Succinate mediates inflammation-induced adrenocortical dysfunction
Curation statements for this article:-
Curated by eLife

eLife assessment
Acute inflammation in mammals activates the hypothalamic pituatary axis leading to increased glucocorticoid release, which is required to restrain the inflammatory response. However, in settings of severe or prolonged inflammation, such as that seen in sepsis, there is reduced adrenal steridogenesis. The studies described in this paper provide a plausible mechanism for adrenal resistance which develops during excessive inflammation.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The hypothalamus-pituitary-adrenal (HPA) axis is activated in response to inflammation leading to increased production of anti-inflammatory glucocorticoids by the adrenal cortex, thereby representing an endogenous feedback loop. However, severe inflammation reduces the responsiveness of the adrenal gland to adrenocorticotropic hormone (ACTH), although the underlying mechanisms are poorly understood. Here, we show by transcriptomic, proteomic, and metabolomic analyses that LPS-induced systemic inflammation triggers profound metabolic changes in steroidogenic adrenocortical cells, including downregulation of the TCA cycle and oxidative phosphorylation, in mice. Inflammation disrupts the TCA cycle at the level of succinate dehydrogenase (SDH), leading to succinate accumulation and disturbed steroidogenesis. Mechanistically, IL-1β reduces SDHB expression through upregulation of DNA methyltransferase 1 (DNMT1) and methylation of the SDHB promoter. Consequently, increased succinate levels impair oxidative phosphorylation and ATP synthesis and enhance ROS production, leading to reduced steroidogenesis. Together, we demonstrate that the IL-1β-DNMT1-SDHB-succinate axis disrupts steroidogenesis. Our findings not only provide a mechanistic explanation for adrenal dysfunction in severe inflammation, but also offer a potential target for therapeutic intervention.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
This study explores the mechanisms responsible for reduced steroidogenesis of adrenocortical cells in a mouse model of systemic inflammation induced by LPS administration. Working from RNA and protein profiling data sets in adrenocortical tissue from LPS-treated mice they report that LPS perturbs the TCA cycle at the level of succinate dehydrogenase B (SDHB) impairing oxidative phosphorylation. Additional studies indicate these events are coupled to increased IL-1β levels which inhibit SDHB expression through DNA methyltransferase-dependent DNA methylation of the SDHB promoter.
In general, these are interesting studies with some novel implications. I do, however, have concerns with some of the author's rather broad conclusions given the limitations of their experimental approach. The …
Author Response
Reviewer #1 (Public Review):
This study explores the mechanisms responsible for reduced steroidogenesis of adrenocortical cells in a mouse model of systemic inflammation induced by LPS administration. Working from RNA and protein profiling data sets in adrenocortical tissue from LPS-treated mice they report that LPS perturbs the TCA cycle at the level of succinate dehydrogenase B (SDHB) impairing oxidative phosphorylation. Additional studies indicate these events are coupled to increased IL-1β levels which inhibit SDHB expression through DNA methyltransferase-dependent DNA methylation of the SDHB promoter.
In general, these are interesting studies with some novel implications. I do, however, have concerns with some of the author's rather broad conclusions given the limitations of their experimental approach. The paper could be improved by addressing the following points:
- The limitations of using LPS as the model for systemic inflammation need to be explicitly described.
We thank the Reviewer for this suggestion. Indeed, the LPS model has several limitations as a preclinical model of sepsis, which are outlined in the revised Discussion. Despite its limitations, we chose this model over other models of sepsis, such as the cecal slurry model, due to its high reproducibility, which enabled the here presented mechanistic studies.
- The initial in vivo findings, which support the proposed metabolic perturbation, are based on descriptive profiling data obtained at one time point following a single dose of LPS. The author's conclusion that the ultimate transcriptional pathway identified hinges critically on knowledge of the time course of this effect following LPS, which is not adequately addressed in the paper. How was this time and dose of LPS established and are there data from different dose and time points?
