Mutant SF3B1 promotes malignancy in PDAC
Curation statements for this article:-
Curated by eLife

eLife assessment
This work examines a role for altered splicing in pancreatic tumorigenesis by interrogating effects of a specific mutation in the Sf3b splicing factor in pancreatic organoid and cell line growth primarily, with some in vivo work also performed. There is significant potential in the study but there is a concern about the lack of in vivo validation of claims that are most relevant to metastatic progression and the focus on one specific mechanism at the expense of other possible effects on splicing of factors important for disease progression.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The splicing factor SF3B1 is recurrently mutated in various tumors, including pancreatic ductal adenocarcinoma (PDAC). The impact of the hotspot mutation SF3B1 K700E on the PDAC pathogenesis, however, remains elusive. Here, we demonstrate that Sf3b1 K700E alone is insufficient to induce malignant transformation of the murine pancreas, but that it increases aggressiveness of PDAC if it co-occurs with mutated KRAS and p53. We further show that Sf3b1 K700E already plays a role during early stages of pancreatic tumor progression and reduces the expression of TGF-β1-responsive epithelial–mesenchymal transition (EMT) genes. Moreover, we found that SF3B1 K700E confers resistance to TGF-β1-induced cell death in pancreatic organoids and cell lines, partly mediated through aberrant splicing of Map3k7 . Overall, our findings demonstrate that SF3B1 K700E acts as an oncogenic driver in PDAC, and suggest that it promotes the progression of early stage tumors by impeding the cellular response to tumor suppressive effects of TGF-β.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
The authors examine the role of the K700E mutation in the Sf3B1 splicing factor in PDAC and report that this Sf3B1 mutation promotes PDAC by decreasing sensitivity to TGF-b resulting in decreased EMT and decreased apoptosis as a result. They propose that the Sf3b1 K700E mutant causes decreased expression of Map3K7, a known mediator of TGF-β signaling and also known to be alternately spliced in other systems by the Sf3b1 K700E mutation. The role of splicing defects in cancer is relatively understudied and could identify novel targets for therapeutic intervention so this work is of potential significance. However, the data is over-interpreted in many instances and it is not clear the authors can make the claims they do based on the data shown. In particular, the data showing that decreased …
Author Response
Reviewer #1 (Public Review):
The authors examine the role of the K700E mutation in the Sf3B1 splicing factor in PDAC and report that this Sf3B1 mutation promotes PDAC by decreasing sensitivity to TGF-b resulting in decreased EMT and decreased apoptosis as a result. They propose that the Sf3b1 K700E mutant causes decreased expression of Map3K7, a known mediator of TGF-β signaling and also known to be alternately spliced in other systems by the Sf3b1 K700E mutation. The role of splicing defects in cancer is relatively understudied and could identify novel targets for therapeutic intervention so this work is of potential significance. However, the data is over-interpreted in many instances and it is not clear the authors can make the claims they do based on the data shown. In particular, the data showing that decreased Map3k7 underlies the effects of the Sf3b1K700E mutant is very weak. Does over-expression of Map3k7 promote the EMT signature and induce apoptosis? Do the Map3k7 expressing organoids form tumors more effectively when transplanted into mice? Also, the novelty of the work is a concern since aberrant Map3k7 splicing due to SF3B1 mutation was seen previously in other systems. The authors also do not address the apparent conundrum of Sf3b1 K700E mutation promoting tumorigenesis despite there being less EMT which is also required for progression to metastasis in PDAC.
Major Concerns.
- The analysis of the effect of Sf3b1K700E expression on normal pancreas and on PanINs in KC mice and PDAC in KPC mice is superficial and could be enhanced by staining for amylase, cytokeratin-19 and insulin. In particular, the data quantified in figure 1L should be accompanied by staining for CK19, Mucin5AC or some other marker of ductal transformation. Also, are any effects seen at older ages in normal mice?
We performed staining of normal and cancerous mouse pancreata using Ck19, MUC5AC and b-amylase antibodies. In line with our hypothesis that Sf3b1K700E mainly plays a role in early stages of PDAC formation, we observed significant differences in CK19 (increase), MUC5AC (increase) and b-amylase (decrease) expression in early stage KPC-Sf3b1K700E vs. KPC tumors (Fig. 1G-J), but not in late stage tumors (see Figure 1-figure supplement 1F-I). In addition, no differences were observed in normal mice. We added these data to the revised manuscript (see Figure 1-figure supplement 1D, E).
