The lingering effects of Neanderthal introgression on human complex traits
Curation statements for this article:-
Curated by eLife

eLife assessment
A small proportion of the genomes of humans whose ancestors lived outside Africa traces back to an interbreeding event with Neanderthals. While we know that selection has generally acted to remove Neanderthal ancestry, intense interest has focused on understanding the contribution to current human phenotypic variation. This paper uses a new set of approaches to carefully quantify this contribution, taking into account various complicating factors. The work will be of interest to colleagues in human evolution and evolutionary biology more generally.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The genetic variants introduced into the ancestors of modern humans from interbreeding with Neanderthals have been suggested to contribute an unexpected extent to complex human traits. However, testing this hypothesis has been challenging due to the idiosyncratic population genetic properties of introgressed variants. We developed rigorous methods to assess the contribution of introgressed Neanderthal variants to heritable trait variation and applied these methods to analyze 235,592 introgressed Neanderthal variants and 96 distinct phenotypes measured in about 300,000 unrelated white British individuals in the UK Biobank. Introgressed Neanderthal variants make a significant contribution to trait variation (explaining 0.12% of trait variation on average). However, the contribution of introgressed variants tends to be significantly depleted relative to modern human variants matched for allele frequency and linkage disequilibrium (about 59% depletion on average), consistent with purifying selection on introgressed variants. Different from previous studies (McArthur et al., 2021), we find no evidence for elevated heritability across the phenotypes examined. We identified 348 independent significant associations of introgressed Neanderthal variants with 64 phenotypes. Previous work (Skov et al., 2020) has suggested that a majority of such associations are likely driven by statistical association with nearby modern human variants that are the true causal variants. Applying a customized fine-mapping led us to identify 112 regions across 47 phenotypes containing 4303 unique genetic variants where introgressed variants are highly likely to have a phenotypic effect. Examination of these variants reveals their substantial impact on genes that are important for the immune system, development, and metabolism.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
In this manuscript, Wei & Robles et al seek to estimate the heritability contribution of Neanderthal Informative Markers (NIM) relative to SNPs that arose in modern humans (MH). This is a question that has received a fair amount of attention in recent studies, but persistent statistical limitations have made some prior results difficult to interpret. Of particular concern is the possibility that heritability (h^2) attributed to Neanderthal markers might be tagging linked variants that arose in modern humans, resulting in overestimation of h^2 due to Neanderthal variants. Neanderthal variants also tend to be rare, and estimating the contribution of rare alleles to h^2 is challenging. In some previous studies, rare alleles have been excluded from h^2 estimates.
Wei & Robles et al develop …
Author Response
Reviewer #1 (Public Review):
In this manuscript, Wei & Robles et al seek to estimate the heritability contribution of Neanderthal Informative Markers (NIM) relative to SNPs that arose in modern humans (MH). This is a question that has received a fair amount of attention in recent studies, but persistent statistical limitations have made some prior results difficult to interpret. Of particular concern is the possibility that heritability (h^2) attributed to Neanderthal markers might be tagging linked variants that arose in modern humans, resulting in overestimation of h^2 due to Neanderthal variants. Neanderthal variants also tend to be rare, and estimating the contribution of rare alleles to h^2 is challenging. In some previous studies, rare alleles have been excluded from h^2 estimates.
Wei & Robles et al develop and assess a method that estimates both total heritability and per-SNP heritability of NIMs, allowing them to test whether NIM contributions to variation in human traits are similar or substantially different than modern human SNPs. They find an overall depletion of heritability across the traits that they studied, and found no traits with enrichment of heritability due to NIMs. They also developed a 'fine-mapping' procedure that aims to find potential causal alleles and report several potentially interesting associations with putatively functional variants.
Strengths of this study include rigorous assessment of the statistical methods employed with simulations and careful design of the statistical approaches to overcome previous limitations due to LD and frequency differences between MH and NIM variants. I found the manuscript interesting and I think it makes a solid contribution to the literature that addresses limitations of some earlier studies.