We thank the Reviewer for raising this question, which we indeed addressed at the beginning of our studies in order to determine a suitable time point and dose of LPS treatment. We chose 6 h as a suitable starting time point to perform transcriptional analyses, based on the fact that LPS triggers transcriptional changes in the adrenal gland and other tissues within the range of few hours (1-3). Confirming our expectations we found 2,609 differentially expressed genes (Figure 1a) in the adrenal cortex of LPS-treated mice among which many were involved in cellular metabolism (Figure 1d,e, 2a-e, Table 1, Table 2). Acute transcriptional changes, which are more likely to reflect direct effects of inflammatory signals compared to changes occurring at later time points (for instance in the range of days), would allow us to mechanistically investigate the effects of inflammation in the adrenal gland, which was the purpose of our studies. Hence, we were guided by the transcriptional changes observed at 6 h of LPS treatment and established the hypothesis that disruption of the TCA cycle in adrenocortical cells is key in the impact of inflammation on adrenal function. Along this line, we analyzed the metabolomic profile of the adrenal gland at 6 and 24 h of LPS treatment. At 6 h succinate levels as well as the succinate / fumarate ratio remained unchanged (Author response image 1A), while at 24 h post-injection these were increased by LPS (Author response image 1B, Figure 2l,o,q). The time delay of the increase in succinate levels (observed at 24 h) following downregulation of Sdhb mRNA expression (at 6 h) can be explained by the time required for reduction of SDHB protein levels, which is dependent on the protein turnover suggested to be approximately 12 h in HeLa cells (4). Based on these findings, all further metabolomic analyses were performed at 24 h of LPS treatment.
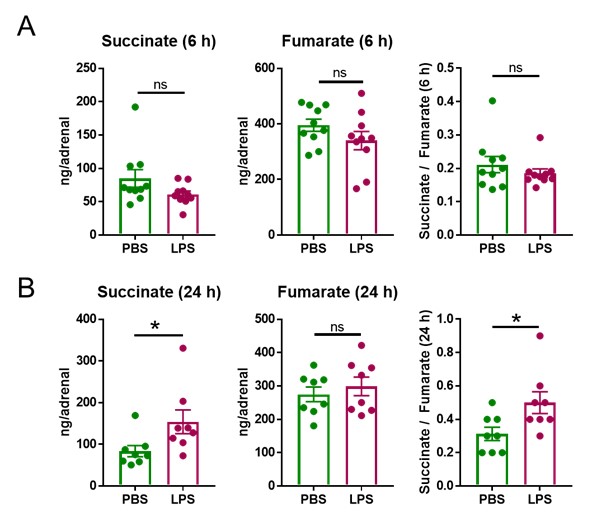
Author response image 1. LPS increases the succinate/fumarate ratio at 24 but not 6h. Mice were i.p. injected with 1 mg/kg LPS and 6 h (A) and 24 h (B) post-injection succinate and fumarate levels were determined by LC-MS/MS in the adrenal gland. n=8-10; data are presented as mean ± s.e.m. Statistical analysis was done with two-tailed Mann-Whitney test. *p < 0.05.
Having established the most suitable time points of LPS treatments to observe induced transcriptional and metabolic changes, we set out to define the LPS dose to be used in subsequent experiments. The data shown in Author response image 1, were acquired after treatment with 1 mg/kg LPS. This is a dose that was previously reported to cause transcriptional re-profiling of the adrenal gland (1, 2). However, 5 mg/kg LPS, similarly to 1 mg/kg LPS, also reduced Sdhb, Idh1 and Idh2 expression at 4 h (Author response image 2A) and increased succinate and isocitrate levels at 24 h (Author response image 2B) in the adrenal gland. Given that the effects of 1 and 5 mg/kg LPS were similar, for animal welfare reasons we continued our studies with the lower dose.