- The invasion assays used are limited and should be complemented by more routine quantification of cell migration and invasion including such assays as a scratch assay, Boyden chamber assays and use of the IncuCyte system to quantify. As it stands the image in Figure 3B is difficult to interpret since it is very poorly described in the figure legend. Additional evidence is needed to make the claims made by the authors.
During the revisions we performed wound healing/scratch assays using PANC-1 cells with inducible SF3B1 WT/K700E overexpression. We observed a significant difference in migratory capacity between SF3B1 WT- and SF3B1 K700E overexpressing cells stimulated with TGF-β. We added this data to the revised manuscript (Fig. 2I, J). We also describe the abovementioned figure 3B in more detail (revised manuscript Fig. 2G, H; line 759-767).
- The authors should show the actual CC3 staining quantified in Suppl. Figure 2G.
We added a representative image of CC3 staining (see Figure 3-figure supplement 1A) for the quantified data (see Figure 3-figure supplement 1B in the revised manuscript).
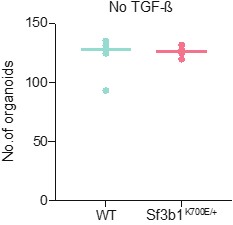
- The graph in Figure 3L should show WT and Sf3b1K700E expressing organoids number both with and without TGF-b.
Since without TGF-b supplementation organoids have to be split in a 1:3 ratio every 5 days, we could not follow the same passaging regimen as in experiments with TGF-b supplementation (split in a 1:2 ratio every 20 days, Fig. 3I). However, we assessed the organoid number grown in control medium without TGF-b for 4 passages (20 days) in a 1:3 ratio, and observe no difference in organoid number in WT and Sf3b1K700E expressing organoids (Author response image 1). In the revised manuscript we show with a highly quantitative read-out (CellTiterGlo) that Sf3b1K700E expressing organoids do not grow faster than Sf3b1 WT expressing organoids in absence of TGF-β (see Figure 3-figure supplement 1E). Taken together, we can exclude that Sf3b1K700E organoids outgrow Sf3b1 WT organoids in medium with TGF-β supplementation because they generally have a growth advantage.
Author response image 1.
Author response image 1. WT and Sf3b1K700E expressing organoids were cultured without TGF-β supplementation. Organoids were split in a 1:3 ratio every 5 days. Data points show organoid number before splitting, assessed for 4 passages.
Reviewer #2 (Public Review):
The manuscript has several areas of strength; it functionally explores a mutant that is detected in a portion of pancreatic cancers; it conducts mechanistic investigation and it uses human cell lines to validate the findings based on mouse models. Some areas for improvement are described below.
- TGF-b is known to act as a tumor suppressor early in carcinogenesis, and as a tumor promoter later. The authors should extend their analysis of mouse models to determine whether the effect of SF3B1K700E is specific to promoting initiation (e.g. more, early acinar ductal metaplasia) or faster progression of PanINs following their formation. Another way to address this could be acinar cultures, to determine whether an increased propensity to ADM exists.
To further detangle the effect KPC-Sf3b1K700E with respect to tumor progression, we analyzed our autochthonous model at an early and late stage of tumor progression: Histological examination at 5 weeks revealed increased propensity to ADM (see Figure 1-figure supplement 1J, K), PanIN formation (shown by Muc5a1 and CK19 IF stainings, Fig. 1G, I, J) and a concomitant decrease of acinar cells (shown by b-amylase staining) in KPC-Sf3b1K700E vs. KPC tumors (Fig. 1G, H). Analyzing tumors at 9 weeks of age did not show differences in CK19 staining and fibrosis. We added these data to the revised manuscript (see Figure 1-figure supplement 1F-I).
- Given that the effect of SF3B1K700E expression is more prominent in KC mice, rather than in KPC mice, the authors should explain the rationale for using the latter for RNA sequencing.
In KC mice, pre-invasive PanIN lesions only infrequently progress to PDAC (spontaneous progression, see Gabriel et al., Pancreatology, 2020 ). Therefore, it would have been difficult to collect enough material for cell sorting and downstream RNA sequencing of tumor cells. The KPC mouse model develops PDAC with a 100% penetrance, allowing the collection of sufficient material.
- Given that this mutation is found in about 3% of human pancreatic cancer, it would be interesting to know whether these tumors have any unique feature, and specifically any characteristic that could be harnessed therapeutically.
