Pre-existing chromosomal polymorphisms in pathogenic E. coli potentiate the evolution of resistance to a last-resort antibiotic
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
This paper combines evolution experiments with genomic analysis of environmental samples to study the evolution of colistin resistance in E. coli. It highlights the importance of pre-existing genomic variations in clinical strains in driving the evolution of antibiotic resistance. The results presented here are relevant for clinical and non-clinical microbiologists studying antibiotic resistance to last-resort drugs like colistin. The design of the research is simple and elegant, and the genomic data analysis connects the in vitro findings to the real world. However, the authors could better align the experimental and clinical data, and better clarify their experimental design choices.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 and Reviewer #2 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
- The Natural History of Model Organisms (eLife)
Abstract
Bacterial pathogens show high levels of chromosomal genetic diversity, but the influence of this diversity on the evolution of antibiotic resistance by plasmid acquisition remains unclear. Here, we address this problem in the context of colistin, a ‘last line of defence’ antibiotic. Using experimental evolution, we show that a plasmid carrying the MCR-1 colistin resistance gene dramatically increases the ability of Escherichia coli to evolve high-level colistin resistance by acquiring mutations in lpxC , an essential chromosomal gene involved in lipopolysaccharide biosynthesis. Crucially, lpxC mutations increase colistin resistance in the presence of the MCR-1 gene, but decrease the resistance of wild-type cells, revealing positive sign epistasis for antibiotic resistance between the chromosomal mutations and a mobile resistance gene. Analysis of public genomic datasets shows that lpxC polymorphisms are common in pathogenic E. coli, including those carrying MCR-1, highlighting the clinical relevance of this interaction. Importantly, lpxC diversity is high in pathogenic E. coli from regions with no history of MCR-1 acquisition, suggesting that pre-existing lpxC polymorphisms potentiated the evolution of high-level colistin resistance by MCR-1 acquisition. More broadly, these findings highlight the importance of standing genetic variation and plasmid/chromosomal interactions in the evolutionary dynamics of antibiotic resistance.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
Most work on antibiotic resistance focuses on particular resistance genes often located on plasmids, but rarely how these genes interact with others located on the chromosome of the host organism. Considering variation in the host genome and its interaction with resistance plasmids can help predict which hosts are more likely to become resistant to a given antibiotic and explain why the same plasmid may not confer the same level of resistance to different strains.
The authors take a clever approach to finding such genetic interactions by designing an evolution experiment using E. coli carrying an MCR-1 plasmid containing resistance genes to colistin. They then select for increased resistance to colistin and sequence the genomes of the most resistant isolates. This allowed them to …
Author Response
Reviewer #1 (Public Review):
Most work on antibiotic resistance focuses on particular resistance genes often located on plasmids, but rarely how these genes interact with others located on the chromosome of the host organism. Considering variation in the host genome and its interaction with resistance plasmids can help predict which hosts are more likely to become resistant to a given antibiotic and explain why the same plasmid may not confer the same level of resistance to different strains.
The authors take a clever approach to finding such genetic interactions by designing an evolution experiment using E. coli carrying an MCR-1 plasmid containing resistance genes to colistin. They then select for increased resistance to colistin and sequence the genomes of the most resistant isolates. This allowed them to identify a particular gene lpxC that confers increased resistance to E. coli when combined with the MCR-1 plasmid (more than the sum of each mutation alone) and find that this is because of decreased membrane surface charge. They then investigate whether this mutation is relevant in wild E. coli isolates by analysing environmental samples from patients and other sources and find that indeed, this mutation is often found in carriers of the MCR-1 plasmid.
The study is very well-designed and presented in a concise and logical manner. The use of evolution experiments to identify the mutations and then engineer them to quantify the epistatic effects and understand the mechanism behind them is very elegant. The real-world relevance is then supported by looking for these mutations in environmental samples. Despite this simplicity and clarity, in some places, the writing could be improved. I particularly found that the second half of the paper was not as easy to follow as the first part and could benefit from some clarifications. The figures could also contain a bit more information to help the reader.
Thank you!
1.1 For example, the abstract starts by talking about standing genetic variation but it's not immediately clear what is meant by that. Standing genetic variation seems to suggest that the resistance gene itself is present in the initial population, rather than variation in other loci that might affect the selection of the resistance gene. This could be better formulated.
We have revised the abstract to be clearer about the source of genetic variation.
1.2 The figures could be improved by being more specific about the datasets: are mutations in Figure 2 in the WT or the MCR-1 positive lines? Are the SNPs in Fig. 4A in lpxC? Do all isolates in Fig. 4 have the MCR-1 plasmid?