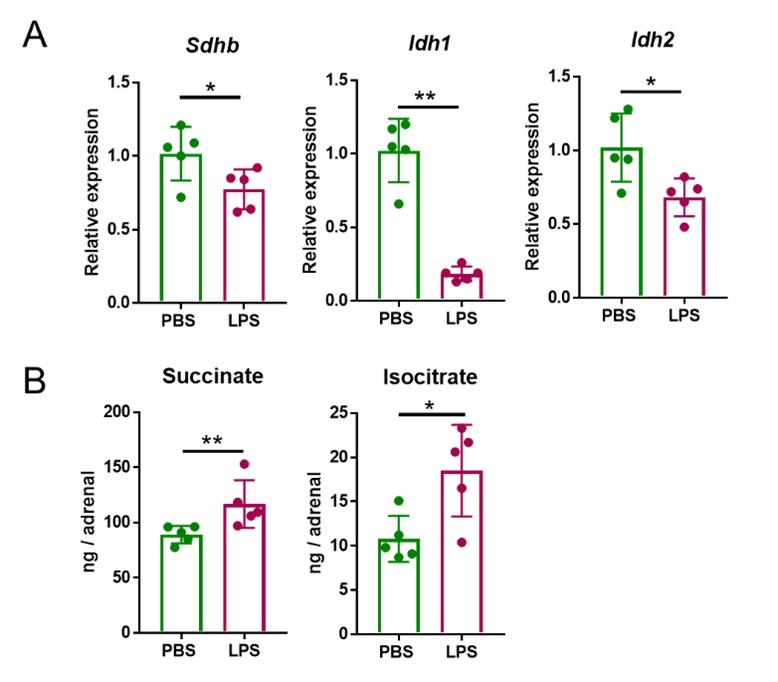
Author response image 2. Five mg/kg LPS downregulate Sdhb, Idh1 and Idh2 expression and increase succinate and isocitrate levels in the adrenal gland of mice. Sdhb, Idh1 and Idh2 expression (A) and succinate and isocitrate levels (B) were assessed in the adrenal gland of mice treated with 5 mg/kg LPS for 4 h (A) and 24 h (B). n=5; data are presented as mean ± s.d. Statistical analysis was done with two-tailed Mann-Whitney test. *p < 0.05, **p < 0.01.
- Related to the point above, the authors data supporting a break in the TCA cycle would be strengthened direct biochemical assessment (metabolic flux analysis) of step kin the TCA cycle process impacted.
We entirely agree with the Reviewer and considered performing TCA cycle metabolic flux analyses in adrenocortical cells. Unfortunately, the low yield of adrenocortical cells per mouse (approx. 3,000- 6,000) does not allow the performance of metabolic flux experiments, which require higher cell numbers per sample, several time points per condition and an adequate number of replicates per experiment. Moreover, NCI-H295R cells being adrenocortical carcinoma cells are expected to have substantially altered metabolic fluxes compared to normal cells. Since we wouldn’t have the capacity to confirm findings from metabolic flux experiments in NCI-H295R cells in primary adrenocortical cells, as we did for the rest of the experiments, we decided to not perform metabolic flux experiments in NCI-H295R cells. However, performing metabolic flux analyses in adrenocortical cells under inflammatory or other stress conditions remains an important future task that we will pursue upon establishment of a more suitable cell culture system.
- The proposed connection of DNMT and IL1 signaling to systemic inflammation and reduced steroidogenesis could be more firmly established by additional studies in adrenal cortical cells lacking these genes.
We thank the Reviewer for this excellent suggestion. In the revised manuscript we strengthened the evidence for an IL-1β –DNMT1 link and show that DNMT1 deficiency blocks the effects of IL-1β on SDHB promoter methylation (Figure 6k), the succinate / fumarate ratio (Figure 6m), the oxygen consumption rate (Figure 6n) and steroidogenesis (Figure 6o-q) in adrenocortical cells. In order to validate the role of IL-1β in vivo, mice were simultaneously treated with LPS and Raleukin, an IL-1R antagonist. Treatment with Raleukin increased the SDH activity (Figure 6r), reduced succinate levels and the succinate / fumarate ratio (Figure 6s,t) and increased corticosterone production in LPS-treated mice (Figure 6u).