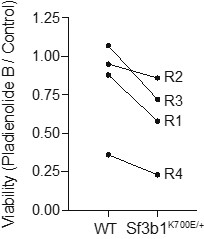
Unfortunately, the size of published datasets is too small for a meaningful differential gene expression analysis of SF3B1-WT vs. SF3B1-K700E PDAC tumors (due to the low occurrence of SF3B1-K700E PDAC). However, harnessing the K700E mutation therapeutically by increasing missplicing through splicing inhibitors has previously been suggested, and it was shown that SF3B1-K700E mutated cancer cells are more prone to apoptosis when splicing is chemically targeted than SF3B1-WT cells. We tested a similar approach in murine pre-cancerous organoids, demonstrating that Sf3b1-WT organoids show higher survival than Sf3b1K700E expressing organoids when treated with the splicing-inhibitor Pladienolide B (Author response image 2). However, since this concept is not novel and not within the topic of our manuscript, we would prefer to not integrate this data into our manuscript.
Author response image 2.
Author response image 2. 33 nM of the splicing inhibitor Pladienolide B was added to the cell culture medium for 48 hours and the viability was assessed by normalizing organoid numbers to untreated control organoids. The line indicates WT and Sf3b1K700E organoids assessed in the same replicate.
- It would be interesting to know whether this mutation mutually exclusive to other mutations affecting response to TGF-b. Further, while the data might not be widely available, it would be interesting to know whether in human patients the mutation occurs in precursor lesions (PanIN might be difficult to assess, but IPMN might be doable) or at later stages.
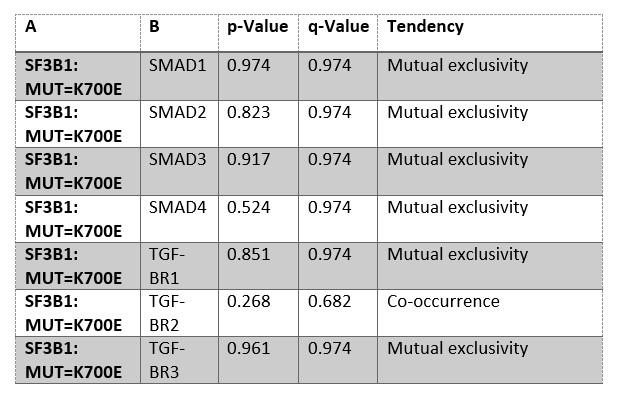
We performed a mutual exclusivity analysis in PDAC samples available at www.cbioportal.org, but did not find mutual exclusivity of SF3B1-K700E to genes of the TGF-β-pathway. Of note, the value of the analysis is limited by the small sample size of SF3B1-K700E PDAC (n=7) Moreover, to our knowledge there is no public tissue biobank for PDAC which would allow us to assess the stage of SF3B1-K700E mutated PDAC tumors. Thus, unfortunately we cannot histologically assess if the mutations already occur in early stages of human tumor development.
Author response table 1.
Author response table 1: Mutual exclusivity analysis of public PDAC databases (ICGC, CPTAC, QCMG, TCGA, UTSW), including 910 patients. Mutation frequency is 25% for SMAD4, 5% for TGF-ΒR2, 3% for SMAD2, 2.6% for TGF-ΒR1, 1.4% for SMAD3, 0.7% for SF3B1-K700E, 0.7% for TGF-ΒR3, 0.4% for SMAD1. Analysis was performed on cbioportal.org.
Reviewer #3 (Public Review):
Alternative splicing as a result of mutations in different components of the splicing machinery has been associated with a variety of cancer types, including hematological malignancies where this has been most extensively studied but also for solid tumors such as breast and pancreatic ductal adenocarcinoma (PDAC). Here the authors analyze genome sequencing data in human PDAC samples and identify a recurring mutation in the SF3B1 subunit that substitutes lysine for glutamate at residue 700 (SF3B1K700E) in PDACs. This mutation has been identified and its' molecular role in disease progression in other diseases has been studied, but the mechanism for promoting disease progression in pancreatic cancer has not been as well characterized.