My main questions for the authors concern potential limitations of their simulation approach. In particular, they describe varying genetic architectures corresponding to the enrichment of effects among rare alleles or common alleles. I agree with the authors that it is important to assess the impact of (unknown) architecture on the inference, but the models employed here are ad hoc and unlikely to correspond to any mechanistic evolutionary model. It is unclear to me whether the contributions of rare and common alleles (and how these correspond with levels of LD) in real data will be close enough to these simulated schemes to ensure good performance of the inference.
In particular, the common allele model employed makes 90% of effect variants have frequencies above 5% -- I am not aware of any evolutionary model that would result in this outcome, which would suggest that more recent mutations are depleted for effects on traits (of course, it is true that common alleles explain much more h^2 under neutral models than rare alleles, but this is driven largely by the effect of frequency on h^2, not the proportion of alleles that are effect alleles). Likewise, the rare allele model has the opposite pattern, with 90% of effect alleles having frequencies under 5%. Since most alleles have frequencies under 5% anyway (~58% of MH SNPs and ~73% of NIM SNPs) this only modestly boosts the prevalence of low frequency effect alleles relative to their proportion. Some selection models suggest that rare alleles should have much bigger effects and a substantially higher likelihood of being effect alleles than common alleles. I'm not sure this situation is well-captured by the simulations performed. With LD and MAF annotations being applied in relatively wide quintile bins, do the authors think their inference procedure will do a good job of capturing such rare allele effects? This seems particularly important to me in the context of this paper, since the claim is that Neanderthal alleles are depleted for overall h^2, but Neanderthal alleles are also disproportionately rare, meaning they could suffer a bigger penalty. This concern could be easily addressed by including some simulations with additional architectures to those considered in the manuscript.
We thank the reviewers for their thoughtful comments regarding rare alleles, and we agree that our RARE simulations only moderately boosted the enrichment of rare alleles in causal mutations. To address this, we added new simulations, ULTRA RARE, in which SNPs with MAF < 0.01 constitute 90% of the causal variants. Similar to our previous simulations, we use 100,000 and 10,000 causal variants to mimic highly polygenic and moderately polygenic phenotypes, and 0.5 and 0.2 for high and moderately heritable phenotypes. We similarly did three replicated simulations for each combination and partitioned the heritability with Ancestry only annotation, Ancestry+MAF annotation, Ancestry+LD annotation, and Ancestry+MAF+LD annotation. Our Ancestry+MAF+LD annotation remains calibrated in this setting (see Figure below). We believe this experiment strengthens our paper and have added it as Fig S2.
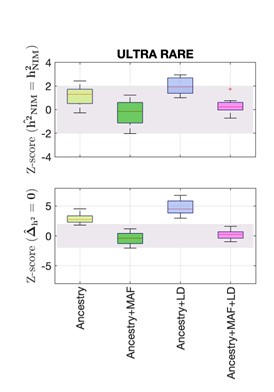
While we agree that these architectures are ad-hoc and are unlikely to correspond to realistic evolutionary scenarios, we have chosen these architectures to span the range of possible architecture so that the skew towards common or rare alleles that we have explored are extreme. The finding that our estimates are calibrated across the range that we have explored leads us to conclude that our inferences should be robust.
More broadly, we concur with the reviewer that our results (as well as others in the field) may need to be revisited as our view of the genetic architecture of complex traits evolves. The methods that we propose in this paper are general enough to explore such architectures in the future by choosing a sufficiently large set of annotations that match the characteristics across NIMs and MH SNPs. A practical limitation to this strategy is that the use of a large number of annotations can result in some annotations being assigned a small number of SNPs which would, in turn, reduce the precision of our estimates. This limitation is particularly relevant due to the smaller number of NIMs compared to MH SNPs (around 250K vs around 8M).
Reviewer #2 (Public Review):
The goal of the work described in this paper is to comprehensively describe the contribution of Neanderthal-informative mutations (NIMs) to complex traits in modern human populations. There are some known challenges in studying these variants, namely that they are often uncommon, and have unusually long haplotype structures. To overcome these, the authors customized a genotyping array to specifically assay putative Neanderthal haplotypes, and used a recent method of estimating heritability that can explicitly account for differences in MAF and LD.