Reviewer #2 (Public Review):
The present manuscript provides a mechanistic explanation for an event in adrenal endocrinology: the resistance which develops during excessive inflammation relative to acute inflammation. The authors identify disturbances in adrenal mitochondria function that differentiate excessive inflammation. During severe inflammation the TCA in the adrenal is disrupted at the level of succinate production producing an accumulation of succinate in the adrenal cortex. The authors also provide a mechanistic explanation for the accumulation of succinate, they demonstrate that IL1b decreases expression of SDH the enzyme that degrades succinate through a methylation event in the SDH promoter. This work presents a solid explanation for an important phenomenon. Below are a few questions that should be resolved experimentally.
- The authors should confirm through direct biochemical assays of enzymatic activity that steroidogenesis enzyme activity is not impaired. Many of these enzymes are located in the mitochondria and their activity may be diminished due to the disturbed, high succinate environment of the cortical cell as opposed to the low ATP production.
We thank the Reviewer for this question. The activity of the first and rate-limiting steroidogenic enzyme, cytochrome P450-side-chain-cleavage (SCC, CYP11A1) which generates pregnenolone from cholesterol, was recently shown to require intact SDH function (5). In agreement with this report we show that production of progesterone, the direct derivative of pregnenolone, is impaired upon SDH inhibition (Figure 5b,e,h). In addition, we assessed the activity of CYP11B1 (steroid 11β-hydroxylase), the enzyme catalyzing the conversion of 11-deoxycorticosterone to corticosterone, i.e. the last step of glucocorticoid synthesis, by determining the corticosterone and 11-deoxycorticosterone levels by LC-MS/MS and calculating the ratio of corticosterone to 11-deoxycorticosterone in ACTH-stimulated adrenocortical cells and explants. The corticosterone / 11-deoxycorticosterone ratio was not affected by Sdhb silencing in adrenocortical cells (Figure 5- Supplement 2g) nor did it change upon LPS treatment in adrenal explants (Figure 5- Supplement 2h), suggesting that CYP11B1 activity may not be altered upon SDH blockage. Hence, we propose that upon inflammation impairment of SDH function may disrupt at least the first steps of steroidogenesis (producing pregnenolone/progesterone), thereby diminishing production of all downstream adrenocortical steroids. This is now discussed in the revised manuscript.
- What is the effect of high ROS production? Is steroidogenesis resolved if ROS is pharmacologically decreased even if the reduction of ATP is not resolved?
We thank the Reviewer for this suggestion, which helped us to broaden our findings. Indeed, ROS scavenging by the vitamin E analog Trolox (Figure 5n) partially reversed the inhibitory effect of DMM on steroidogenesis (Figure 5o,p), suggesting that impairment of SDH function impacts steroidogenesis also via enhanced ROS production (Figure 4g).
- Does increased intracellular succinate (through cell permeable succinate treatment) inhibit steroidogenesis even if there is not a blockage of OXPHOS?
We suggest that SDH inhibition and succinate accumulation lead to reduced steroidogenesis due to impaired oxidative phosphorylation (Figure 4c,e, 5i), reduced ATP synthesis (Figure 4d, 5j-m) and increased ROS production (Figure 4g, 5o,p). Since SDH is part (complex II) of the electron chain transfer it cannot be decoupled from oxidative phosphorylation, thereby limiting the experimental means for addressing this question.
- It should be demonstrated the genetic loss of IL1 signaling in adrenal cortical cells results in a loss of the effect of LPS on reduced steroidogenesis and increased succinate accumulation.
We thank the Reviewer for this suggestion. Development of a mouse line with genetic loss of Il-1r in adrenocortical cells was rather impossible during the short time of revisions. Instead, mice under LPS treatment were treated with the IL-1R antagonist, Raleukin, to study the in vivo effects of IL-1β in the adrenal gland. IL-1R antagonism increased SDH activity in the adrenal cortex (Figure 6r), decreased succinate levels and the succinate/fumarate ratio in the adrenal gland (Figure 6s,t) and enhanced corticosterone production (Figure 6u) in LPS-treated mice, supporting our hypothesis that IL-1β mediates the effects of systemic inflammation in the adrenal cortex.