To study how SF3B1K700E contributes to PDAC pathology, the authors generate a novel genetically modified mouse model of a pancreas specific SF3B1K700E mutation and explore its oncogenicity and tumor promoting potential. The authors find that SF3B1K700E is not oncogenic, but potentiates the oncogenic potential of Kras and p53 (KP) driver mutations commonly found in PDAC tumors. The authors then proceed to characterize the molecular mechanisms that might drive this phenotype. By transcriptomic analysis, the authors find KP-SF3B1K700E tumors have downregulation of epithelial-to-mesenchymal transition (EMT) genes compared to KP tumors. The cytokine TGFβ has previously been found to limit PDAC initiation and progression by causing lethal EMT in PDAC and PDAC precursor cells. Thus, the authors propose SF3B1K700E inhibition of EMT blocks the tumor suppressive activity of TGFβ and this underpins the tumor promoting role of SF3B1K700E mutation in PDAC. Consistent with this finding, SF3B1K700E mutation blocks TGFβ-induced toxicity in a variety of cell culture models of PDAC and PDAC precursor models.
Lastly, the authors seek to identify how altered splicing reduces EMT activity in PDAC cells. The authors identify misspliced genes consistent in both KP and human SF3B1K700E mutant cancer samples and find Map3k7 as one of 11 consistently misspliced genes. MAP3K7 has previously been identified as a positive regulator of EMT. Thus the authors speculated Map3k7 missplicing would lead to reduced MAP3K7 activity and a reduction EMT and that this underpins the TGFβ in SF3B1K700E mutant PDAC cells. Consistent with this, the authors find inhibition of MAP3K7 reduces TGFβ toxicity in SF3B1K700E WT cells and overexpression of MAP3K7 in SF3B1K700E mutant PDAC cells induces TGFβ toxicity. Altogether, this suggests activity of Map3k7 is responsible for altered EMT activity and TGFβ sensitivity in SF3B1K700E mutant PDAC.
Altogether, the authors generate a valuable model to study the role of a recurring splicing mutation in PDAC and provide compelling evidence that this mutation is accelerates disease. The authors then perform both: (1) an open-ended investigation of how this mutation alters PDAC cell biology where they identify altered EMT activity and (2) rigorous mechanistic studies showing suppressed EMT provides PDAC cells with resistance to TGFβ, which has previously been shown to be tumor suppressive in PDAC, suggesting a possible mechanism by which SF3B1K700E mutation is oncogenic in PDAC that future animal studies can confirm. This work generates valuable models and datasets to advance the understanding of how mutations in the splicing machinery can promote PDAC progression and suggests alternative splicing of MAP3K7 is one such possible mechanism that altered splicing promotes PDAC progression in vivo.
- One major concern about the manuscript is that the proposed mechanism by which SF3B1K700E mutation accelerates PDAC progression (MAP3K7 inhibition -> EMT inhibition -> reduced TGF-β toxicity) is only tested in ex vivo culture models and there is very limited and correlative data to suggest that this is the operative mechanism by which SF3B1K700E mutant tumors are accelerated. This is especially important because of recent findings that IFN-α signaling, which the authors also found to be high in SF3B1K700E mutant tumors, also promotes PDAC progression (https://www.biorxiv.org/content/10.1101/2022.06.29.497540v1). Thus, while thoroughly convinced by the rigorous ex vivo work that SF3B1K700E does lead to MAP3K7 inhibition -> EMT inhibition -> reduced TGF-β toxicity, further experiments to confirm this mechanism is critical in vivo would be needed to convince me that this mechanism is critical to tumor progression in vivo. For example, would forced expression of MAP3K7 slow orthotopic KP-SF3B1K700E tumor growth while leaving IFN-α signaling unperturbed?
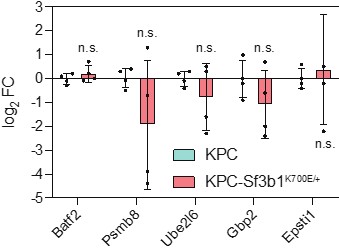
We thank the reviewer for raising these important points. To first test if the upregulation of IFN-α signaling, seen in our RNA-seq data of sorted KPC-Sf3b1K700E cells, was directly caused by the Sf3b1-K700E mutation, we assessed the 5 most deregulated genes of the IFN-α signature in in-vitro activated KPC and KPC-Sf3b1K700E organoids (analogous to the experiments on the EMT gene signature in see Figure 2-figure supplement 1D). However, in contrast to EMT marker genes, INFa signature genes were not differently expressed in KPC-Sf3b1K700E vs. KPC organoids (Author response image 3). Thus, increased IFN-α signaling in KPC-Sf3b1K700E tumors in mice is likely an indirect consequence of further progressed cancers rather than an effect directly caused by Sf3b1K700E mediated missplicing.