Thank you for the comment. We have edited the figure legend (line 128, page 5). Yes, Fig. 4A shows SNPs in lpxC, and all the isolates in Fig 4 have the MCR-1 plasmid. We have now clarified this in the figure legend (line 230, page 9).
1.3 Finally, the arguments being made about diversity in the different phylogroups were not very clear. This could be made more explicit at first mention, rather than later in the discussion section.
We have revised this section to clarify theses points (lines 242-245, page 10).
Reviewer #3 (Public Review):
Jangir et al. used an 'evolutionary ramp' experiment to evolve E. coli strains under the selection pressure of increasing colistin concentrations wherein the surviving fractions were collected for genomic analysis. They report that the mcr-1 carrying strain evolved higher colistin resistance much faster only in presence of lpxC mutations in the genome. They identify the mcr-1 and lpxC interactions to be positively epistatic and mutations only in lpxC do not lead to resistance to colistin. Taking a cue from their evolution experiments, they looked for the variations in lpxC sequences in the genomic datasets of clinical E. coli strains. They found many such variations in the genomes of clinical isolates. Importantly, they found those variations to be present even in non-resistant strains which might predispose those strains to gain untreatable levels of colistin resistance.
Strengths:
The study focuses on two key aspects of antibiotic resistance in clinical settings. First, is the antibiotic colistin itself which is part of the last line of defense. Second, is the importance of genomic variations in clinical isolates that have not been linked to any antibiotic resistance mechanisms. The data were presented in a logical sequence and maintained brevity. The link of lpxC to mcr-1 resistance is convincing.
Thank you!
Weaknesses:
The basic premise of the paper is solid but the following should be addressed.
3.1 In Figure 1, the authors applied the 'evolutionary ramp' method to isolate evolved strains with higher MIC to colistin; but, the conditions for the evolution of WT and strain carrying mcr-1 are different.Maintaining mcr-1 requires antibiotic selection which WT cannot withstand. Hence, if I am not mistaken, WT was not grown in the presence of any antibiotic.
The referee’s assertion that the selective pressures experienced by the WT and MCR+ populations were different is incorrect. We increased relative antibiotic dose (i.e., as a fraction of the MIC of the parental strains) at the same rate for both the WT and MCR+ populations. This is clearly explained in the text (lines 98-100, page 3), and the absolute colistin doses are shown in Figure 1. Please also see response 2.4 above.
In our study, we used a naturally occurring MCR-1 carrying plasmid from the IncX4 family. This plasmid is actually very stable (in the short term at least) in the absence of colistin, in spite of the costs imposed by MCR-1. We speculate that this stability in part reflect the high conjugation rate of the plasmids and the presence of a toxin-antitoxin module.
3.2 Not only that, maintaining a ~32 Kb plasmid itself can have different selective landscapes. The authors may replicate the experiment with their low-copy clone of mcr-1 which would make it easier for the authors to have an empty vector in WT as a proper control. Since now they know the expected mutations to be in lpxC, they might sequence a PCR amplicon of that region for validation of their hypothesis.
This is an interesting idea for a future study. We agree with the referee that the presence of the MCR-1 plasmid may impose additional selective pressures that could potentially lead to bacteria-plasmid co-evolution. However, our data suggests that bacteria-plasmid interactions were not an important selective force over the course of our experiment: we detected no mutations in the plasmid and almost all of the chromosomal mutations that we detected could be easily associated with selective pressures imposed by colistin.
3.3 In Figure 2, what are the effects of these mutations in lpxC? The authors state that many mutations map on to the metal binding domain; but are those significant changes? LpxC is relatively well characterized and authors may want to comment on these mutations a little more.
Yes, most of the evolved lines had mutations in the metal-binding domain site, and it is known that this site is very important for lpxC activity. For example, mutations at positions 79, 238, 242 and 246 lead to a hundred to thousand-fold decrease in lpxC activity (PMID: 24117400, 24108127, and 11148046), and many of our mutations map close to these sites (lines 140-143, page 6, and Figure 2b).
3.4 Also, lpxC mutations showed enrichment but lpxA did not. Is this suggestive of the type of Lipid A that is more preferred for the epistatic interactions? The authors may want to comment on that.
Interestingly, this could be the case that the epistatic interactions depend on the type of lipid A modification and the associated pleiotropic effects. Because mutations in LPS biosynthesis genes can have diverse adverse effects as it alters the membrane properties. However, in-depth future work is required to understand how the different types of changes in lipid A influence interactions with MCR. We chose not to further explore this in the paper, because lpxA was rarely mutated (2/17 clones) compared to lpxC (11/17 clones).