This study is well thought-out, and the ability to specifically target the genotyping array to the variants in question and then use that information to properly control for population structure is a massive benefit. The methodology also allowed them to include rarer alleles that were generally excluded from previous studies. The simulations are thorough and convincingly show the importance of accounting for both MAF and LD in addition to ancestry. The fine-mapping done to disentangle effects between actual Neanderthal variants and Modern human ones on the same haplotype also seems reasonable. They also strike a good balance between highlighting potentially interesting examples of Neanderthal variants having an effect on phenotype without overinterpreting association-based findings.
The main weakness of the paper is in its description of the work, not the work itself. The paper currently places a lot of emphasis on comparing these results to prior studies, particularly on its disagreement with McArthur, et al. (2021), a study on introgressed variant heritability that was also done primarily in UK Biobank. While they do show that the method used in that study (LDSR) does not account for MAF and LD as effectively as this analysis, this work does not support the conclusion that this is a major problem with previous heritability studies. McArthur et al. in fact largely replicate these results that Neanderthal variants (and more generally regions with Neanderthal variants) are depleted of heritability, and agree with the interpretation that this is likely due to selection against Neanderthal alleles. I actually find this a reassuring point, given the differences between the variant sets and methods used by the two studies, but it isn't mentioned in the text. Where the two studies differ is in specifics, mainly which loci have some association with human phenotypes; McArthur et al. also identified a couple groups of traits that were exceptions to the general rule of depleted heritability. While this work shows that not accounting for MAF and LD can lead to underestimating NIM heritability, I don't follow the logic behind the claim that this could lead to a false positive in heritability enrichment (a false negative would be more likely, surely?). There are also more differences between this and previous heritability studies than just the method used to estimate heritability, and the comparisons done here do not sufficiently account for these. A more detailed discussion to reconcile how, despite its weaknesses, LDSR picks up similar broad patterns while disagreeing in specifics is merited.
We agree with the reviewer that our results are generally concordant with those of McArthur et al. 2021 and this concordance is reassuring given the differences across our studies. The differences across the studies, wherein McArthur et al. 2021 identify a few traits with elevated heritability while we do not, could arise due to reasons beyond the methodological differences such as differences in the sets of variants analyzed. We have partially explored this possibility in the revised manuscript by analyzing the set of introgressed variants identified by the Sprime method (which was used in McArthur et al. 2021) using our method: we continue to observe a pattern of depletion with no evidence for enrichment. We hypothesize that the reason why LDSR picks up similar overall patterns despite its limitations is indicative of the nature of selection on introgressed alleles (which, in turn, influences the dependence of effect size on allele frequency and LD). Investigating this hypothesis will require a detailed understanding of the LDSR results on parameters such as the MAF threshold on the regression SNPs and the LD reference SNPs and the choice of the LD reference panel.
Not accounting for MAF and LD can underestimate NIM heritability but can both underestimate and overestimate heritability at MH SNPs. Hence, tests that compare per-SNP heritability at NIMs to MH SNPs can therefore lead to false positives both in the direction of enrichment and depletion.
We have now written in the Discussion: “In spite of these differences in methods and NIMs analyzed, our observation of an overall pattern of depletion in the heritability of introgressed alleles is consistent with the findings of McArthur et al. The robustness of this pattern might provide insights into the nature of selection against introgressed alleles”
In general this work agrees with the growing consensus in the field that introgressed Neanderthal variants were selected against, such that those that still remain in human populations do not generally have large effects on phenotypes. There are exceptions to this, but for the most part observed phenotypic associations depend on the exact set of variants being considered, and, like those highlighted in this study, still lack more concrete validation. While this paper does not make a significant advance in this general understanding of introgressed regions in modern populations, it does increase our knowledge in how best to study them, and makes a good attempt at addressing issues that are often just mentioned as caveats in other studies. It includes a nice quantification of how important these variables are in interpreting heritability estimates, and will be useful for heritability studies going forward.