Author response image 3.
Author response image 3. Expression of the 5 most deregulated genes of the IFN-α gene set identified in sorted KPC-Sf3b1K700E cells in in-vitro activated KPC-Sf3b1K700E and KPC organoids. 4 biological replicates were performed. For analysis, Ct-values of the indicated genes were normalized to Actb and a two-tailed unpaired t-test was used to compute the indicated p-values.
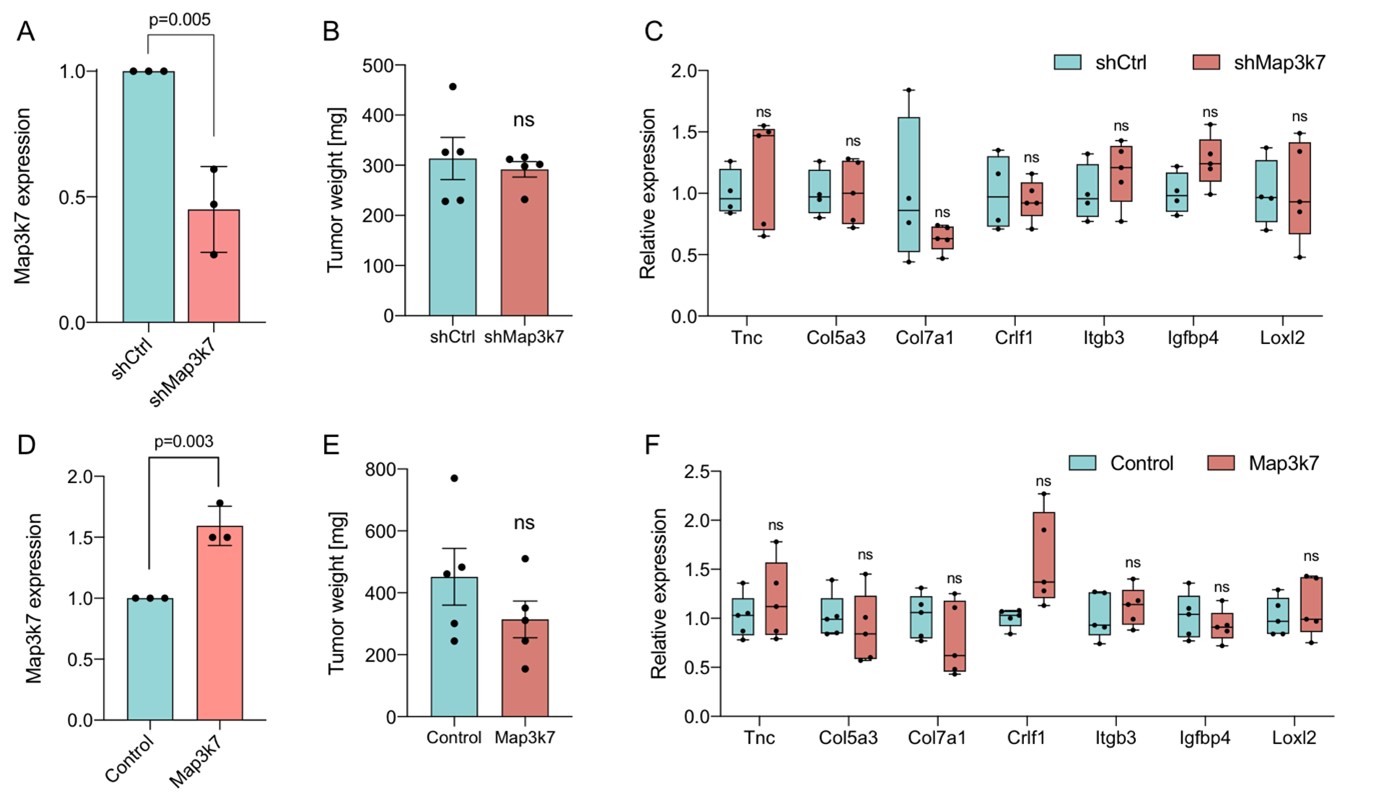
To next examine the effect of Map3k7 on tumors in vivo, we established orthotopic transplantation models with KPC and KPC-Sf3b1K700E cells, with overexpression or knockdown of Map3k7 (Author response image 4). However, in contrast to the autochthonous mouse model, already orthotopically transplanted KPC vs. KPC-Sf3b1K700E cells did not show differences in tumor size (see Figure 1-figure supplement 1M, N). These data support our hypothesis that Sf3b1-K700E rather plays an important role during early stages of PDAC (KPC cells are isolated from fully developed PDAC tumors and orthotopic KPC transplantation thus represents a late-stage PDAC model).