3.5 In Figure 3, the lpxC mutant shows a reduction in fitness in a competition assay. What is the growth pattern of individual strains?
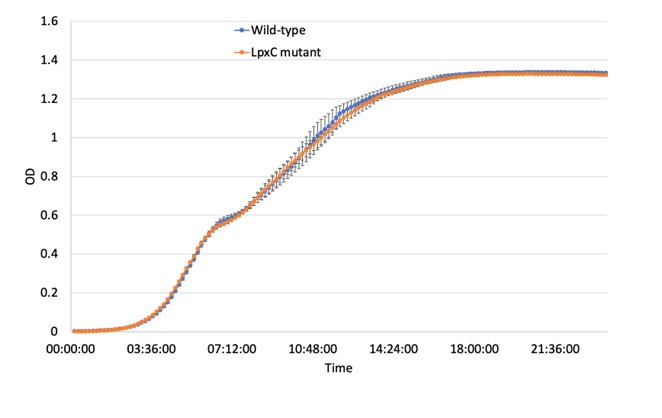
The standard growth curve assay shows no significant difference in growth rate between LpxC mutant and wild-type strain (figure below). This is evident by the fact that standard growth curves are not ideal for capturing small differences in growth/fitness. Therefore, we emphasize the results of the competition experiment as this is gold standard method for measuring fitness effects (Figure 3c).
3.6 There is a possibility that slow growth of lpxC mutant provides benefit under antibiotic stress.
This is an interesting idea, but in this case, the slow growth of the lpxC mutant is clearly associated with a small decrease in colistin resistance (Figure 3A).
3.7 Minor comment: the three individual replicates shown in Figure 3a are all identical within a sample and do not add to the figure where n=3. The authors can simply show SD or report correct values of replicates.
We chose to show the raw data points, as this is the style of presentation that is being increasingly used by journals (i.e., many journals now say show all raw data points when n<6 or 10). It would not make sense to show a standard error as this was equal to 0.
3.8 In Figure 4, as the authors themselves have stated, the difference in heterogeneity could be simply due to variation within phylogroups and subsequent compositional differences within the populations. The authors must check if mutations were found in the same location of lpxA as found in their own evolved strains. Without this information, the heterogeneity data would be speculative. Adding the lpxC variants reported in figure 2 to the trees of figure 4 (right) will make it clear if their conclusion is justified.
This is an interesting point. We found no overlap between our experimentally evolved mutations and naturally occurring lpxC mutations, either at the level of nucleotides or codons. However, it is unclear if we should expect to see an overlap for two reasons:
- The mutations present in natural isolates likely reflect a combination of beneficial mutations, neutral mutations, and weakly deleterious mutations. The mutations found in our evolved isolates, on the other hand, are all mutations that were beneficial under colistin selection. As such, it is probably not reasonable to expect a strong overlap between the two sets of mutations.
- The lpxC mutations that we observed in our 11 lpxC mutated isolates are highly diverse – we found no cases of parallel evolution at the nucleotide level, and only a single example of parallel evolution at the codon level. Given this, our data suggest that a very wide diversity of sites of lpxC can interact epistatically with MCR-1 to increase colistin resistance. Again, this high diversity of potential lpxC mutations should give a weak association between lab evolved and clinical isolates.
We have added these points in the text (lines 278-304, pages 11-12).
3.9 The authors can perform a confirmatory experiment for the pre-existing part of their hypothesis. If they perform the evolutionary ramp experiment with a strain carrying lpxC mutant strain, will they see faster evolution of high MIC mutants?
This is an interesting idea, our results suggest that more rapid evolution of high level colistin resistance would occur in the lpxC mutant compared to a wild-type strain (assuming that both had an equivalent opportunity to acquire MCR-1 by horizontal gene transfer).
4.0 The rationale of how the presence of lpxC mutations can cause a strain without any colistin resistance to acquire mcr-1 is not addressed. The authors may want to comment on that.
MCR-1 is carried on conjugative plasmids, and the main plasmid families that carry MCR-1 (IncI2 and IncX4) have high conjugative rates. We have changed the text of introduction to emphasize that MCR-1 is carried on conjugative plasmids, and we have linked MCR-1 acquisition to plasmid conjugation (lines 327-328, page 13).