-
eLife assessment
A small proportion of the genomes of humans whose ancestors lived outside Africa traces back to an interbreeding event with Neanderthals. While we know that selection has generally acted to remove Neanderthal ancestry, intense interest has focused on understanding the contribution to current human phenotypic variation. This paper uses a new set of approaches to carefully quantify this contribution, taking into account various complicating factors. The work will be of interest to colleagues in human evolution and evolutionary biology more generally.
-
Reviewer #1 (Public Review):
In this manuscript, Wei & Robles et al seek to estimate the heritability contribution of Neanderthal Informative Markers (NIM) relative to SNPs that arose in modern humans (MH). This is a question that has received a fair amount of attention in recent studies, but persistent statistical limitations have made some prior results difficult to interpret. Of particular concern is the possibility that heritability (h^2) attributed to Neanderthal markers might be tagging linked variants that arose in modern humans, resulting in overestimation of h^2 due to Neanderthal variants. Neanderthal variants also tend to be rare, and estimating the contribution of rare alleles to h^2 is challenging. In some previous studies, rare alleles have been excluded from h^2 estimates.
Wei & Robles et al develop and assess a method …
Reviewer #1 (Public Review):
In this manuscript, Wei & Robles et al seek to estimate the heritability contribution of Neanderthal Informative Markers (NIM) relative to SNPs that arose in modern humans (MH). This is a question that has received a fair amount of attention in recent studies, but persistent statistical limitations have made some prior results difficult to interpret. Of particular concern is the possibility that heritability (h^2) attributed to Neanderthal markers might be tagging linked variants that arose in modern humans, resulting in overestimation of h^2 due to Neanderthal variants. Neanderthal variants also tend to be rare, and estimating the contribution of rare alleles to h^2 is challenging. In some previous studies, rare alleles have been excluded from h^2 estimates.
Wei & Robles et al develop and assess a method that estimates both total heritability and per-SNP heritability of NIMs, allowing them to test whether NIM contributions to variation in human traits are similar or substantially different than modern human SNPs. They find an overall depletion of heritability across the traits that they studied, and found no traits with enrichment of heritability due to NIMs. They also developed a 'fine-mapping' procedure that aims to find potential causal alleles and report several potentially interesting associations with putatively functional variants.
Strengths of this study include rigorous assessment of the statistical methods employed with simulations and careful design of the statistical approaches to overcome previous limitations due to LD and frequency differences between MH and NIM variants. I found the manuscript interesting and I think it makes a solid contribution to the literature that addresses limitations of some earlier studies.
My main questions for the authors concern potential limitations of their simulation approach. In particular, they describe varying genetic architectures corresponding to the enrichment of effects among rare alleles or common alleles. I agree with the authors that it is important to assess the impact of (unknown) architecture on the inference, but the models employed here are ad hoc and unlikely to correspond to any mechanistic evolutionary model. It is unclear to me whether the contributions of rare and common alleles (and how these correspond with levels of LD) in real data will be close enough to these simulated schemes to ensure good performance of the inference.
In particular, the common allele model employed makes 90% of effect variants have frequencies above 5% -- I am not aware of any evolutionary model that would result in this outcome, which would suggest that more recent mutations are depleted for effects on traits (of course, it is true that common alleles explain much more h^2 under neutral models than rare alleles, but this is driven largely by the effect of frequency on h^2, not the proportion of alleles that are effect alleles). Likewise, the rare allele model has the opposite pattern, with 90% of effect alleles having frequencies under 5%. Since most alleles have frequencies under 5% anyway (~58% of MH SNPs and ~73% of NIM SNPs) this only modestly boosts the prevalence of low frequency effect alleles relative to their proportion. Some selection models suggest that rare alleles should have much bigger effects and a substantially higher likelihood of being effect alleles than common alleles. I'm not sure this situation is well-captured by the simulations performed. With LD and MAF annotations being applied in relatively wide quintile bins, do the authors think their inference procedure will do a good job of capturing such rare allele effects? This seems particularly important to me in the context of this paper, since the claim is that Neanderthal alleles are depleted for overall h^2, but Neanderthal alleles are also disproportionately rare, meaning they could suffer a bigger penalty. This concern could be easily addressed by including some simulations with additional architectures to those considered in the manuscript.