Unfortunately, these data also demonstrate that orthotopic transplantation of KPC cells is not a suitable model for studying the impact of Map3k7 in PDAC development, and as expected, neither Map3k7 overexpression in transplanted KPC-Sf3b1K700E cells nor shRNA mediated knockdown of Map3k7 (shMap3k7) in transplanted KPC cells led to differences in growth compared to their control groups (Author response image 4). In line with these results, the EMT genes that were found to be differentially expressed in our autochthonous mouse model (KPC vs. KPC-Sf3b1K700E) were expressed at similar levels upon Map3K7 downregulation or overexpression.
Since establishment of an autochthonous KPC PDAC mouse model with a knock-down of MAP3K7 is out of scope for a revision, in the revised manuscript we discuss the limitation of our study that the molecular link between Sf3b1K700E, Map3k7 and Tgfb resistance has only been studied in vitro in organoids and cell lines. We also adapted the abstract and the title of the manuscript accordingly (formerly “Mutant SF3B1 promotes PDAC malignancy through TGF-β resistance”, now “Mutant SF3B1 promotes malignancy in PDAC”).
Author response image 4.
Author response image 4. (A) Relative gene expression of Map3k7 in KPC cells transduced with shRNA targeting Map3k7 (shMap3k7), normalized to KPC cells transduced with scrambled control shRNA (shCtrl). 3 biological replicates are shown. (B) Weight of tumors derived by orthotopical transplantation of shMap3k7 and shCtrl KPC cells. 5 biological replicates are shown. (C) Relative gene expression of EMT genes in tumors derived by orthotopic transplantation of shCtrl and shMap3k7 cells. 4 biological replicates are shown. (D) Relative gene expression of Map3k7 in KPC-Sf3b1K700E cells transduced with an overexpression vector of Map3k7 (OE Map3k7), normalized to control KPC cells without Map3k7 overexpression. 3 biological replicates are shown, a two-sided student’s t-test was used to calculate significance. (E) Weight of tumors derived by orthotopical transplantation of Map3k7 overexpressing KPC-Sf3b1K700E cells (n=5) and control KPC-Sf3b1K700E cells (n=4). (F) Relative gene expression of EMT genes in tumors derived by orthotopic transplantation of KPC-Sf3b1K700E cells with- and without overexpression of Map3k7. 4 biological replicates are shown. A two-sided student’s t-test was used to calculate significance in Fig. 2A-F.
-
eLife assessment
This work examines a role for altered splicing in pancreatic tumorigenesis by interrogating effects of a specific mutation in the Sf3b splicing factor in pancreatic organoid and cell line growth primarily, with some in vivo work also performed. There is significant potential in the study but there is a concern about the lack of in vivo validation of claims that are most relevant to metastatic progression and the focus on one specific mechanism at the expense of other possible effects on splicing of factors important for disease progression.
-
Reviewer #1 (Public Review):
The authors examine the role of the K700E mutation in the Sf3B1 splicing factor in PDAC and report that this Sf3B1 mutation promotes PDAC by decreasing sensitivity to TGF-b resulting in decreased EMT and decreased apoptosis as a result. They propose that the Sf3b1 K700E mutant causes decreased expression of Map3K7, a known mediator of TGFb signaling and also known to be alternately spliced in other systems by the Sf3b1 K700E mutation. The role of splicing defects in cancer is relatively understudied and could identify novel targets for therapeutic intervention so this work is of potential significance. However, the data is over-interpreted in many instances and it is not clear the authors can make the claims they do based on the data shown. In particular, the data showing that decreased Map3k7 underlies the …
Reviewer #1 (Public Review):
The authors examine the role of the K700E mutation in the Sf3B1 splicing factor in PDAC and report that this Sf3B1 mutation promotes PDAC by decreasing sensitivity to TGF-b resulting in decreased EMT and decreased apoptosis as a result. They propose that the Sf3b1 K700E mutant causes decreased expression of Map3K7, a known mediator of TGFb signaling and also known to be alternately spliced in other systems by the Sf3b1 K700E mutation. The role of splicing defects in cancer is relatively understudied and could identify novel targets for therapeutic intervention so this work is of potential significance. However, the data is over-interpreted in many instances and it is not clear the authors can make the claims they do based on the data shown. In particular, the data showing that decreased Map3k7 underlies the effects of the Sf3b1K700E mutant is very weak. Does over-expression of Map3k7 promote the EMT signature and induce apoptosis? Do the Map3k7 expressing organoids form tumors more effectively when transplanted into mice? Also, the novelty of the work is a concern since aberrant Map3k7 splicing due to SF3B1 mutation was seen previously in other systems. The authors also do not address the apparent conundrum of Sf3b1 K700E mutation promoting tumorigenesis despite there being less EMT which is also required for progression to metastasis in PDAC.
