort of resected early-stage (pT1M0) CRCs was analyzed by rigorous methods including careful quantitative analysis. The data generated are unbiased and potentially useful. A number of proteins are found to be different between CRCs with and without lymph node metastases, which are used to train a machine learning model that performs flawlessly in predicting LNM in the training cohort and very well in predicting LNM in two validation cohorts. The authors then develop two simplified classifiers that might be more readily extended into clinical care: a 9-protein proteomic assay and a 5-protein immunohistochemical assay; both of these also perform well in predicting LNM. Because LNM is a key prognostic factor, and colectomy (which includes removal of lymph nodes needed to assess LNM) carries significant risk and morbidity, particularly in rectal cancer, classifiers like these are potentially interesting. Finally, the authors identify the loss of expression of RHOT2 as a novel prognostic factor.Weaknesses:
Major points:
The data are limited by a number of assumptions about metastasis, minimal contextualization of the results, and claims that are too strong given the data. Critically, the authors use the presence or absence of LNM as the study's only outcome; while LNM is a key predictor in CRC, it is uncommon in T1 CRC (generally 3-10%, 12% in this study), stochastic, inefficient, and incompletely identified by histologic evaluation. Larger resection (here, colectomy) removes both identified and occult LNM, which is probably best studied in randomized trials of lymphadenectomy in Japanese gastric cancer cohorts and should be better discussed. Critically, patient survival or disease-free survival would be more relevant outcomes. Further, absent longer-term data, many patients without identified LNM might nonetheless be high-risk and skew the cohorts. It is also not clear whether these findings would be generalizable to other early-stage colon cancers.The data are also not correlated with the genetics of the cases, which were not discussed. The results would benefit from the inclusion of standard-of-care MSI status. The classifiers would also be much more impactful if they were generalizable beyond T1 CRCs; this could be readily tested in public datasets.
The authors explain the data as mechanistic, but, aside from one experiment modulating RHOT2 levels, they are fundamentally correlative and should be described as such.
Although they focused on areas containing >80% tumor as judged by the reading pathologist, it is unclear whether the identified proteomic changes originate from the tumor or the microenvironment.
The authors fail to properly contextualize the results or overstate the novelty of their study. A number of examples - the study is claimed as "the first proteomic study of T1 CRC" and "the first comprehensive proteomics study to focus on LNM in patients with submucosal T1 CRCs"; neither of these appears to be true, for example, Steffen et al. (Journal of Proteome Research, 2021, reference 18) may satisfy both of these, although the numbers are smaller. Many other results are reported without context, for example, proteomic characterization of mucinous carcinomas has been performed previously, a modest correlation in mucinous carcinoma is ascribed a large mechanistic role, and PDPN is discussed but is not contextualized as a protein that has been well-studied in the context of metastasis.
The data on RHOT2 are promising but very preliminary. RHOT2 is described as ubiquitous in colorectal cancer cell lines; a brief search in Human Protein Atlas shows RHOT2 RNA and proteins are ubiquitously expressed throughout the body. While its loss appears potentially prognostic, it is unclear whether this is simply a surrogate for other features, such as loss of differentiation state, and whether this is unique to CRC; multivariate analysis would be important.
-