- It should be demonstrated the genetic loss of IL1 signaling in adrenal cortical cells results in a loss of the effect of LPS on SDH activity and ATP production and SDH promoter methylation
As outlined above, Raleukin treatment increased SDH activity in the adrenal cortex (Figure 6r) and decreased succinate levels and the succinate/fumarate ratio in the adrenal gland (Figure 6s,t) of mice treated with LPS. Furthermore, IL-1β reduced the ATP/ADP ratio (Figure 6e) and enhanced SDHB promoter methylation in NCI-H295R cells (Figure 6k).
- It should be shown that the silencing of DNMT eliminates or diminishes the effect of LPS on reduced steroidogenesis and increased succinate accumulation.
We thank the Reviewer for this suggestion, which prompted us to strengthen the evidence for the implication of DNMT1 in the effects of LPS on adrenocortical cell metabolism and function. As mentioned above, development of a new mouse line, in this case bearing genetic loss of DNMT1 in adrenocortical cells, was considered impossible during the short time of revisions. Therefore, we assessed the role of DNMT1 by silencing it via siRNA transfections in primary adrenocortical cells and NCI-H295R cells. We show that DNMT1 silencing inhibits the effect of IL-1β on SDHB promoter methylation (Figure 6k), restores Sdhb expression (Figure 6l) and reduces the succinate/fumarate ratio in IL-1β treated adrenocortical cells (Figure 6m). Accordingly, DNMT1 silencing restores ACTH-induced production of corticosterone, 11-deoxycorticosterone and progesterone in IL-1β treated adrenocortical cells (Figure 6o-q). We chose to stimulate adrenocortical cells with IL-1β instead of LPS, as in vitro the effects of IL-1β were more robust than these of LPS (possibly due to a reduction of TLR4 expression or function in cultured adrenocortical cells) and in order to show the link between IL-1β and DNMT1.
- Does silencing of DNMT reduce OXPHOS in adrenal cortical cells?
We measured the oxygen consumption rate in NCI-H295R cells, which were transfected with siRNA against DNMT1 and treated or not with IL-1β. IL-1β reduced the OCR in cells transfected with control siRNA, while DNMT1 silencing blunted the effect of IL-1β (Figure 6n).
- The effects of LPS on reduced adrenal steroidogenesis are not elaborated at the physiological level. The manuscript should demonstrate the ramifications of the adrenal function decreasing after LPS. Does CORT release become less pronounced after subsequent challenges? Does baseline CORT decrease at some point? No physiological consequences are shown. Similarly, these physiological consequences of decreased adrenal function should be dependent on decreased SDH activity and OXPHOS in adrenal cells and this should be demonstrated experimentally.
We thank the Reviewer for raising this excellent question. Inflammation is a potent inducer of the Hypothalamus-Pituitary-Adrenal gland (HPA) axis, causing increased glucocorticoid production, a stress response leading to vital immune and metabolic adaptations. Accordingly, LPS treatment rapidly increases glucocorticoid production in mice (1, 6, 7). Reduced adrenal gland responsiveness to ACTH associates with decreased survival of septic mice (8). These preclinical findings stand in accordance with observations in septic patients, in which impairment of adrenal function correlates with high risk for death (9). Along this line, ACTH test was suggested to have prognostic value for identification of septic patients with high mortality risk (9, 10).
In order to confirm impairment of the adrenal gland function in septic mice, animals were subjected to sepsis via administration of a high LPS dose (10 mg / kg) and treated with ACTH 24 h later. Indeed, the ACTH-induced increase in corticosterone levels was diminished in LPS-treated mice (Author response image 3). This finding was further confirmed in adrenal explants, in which LPS pre-treatment also blunted ACTH-stimulated corticosterone production (Figure 5s).