Major Concerns.
1. The analysis of the effect of Sf3b1K700E expression on normal pancreas and on PanINs in KC mice and PDAC in KPC mice is superficial and could be enhanced by staining for amylase, cytokeratin-19 and insulin. In particular, the data quantified in figure 1L should be accompanied by staining for CK19, Mucin5AC or some other marker of ductal transformation. Also, are any effects seen at older ages in normal mice?
2. The invasion assays used are limited and should be complemented by more routine quantification of cell migration and invasion including such assays as a scratch assay, Boyden chamber assays and use of the IncuCyte system to quantify. As it stands the image in Figure 3B is difficult to interpret since it is very poorly described in the figure legend. Additional evidence is needed to make the claims made by the authors.
3. The authors should show the actual CC3 staining quantified in Suppl. Figure 2G.
4. The graph in Figure 3L should show WT and Sf3b1K700E expressing organoids number both with and without TGF-b. -
Reviewer #2 (Public Review):
The manuscript has several areas of strength; it functionally explores a mutant that is detected in a portion of pancreatic cancers; it conducts mechanistic investigation and it uses human cell lines to validate the findings based on mouse models. Some areas for improvement are described below.
TGF-b is known to act as a tumor suppressor early in carcinogenesis, and as a tumor promoter later. The authors should extend their analysis of mouse models to determine whether the effect of SF3B1K700E is specific to promoting initiation (e.g. more, early acinar ductal metaplasia) or faster progression of PanINs following their formation. Another way to address this could be acinar cultures, to determine whether an increased propensity to ADM exists.
Given that the effect of SF3B1K700E expression is more prominent in …
Reviewer #2 (Public Review):
The manuscript has several areas of strength; it functionally explores a mutant that is detected in a portion of pancreatic cancers; it conducts mechanistic investigation and it uses human cell lines to validate the findings based on mouse models. Some areas for improvement are described below.
TGF-b is known to act as a tumor suppressor early in carcinogenesis, and as a tumor promoter later. The authors should extend their analysis of mouse models to determine whether the effect of SF3B1K700E is specific to promoting initiation (e.g. more, early acinar ductal metaplasia) or faster progression of PanINs following their formation. Another way to address this could be acinar cultures, to determine whether an increased propensity to ADM exists.
Given that the effect of SF3B1K700E expression is more prominent in KC mice, rather than in KPC mice, the authors should explain the rationale for using the latter for RNA sequencing.
Given that this mutation is found in about 3% of human pancreatic cancer, it would be interesting to know whether these tumors have any unique feature, and specifically any characteristic that could be harnessed therapeutically.
It would be interesting to know whether this mutation mutually exclusive to other mutations affecting response to TGF-b. Further, while the data might not be widely available, it would be interesting to know whether in human patients the mutation occurs in precursor lesions (PanIN might be difficult to assess, but IPMN might be doable) or at later stages.
-
Reviewer #3 (Public Review):
Alternative splicing as a result of mutations in different components of the splicing machinery has been associated with a variety of cancer types, including hematological malignancies where this has been most extensively studied but also for solid tumors such as breast and pancreatic ductal adenocarcinoma (PDAC). Here the authors analyze genome sequencing data in human PDAC samples and identify a recurring mutation in the SF3B1 subunit that substitutes lysine for glutamate at residue 700 (SF3B1K700E) in PDACs. This mutation has been identified and its' molecular role in disease progression in other diseases has been studied, but the mechanism for promoting disease progression in pancreatic cancer has not been as well characterized.