-
Reviewer #2 (Public Review):
The goal of the work described in this paper is to comprehensively describe the contribution of Neanderthal-informative mutations (NIMs) to complex traits in modern human populations. There are some known challenges in studying these variants, namely that they are often uncommon, and have unusually long haplotype structures. To overcome these, the authors customized a genotyping array to specifically assay putative Neanderthal haplotypes, and used a recent method of estimating heritability that can explicitly account for differences in MAF and LD.
This study is well thought-out, and the ability to specifically target the genotyping array to the variants in question and then use that information to properly control for population structure is a massive benefit. The methodology also allowed them to include …
Reviewer #2 (Public Review):
The goal of the work described in this paper is to comprehensively describe the contribution of Neanderthal-informative mutations (NIMs) to complex traits in modern human populations. There are some known challenges in studying these variants, namely that they are often uncommon, and have unusually long haplotype structures. To overcome these, the authors customized a genotyping array to specifically assay putative Neanderthal haplotypes, and used a recent method of estimating heritability that can explicitly account for differences in MAF and LD.
This study is well thought-out, and the ability to specifically target the genotyping array to the variants in question and then use that information to properly control for population structure is a massive benefit. The methodology also allowed them to include rarer alleles that were generally excluded from previous studies. The simulations are thorough and convincingly show the importance of accounting for both MAF and LD in addition to ancestry. The fine-mapping done to disentangle effects between actual Neanderthal variants and Modern human ones on the same haplotype also seems reasonable. They also strike a good balance between highlighting potentially interesting examples of Neanderthal variants having an effect on phenotype without overinterpreting association-based findings.
The main weakness of the paper is in its description of the work, not the work itself. The paper currently places a lot of emphasis on comparing these results to prior studies, particularly on its disagreement with McArthur, et al. (2021), a study on introgressed variant heritability that was also done primarily in UK Biobank. While they do show that the method used in that study (LDSR) does not account for MAF and LD as effectively as this analysis, this work does not support the conclusion that this is a major problem with previous heritability studies. McArthur et al. in fact largely replicate these results that Neanderthal variants (and more generally regions with Neanderthal variants) are depleted of heritability, and agree with the interpretation that this is likely due to selection against Neanderthal alleles. I actually find this a reassuring point, given the differences between the variant sets and methods used by the two studies, but it isn't mentioned in the text. Where the two studies differ is in specifics, mainly which loci have some association with human phenotypes; McArthur et al. also identified a couple groups of traits that were exceptions to the general rule of depleted heritability. While this work shows that not accounting for MAF and LD can lead to underestimating NIM heritability, I don't follow the logic behind the claim that this could lead to a false positive in heritability enrichment (a false negative would be more likely, surely?). There are also more differences between this and previous heritability studies than just the method used to estimate heritability, and the comparisons done here do not sufficiently account for these. A more detailed discussion to reconcile how, despite its weaknesses, LDSR picks up similar broad patterns while disagreeing in specifics is merited.
In general this work agrees with the growing consensus in the field that introgressed Neanderthal variants were selected against, such that those that still remain in human populations do not generally have large effects on phenotypes. There are exceptions to this, but for the most part observed phenotypic associations depend on the exact set of variants being considered, and, like those highlighted in this study, still lack more concrete validation. While this paper does not make a significant advance in this general understanding of introgressed regions in modern populations, it does increase our knowledge in how best to study them, and makes a good attempt at addressing issues that are often just mentioned as caveats in other studies. It includes a nice quantification of how important these variables are in interpreting heritability estimates, and will be useful for heritability studies going forward.
-