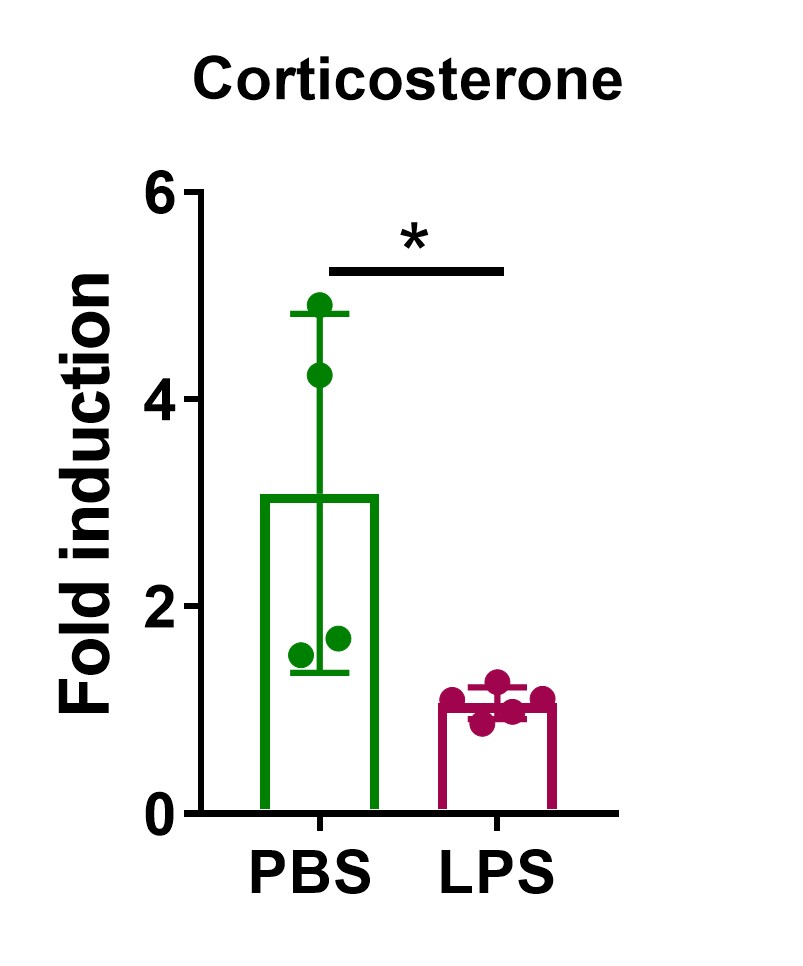
Author response image 3. High LPS dose blunts the ACTH response in mice. C57BL/6J mice were i.p. injected with 10 mg/kg LPS or PBS and 24 h later they were i.p. injected with 1 mg/kg ACTH. One hour after ACTH administration blood was retroorbitally collected and corticosterone plasma levels were determined by LC-MS/MS. n=4-5; data are presented as mean ± s.d. Statistical analysis was done with two-tailed Mann-Whitney test. *p < 0.05.
Given that purpose of our studies was to dissect the mechanisms underlying adrenal gland dysfunction in inflammation rather than analyzing the physiological consequences thereof, we chose not to follow these lines of investigations and concentrate on the role of cell metabolism in adrenocortical cells in the context of inflammation.
References
- W. Kanczkowski, A. Chatzigeorgiou, M. Samus, N. Tran, K. Zacharowski, T. Chavakis, S. R. Bornstein, Characterization of the LPS-induced inflammation of the adrenal gland in mice. Mol Cell Endocrinol 371, 228-235 (2013).
- L. S. Chen, S. P. Singh, M. Schuster, T. Grinenko, S. R. Bornstein, W. Kanczkowski, RNA-seq analysis of LPS-induced transcriptional changes and its possible implications for the adrenal gland dysregulation during sepsis. J Steroid Biochem Mol Biol 191, 105360 (2019).
- V. I. Alexaki, G. Fodelianaki, A. Neuwirth, C. Mund, A. Kourgiantaki, E. Ieronimaki, K. Lyroni, M. Troullinaki, C. Fujii, W. Kanczkowski, A. Ziogas, M. Peitzsch, S. Grossklaus, B. Sonnichsen, A. Gravanis, S. R. Bornstein, I. Charalampopoulos, C. Tsatsanis, T. Chavakis, DHEA inhibits acute microglia-mediated inflammation through activation of the TrkA-Akt1/2-CREB-Jmjd3 pathway. Mol Psychiatry 23, 1410-1420 (2018).