-
Evaluation Summary:
This paper combines evolution experiments with genomic analysis of environmental samples to study the evolution of colistin resistance in E. coli. It highlights the importance of pre-existing genomic variations in clinical strains in driving the evolution of antibiotic resistance. The results presented here are relevant for clinical and non-clinical microbiologists studying antibiotic resistance to last-resort drugs like colistin. The design of the research is simple and elegant, and the genomic data analysis connects the in vitro findings to the real world. However, the authors could better align the experimental and clinical data, and better clarify their experimental design choices.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private …
Evaluation Summary:
This paper combines evolution experiments with genomic analysis of environmental samples to study the evolution of colistin resistance in E. coli. It highlights the importance of pre-existing genomic variations in clinical strains in driving the evolution of antibiotic resistance. The results presented here are relevant for clinical and non-clinical microbiologists studying antibiotic resistance to last-resort drugs like colistin. The design of the research is simple and elegant, and the genomic data analysis connects the in vitro findings to the real world. However, the authors could better align the experimental and clinical data, and better clarify their experimental design choices.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 and Reviewer #2 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
Most work on antibiotic resistance focuses on particular resistance genes often located on plasmids, but rarely how these genes interact with others located on the chromosome of the host organism. Considering variation in the host genome and its interaction with resistance plasmids can help predict which hosts are more likely to become resistant to a given antibiotic and explain why the same plasmid may not confer the same level of resistance to different strains.
The authors take a clever approach to finding such genetic interactions by designing an evolution experiment using E. coli carrying an MCR-1 plasmid containing resistance genes to colistin. They then select for increased resistance to colistin and sequence the genomes of the most resistant isolates. This allowed them to identify a particular gene …
Reviewer #1 (Public Review):
Most work on antibiotic resistance focuses on particular resistance genes often located on plasmids, but rarely how these genes interact with others located on the chromosome of the host organism. Considering variation in the host genome and its interaction with resistance plasmids can help predict which hosts are more likely to become resistant to a given antibiotic and explain why the same plasmid may not confer the same level of resistance to different strains.
The authors take a clever approach to finding such genetic interactions by designing an evolution experiment using E. coli carrying an MCR-1 plasmid containing resistance genes to colistin. They then select for increased resistance to colistin and sequence the genomes of the most resistant isolates. This allowed them to identify a particular gene lpxC that confers increased resistance to E. coli when combined with the MCR-1 plasmid (more than the sum of each mutation alone) and find that this is because of decreased membrane surface charge. They then investigate whether this mutation is relevant in wild E. coli isolates by analysing environmental samples from patients and other sources and find that indeed, this mutation is often found in carriers of the MCR-1 plasmid.
The study is very well-designed and presented in a concise and logical manner. The use of evolution experiments to identify the mutations and then engineer them to quantify the epistatic effects and understand the mechanism behind them is very elegant. The real-world relevance is then supported by looking for these mutations in environmental samples. Despite this simplicity and clarity, in some places, the writing could be improved. I particularly found that the second half of the paper was not as easy to follow as the first part and could benefit from some clarifications. The figures could also contain a bit more information to help the reader.
For example, the abstract starts by talking about standing genetic variation but it's not immediately clear what is meant by that. Standing genetic variation seems to suggest that the resistance gene itself is present in the initial population, rather than variation in other loci that might affect the selection of the resistance gene. This could be better formulated.
The figures could be improved by being more specific about the datasets: are mutations in Figure 2 in the WT or the MCR-1 positive lines? Are the SNPs in Fig. 4A in lpxC? Do all isolates in Fig. 4 have the MCR-1 plasmid?
Finally, the arguments being made about diversity in the different phylogroups were not very clear. This could be made more explicit at first mention, rather than later in the discussion section.
-
Reviewer #2 (Public Review):
Jangir and Yang, et al.'s manuscript presents highly interesting datasets aiming to shed light on the role of standing genetic diversity in the evolution of antibiotic resistance via epistatic interactions between chromosomal and plasmid genes. It combines a set of elegant evolution and downstream experiments with genomics and epidemiological genomic data with datasets from China and the UK. The authors focus on colistin resistance mediated by the MCR-1 colistin resistance gene, and its ability to increase E. coli's capacity to evolve higher levels of colistin resistance by adding mutations in lpxC.
The data presented is interesting and well presented. It also adds to the question of how beneficial foreign material interacts with the bacterial chromosome and how it can be sustained. The results presented …
Reviewer #2 (Public Review):
Jangir and Yang, et al.'s manuscript presents highly interesting datasets aiming to shed light on the role of standing genetic diversity in the evolution of antibiotic resistance via epistatic interactions between chromosomal and plasmid genes. It combines a set of elegant evolution and downstream experiments with genomics and epidemiological genomic data with datasets from China and the UK. The authors focus on colistin resistance mediated by the MCR-1 colistin resistance gene, and its ability to increase E. coli's capacity to evolve higher levels of colistin resistance by adding mutations in lpxC.