To study how SF3B1K700E contributes to PDAC pathology, the authors generate …
Reviewer #3 (Public Review):
Alternative splicing as a result of mutations in different components of the splicing machinery has been associated with a variety of cancer types, including hematological malignancies where this has been most extensively studied but also for solid tumors such as breast and pancreatic ductal adenocarcinoma (PDAC). Here the authors analyze genome sequencing data in human PDAC samples and identify a recurring mutation in the SF3B1 subunit that substitutes lysine for glutamate at residue 700 (SF3B1K700E) in PDACs. This mutation has been identified and its' molecular role in disease progression in other diseases has been studied, but the mechanism for promoting disease progression in pancreatic cancer has not been as well characterized.
To study how SF3B1K700E contributes to PDAC pathology, the authors generate a novel genetically modified mouse model of a pancreas specific SF3B1K700E mutation and explore its oncogenicity and tumor promoting potential. The authors find that SF3B1K700E is not oncogenic, but potentiates the oncogenic potential of Kras and p53 (KP) driver mutations commonly found in PDAC tumors. The authors then proceed to characterize the molecular mechanisms that might drive this phenotype. By transcriptomic analysis, the authors find KP-SF3B1K700E tumors have downregulation of epithelial-to-mesenchymal transition (EMT) genes compared to KP tumors. The cytokine TGFβ has previously been found to limit PDAC initiation and progression by causing lethal EMT in PDAC and PDAC precursor cells. Thus, the authors propose SF3B1K700E inhibition of EMT blocks the tumor suppressive activity of TGFβ and this underpins the tumor promoting role of SF3B1K700E mutation in PDAC. Consistent with this finding, SF3B1K700E mutation blocks TGFβ-induced toxicity in a variety of cell culture models of PDAC and PDAC precursor models.
Lastly, the authors seek to identify how altered splicing reduces EMT activity in PDAC cells. The authors identify misspliced genes consistent in both KP and human SF3B1K700E mutant cancer samples and find Map3k7 as one of 11 consistently misspliced genes. MAP3K7 has previously been identified as a positive regulator of EMT. Thus the authors speculated Map3k7 missplicing would lead to reduced MAP3K7 activity and a reduction EMT and that this underpins the TGFβ in SF3B1K700E mutant PDAC cells. Consistent with this, the authors find inhibition of MAP3K7 reduces TGFβ toxicity in SF3B1K700E WT cells and overexpression of MAP3K7 in SF3B1K700E mutant PDAC cells induces TGFβ toxicity. Altogether, this suggests activity of Map3k7 is responsible for altered EMT activity and TGFβ sensitivity in SF3B1K700E mutant PDAC.
Altogether, the authors generate a valuable model to study the role of a recurring splicing mutation in PDAC and provide compelling evidence that this mutation is accelerates disease. The authors then perform both: (1) an open-ended investigation of how this mutation alters PDAC cell biology where they identify altered EMT activity and (2) rigorous mechanistic studies showing suppressed EMT provides PDAC cells with resistance to TGFβ, which has previously been shown to be tumor suppressive in PDAC, suggesting a possible mechanism by which SF3B1K700E mutation is oncogenic in PDAC that future animal studies can confirm. This work generates valuable models and datasets to advance the understanding of how mutations in the splicing machinery can promote PDAC progression and suggests alternative splicing of MAP3K7 is one such possible mechanism that altered splicing promotes PDAC progression in vivo.
- One major concern about the manuscript is that the proposed mechanism by which SF3B1K700E mutation accelerates PDAC progression (MAP3K7 inhibition -> EMT inhibition -> reduced TGFb toxicity) is only tested in ex vivo culture models and there is very limited and correlative data to suggest that this is the operative mechanism by which SF3B1K700E mutant tumors are accelerated. This is especially important because of recent findings that IFNa signaling, which the authors also found to be high in SF3B1K700E mutant tumors, also promotes PDAC progression (https://www.biorxiv.org/content/10.1101/2022.06.29.497540v1). Thus, while thoroughly convinced by the rigorous ex vivo work that SF3B1K700E does lead to MAP3K7 inhibition -> EMT inhibition -> reduced TGFb toxicity, further experiments to confirm this mechanism is critical in vivo would be needed to convince me that this mechanism is critical to tumor progression in vivo. For example, would forced expression of MAP3K7 slow orthotopic KP-SF3B1K700E tumor growth while leaving IFNa signaling unperturbed?
-