- C. Yang, J. C. Matro, K. M. Huntoon, D. Y. Ye, T. T. Huynh, S. M. Fliedner, J. Breza, Z. Zhuang, K. Pacak, Missense mutations in the human SDHB gene increase protein degradation without altering intrinsic enzymatic function. FASEB J 26, 4506-4516 (2012).
- H. S. Bose, B. Marshall, D. K. Debnath, E. W. Perry, R. M. Whittal, Electron Transport Chain Complex II Regulates Steroid Metabolism. iScience 23, 101295 (2020).
- W. Kanczkowski, V. I. Alexaki, N. Tran, S. Grossklaus, K. Zacharowski, A. Martinez, P. Popovics, N. L. Block, T. Chavakis, A. V. Schally, S. R. Bornstein, Hypothalamo-pituitary and immune-dependent adrenal regulation during systemic inflammation. Proc Natl Acad Sci U S A 110, 14801-14806 (2013).
- W. Kanczkowski, A. Chatzigeorgiou, S. Grossklaus, D. Sprott, S. R. Bornstein, T. Chavakis, Role of the endothelial-derived endogenous anti-inflammatory factor Del-1 in inflammation-mediated adrenal gland dysfunction. Endocrinology 154, 1181-1189 (2013).
- C. Jennewein, N. Tran, W. Kanczkowski, L. Heerdegen, A. Kantharajah, S. Drose, S. Bornstein, B. Scheller, K. Zacharowski, Mortality of Septic Mice Strongly Correlates With Adrenal Gland Inflammation. Crit Care Med 44, e190-199 (2016).
- D. Annane, V. Sebille, G. Troche, J. C. Raphael, P. Gajdos, E. Bellissant, A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA 283, 1038-1045 (2000).
- E. Boonen, S. R. Bornstein, G. Van den Berghe, New insights into the controversy of adrenal function during critical illness. Lancet Diabetes Endocrinol 3, 805-815 (2015).
- C. C. Huang, Y. Kang, The transient cortical zone in the adrenal gland: the mystery of the adrenal X-zone. J Endocrinol 241, R51-R63 (2019).
-
eLife assessment
Acute inflammation in mammals activates the hypothalamic pituatary axis leading to increased glucocorticoid release, which is required to restrain the inflammatory response. However, in settings of severe or prolonged inflammation, such as that seen in sepsis, there is reduced adrenal steridogenesis. The studies described in this paper provide a plausible mechanism for adrenal resistance which develops during excessive inflammation.
-
Reviewer #1 (Public Review):
This study explores the mechanisms responsible for reduced steroidogenesis of adrenocortical cells in a mouse model of systemic inflammation induced by LPS administration. Working from RNA and protein profiling data sets in adrenocortical tissue from LPS-treated mice they report that LPS perturbs the TCA cycle at the level of succinate dehydrogenase B (SDHB) impairing oxidative phosphorylation. Additional studies indicate these events are coupled to increased IL-1β levels which inhibit SDHB expression through DNA methyltransferase-dependent DNA methylation of the SDHB promoter.
In general, these are interesting studies with some novel implications. I do, however, have concerns with some of the author's rather broad conclusions given the limitations of their experimental approach. The paper could be improved …
Reviewer #1 (Public Review):
This study explores the mechanisms responsible for reduced steroidogenesis of adrenocortical cells in a mouse model of systemic inflammation induced by LPS administration. Working from RNA and protein profiling data sets in adrenocortical tissue from LPS-treated mice they report that LPS perturbs the TCA cycle at the level of succinate dehydrogenase B (SDHB) impairing oxidative phosphorylation. Additional studies indicate these events are coupled to increased IL-1β levels which inhibit SDHB expression through DNA methyltransferase-dependent DNA methylation of the SDHB promoter.
In general, these are interesting studies with some novel implications. I do, however, have concerns with some of the author's rather broad conclusions given the limitations of their experimental approach. The paper could be improved by addressing the following points:
1. The limitations of using LPS as the model for systemic inflammation need to be explicitly described.
2. The initial in vivo findings, which support the proposed metabolic perturbation, are based on descriptive profiling data obtained at one time point following a single dose of LPS. The author's conclusion that the ultimate transcriptional pathway identified hinges critically on knowledge of the time course of this effect following LPS, which is not adequately addressed in the paper. How was this time and dose of LPS established and are there data from different dose and time points?