The data presented is interesting and well presented. It also adds to the question of how beneficial foreign material interacts with the bacterial chromosome and how it can be sustained. The results presented here are interesting and important to expand our understanding of antibiotic resistance evolution, but I do have some concerns about the data and the way it is presented.
-
Reviewer #3 (Public Review):
Jangir et al. used an 'evolutionary ramp' experiment to evolve E. coli strains under the selection pressure of increasing colistin concentrations wherein the surviving fractions were collected for genomic analysis. They report that the mcr-1 carrying strain evolved higher colistin resistance much faster only in presence of lpxC mutations in the genome. They identify the mcr-1 and lpxC interactions to be positively epistatic and mutations only in lpxC do not lead to resistance to colistin. Taking a cue from their evolution experiments, they looked for the variations in lpxC sequences in the genomic datasets of clinical E. coli strains. They found many such variations in the genomes of clinical isolates. Importantly, they found those variations to be present even in non-resistant strains which might predispose …
Reviewer #3 (Public Review):
Jangir et al. used an 'evolutionary ramp' experiment to evolve E. coli strains under the selection pressure of increasing colistin concentrations wherein the surviving fractions were collected for genomic analysis. They report that the mcr-1 carrying strain evolved higher colistin resistance much faster only in presence of lpxC mutations in the genome. They identify the mcr-1 and lpxC interactions to be positively epistatic and mutations only in lpxC do not lead to resistance to colistin. Taking a cue from their evolution experiments, they looked for the variations in lpxC sequences in the genomic datasets of clinical E. coli strains. They found many such variations in the genomes of clinical isolates. Importantly, they found those variations to be present even in non-resistant strains which might predispose those strains to gain untreatable levels of colistin resistance.
Strengths:
The study focuses on two key aspects of antibiotic resistance in clinical settings. First, is the antibiotic colistin itself which is part of the last line of defense. Second, is the importance of genomic variations in clinical isolates that have not been linked to any antibiotic resistance mechanisms. The data were presented in a logical sequence and maintained brevity. The link of lpxC to mcr-1 resistance is convincing.
Weaknesses:
The basic premise of the paper is solid but the following should be addressed.
1. In Figure 1, the authors applied the 'evolutionary ramp' method to isolate evolved strains with higher MIC to colistin; but, the conditions for the evolution of WT and strain carrying mcr-1 are different. Maintaining mcr-1 requires antibiotic selection which WT cannot withstand. Hence, if I am not mistaken, WT was not grown in the presence of any antibiotic. Not only that, maintaining a ~32 Kb plasmid itself can have different selective landscapes. The authors may replicate the experiment with their low-copy clone of mcr-1 which would make it easier for the authors to have an empty vector in WT as a proper control. Since now they know the expected mutations to be in lpxC, they might sequence a PCR amplicon of that region for validation of their hypothesis.
2. In Figure 2, what are the effects of these mutations in lpxC? The authors state that many mutations map on to the metal binding domain; but are those significant changes? LpxC is relatively well characterized and authors may want to comment on these mutations a little more.
Also, lpxC mutations showed enrichment but lpxA did not. Is this suggestive of the type of Lipid A that is more preferred for the epistatic interactions? The authors may want to comment on that.
3. In Figure 3, the lpxC mutant shows a reduction in fitness in a competition assay. What is the growth pattern of individual strains? There is a possibility that slow growth of lpxC mutant provides benefit under antibiotic stress.
Minor comment: the three individual replicates shown in Figure 3a are all identical within a sample and do not add to the figure where n=3. The authors can simply show SD or report correct values of replicates.
4. In Figure 4, as the authors themselves have stated, the difference in heterogeneity could be simply due to variation within phylogroups and subsequent compositional differences within the populations. The authors must check if mutations were found in the same location of lpxA as found in their own evolved strains. Without this information, the heterogeneity data would be speculative. Adding the lpxC variants reported in figure 2 to the trees of figure 4 (right) will make it clear if their conclusion is justified.
5. The authors can perform a confirmatory experiment for the pre-existing part of their hypothesis. If they perform the evolutionary ramp experiment with a strain carrying lpxC mutant strain, will they see faster evolution of high MIC mutants?
6. The rationale of how the presence of lpxC mutations can cause a strain without any colistin resistance to acquire mcr-1 is not addressed. The authors may want to comment on that. -