3. Related to the point above, the authors data supporting a break in the TCA cycle would be strengthened direct biochemical assessment (metabolic flux analysis) of step kin the TCA cycle process impacted.
4. The proposed connection of DNMT and IL1 signaling to systemic inflammation and reduced steriodogenesis could be more firmly established by additional studies in adrenal cortical cells lacking these genes. -
Reviewer #2 (Public Review):
The present manuscript provides a mechanistic explanation for an event in adrenal endocrinology: the resistance which develops during excessive inflammation relative to acute inflammation. The authors identify disturbances in adrenal mitochondria function that differentiate excessive inflammation. During severe inflammation the TCA in the adrenal is disrupted at the level of succinate production producing an accumulation of succinate in the adrenal cortex. The authors also provide a mechanistic explanation for the accumulation of succinate, they demonstrate that IL1b decreases expression of SDH the enzyme that degrades succinate through a methylation event in the SDH promoter. This work presents a solid explanation for an important phenomenon. Below are a few questions that should be resolved experimentally.
Reviewer #2 (Public Review):
The present manuscript provides a mechanistic explanation for an event in adrenal endocrinology: the resistance which develops during excessive inflammation relative to acute inflammation. The authors identify disturbances in adrenal mitochondria function that differentiate excessive inflammation. During severe inflammation the TCA in the adrenal is disrupted at the level of succinate production producing an accumulation of succinate in the adrenal cortex. The authors also provide a mechanistic explanation for the accumulation of succinate, they demonstrate that IL1b decreases expression of SDH the enzyme that degrades succinate through a methylation event in the SDH promoter. This work presents a solid explanation for an important phenomenon. Below are a few questions that should be resolved experimentally.
The authors should confirm through direct biochemical assays of enzymatic activity that steroidogenesis enzyme activity is not impaired. Many of these enzymes are located in the mitochondria and their activity may be diminished due to the disturbed, high succinate environment of the cortical cell as opposed to the low ATP production.
What is the effect of high ROS production. Is steroidogenesis resolved if ROS is pharmacologically decreased even if the reduction of ATP is not resolved?
Does increased intracellular succinate (through cell permeable succinate treatment) inhibit steroidogenesis even if there is not a blockage of OXPHOS?
It should be demonstrated the genetic loss of IL1 signaling in adrenal cortical cells results in a loss of the effect of LPS on reduced steroidogenesis and increased succinate accumulation.
It should be demonstrated the genetic loss of IL1 signaling in adrenal cortical cells results in a loss of the effect of LPS on SDH activity and ATP production and SDH promoter methylation
It should be shown that the silencing of DNMT eliminates or diminishes the effect of LPS on reduced steroidogenesis and increased succinate accumulation.
Does silencing of DNMT reduce OXPHOS in adrenal cortical cells?
The effects of LPS on reduced adrenal steroidogenesis are not elaborated at the physiological level. The manuscript should demonstrate the ramifications of the adrenal function decreasing after LPS. Does CORT release become less pronounced after subsequent challenges? Does baseline CORT decrease at some point? No physiological consequences are shown. Similarly, these physiological consequences of decreased adrenal function should be dependent on decreased SDH activity and OXPHOS in adrenal cells and this should be demonstrated experimentally.
-
Reviewer #3 (Public Review):
The authors attempted to elucidate mechanisms underlying adrenal dysfunction in severe inflammation.
Utilizing transcriptomic, proteomic and metabolomic analyses of adrenocortical cells in male mice after lipopolysaccharid induced systemic inflammation is a major strength of this study.
The use of sophisticated methods and the results support the conclusion of the authors that the Interleukin 1beta - DNA methyltransferase 1 - succinate dehydrogenase b axis with increased succinate and reduced ATP levels disrupts steroid production in lipopolysaccharid induced systemic inflammation.
Various inflammatory conditions in humans are treated with steroids and this animal based study may help identify future therapeutic targets besides the administration of glucocorticoids.
-
