Cd59 and inflammation regulate Schwann cell development
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
This paper by Wiltbank et al. "Cd59 and inflammation orchestrate Schwann cell development" investigates the function of the small GPI-anchored protein Cd59, a protein known to suppress complement mediated inflammation, in Schwann cell development and myelination, using zebrafish as a model system. This paper will be of broad interest to developmental biologists, glial biologists and immunologists, as it suggests the interesting and novel findings that Cd59 regulates Schwann cell development, mainly by modulating Schwann cell proliferation.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. The reviewers remained anonymous to the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
- @maria_eichel's saved articles (maria_eichel)
Abstract
Efficient neurotransmission is essential for organism survival and is enhanced by myelination. However, the genes that regulate myelin and myelinating glial cell development have not been fully characterized. Data from our lab and others demonstrates that cd59 , which encodes for a small GPI-anchored glycoprotein, is highly expressed in developing zebrafish, rodent, and human oligodendrocytes (OLs) and Schwann cells (SCs), and that patients with CD59 dysfunction develop neurological dysfunction during early childhood. Yet, the function of Cd59 in the developing nervous system is currently undefined. In this study, we demonstrate that cd59 is expressed in a subset of developing SCs. Using cd59 mutant zebrafish, we show that developing SCs proliferate excessively and nerves may have reduced myelin volume, altered myelin ultrastructure, and perturbed node of Ranvier assembly. Finally, we demonstrate that complement activity is elevated in cd59 mutants and that inhibiting inflammation restores SC proliferation, myelin volume, and nodes of Ranvier to wildtype levels. Together, this work identifies Cd59 and developmental inflammation as key players in myelinating glial cell development, highlighting the collaboration between glia and the innate immune system to ensure normal neural development.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
This study focuses on elucidating the function of CD59, a small GPI-anchored glycoprotein, in Schwann cell development. Patients with CD59 deficiency suffer from neurological dysfunctions, but the link between CD59 deficiency and the development of neurological dysfunctions remains unclear. To clarify this link, the authors used zebrafish as an animal model. They generated cd59 mutant zebrafish and studied their Schwann cell development. The authors started this study by showing CD59 expression data from different sources in the Schwann cell and oligodendrocyte lineages in zebrafish and mice. They continued by demonstrating that CD59 is expressed only by a subset of developing Schwann cells, which is very interesting conceptually for the identification of different Schwann cell populations …
Author Response
Reviewer #1 (Public Review):
This study focuses on elucidating the function of CD59, a small GPI-anchored glycoprotein, in Schwann cell development. Patients with CD59 deficiency suffer from neurological dysfunctions, but the link between CD59 deficiency and the development of neurological dysfunctions remains unclear. To clarify this link, the authors used zebrafish as an animal model. They generated cd59 mutant zebrafish and studied their Schwann cell development. The authors started this study by showing CD59 expression data from different sources in the Schwann cell and oligodendrocyte lineages in zebrafish and mice. They continued by demonstrating that CD59 is expressed only by a subset of developing Schwann cells, which is very interesting conceptually for the identification of different Schwann cell populations and their specific functions and also for the potential development of future techniques targeting specific Schwann cell populations. However, since the authors focused in the following parts of the article on Schwann cell development, it is unclear why they have included data on oligodendrocytes at the start of the manuscript.
Thank you for this question. We included the data on oligodendrocytes because we wanted to be thorough and transparent. Additionally, because some of our own expression data show oligodendrocyte expression, we felt it was prudent to confirm this expression in published RNAseq datasets. Finally, we created and/or used tools to label cd59-positive cells, and we often used expression in both oligodendrocytes and Schwann cells as a readout of complete expression of these tools.
In this study, the authors show that cd59 ablation in zebrafish leads to increased Schwann cell proliferation between 48 and 55 hpf (hours post fertilization), which is quite convincing. However, they claim that this transient increase in proliferation leads to impaired myelination and node of Ranvier formation. Unfortunately, these findings remain correlative and it appears unclear why an increased number of Schwann cells that stop proliferating at the same time-point as wild type Schwann cells would impair myelination and node of Ranvier formation. This phenotype is attributed by the authors to increased proliferation of Schwann cells between 48 and 55 hpf, which seems rather unlikely or not supported by the data currently presented. The hypomyelination phenotype is rather mild, while the impairment of node of Ranvier formation seems quite strong - however, the data currently presented is not very convincing and needs improvement.
Thank you for your observations. With regards to how an increase in SC proliferation could impact myelination and node of Ranvier formation, although the rate of proliferation transiently increases, these excess SCs persist on the nerve. So, even though the mutants can stop developmental proliferation at the same stage, the mutants ultimately have more SCs on the nerve after proliferation has ceased. This raises the interesting question of how could more SCs lead to less myelin? To address this question, we added to the discussion to speculate on possible hypotheses as to why this is the case (please see line 510).
With regards to comparing the strength of the myelin phenotype and the node of Ranvier phenotype, there is no reason to suspect that there is a linear relationship between myelin volume and node of Ranvier assembly. We do know that myelination and SCs are necessary for node of Ranvier assembly. So, it is very possible that any perturbation in myelination could drastically affect node of Ranvier assembly. That said, this relationship is very interesting, and we hope that the cd59 mutant model can be utilized to further investigate these questions in future studies.
In regards to the node of Ranvier data itself, we have provided co-labeling of NF186 and NaV channels on mbpa:tagrfp-caax-positive nerves (see Figure 5 – figure supplement 1D). Using Imaris, we demonstrate that each NF186 cluster colocalizes with a NaV channel cluster. Furthermore, this colocalization only occurs within the myelinated nerve. Collectively, this data demonstrates that our quantification of nodes of Ranvier is reliable.
The data showing an increase of complement activation in cd59 mutants is also not very convincing and should be improved.
Thank you for sharing your concern. To address this issue, we have used Imaris to show MACs that are bound to SC membranes (see Figure 6B) for a clearer view of the data. Comparing wildtype and mutant larvae, there is a visible and significant increase in MAC binding to SC membranes when cd59 is perturbed. Additionally, we have included controls for these antibody labeling experiments to show specificity of these tools.
In addition, the link between increased complement activation and increased proliferation remains to be proven in the context of this study, and the choice of dexamethasone as an inhibitor of complement activation does not appear to be the best choice since it is not specific to the complement.
Thank you for sharing your concern. We agree that dexamethasone impacts other aspects of immune activation other than complement. With this in mind, we did test another drug called compstatin, which inhibits complement protein 3 (C3). Inhibition of C3 impairs all three complement pathways and would abrogate downstream assembly of MACs. Our preliminary data was very promising, demonstrating the same relationship that we see with our dexamethasone treatment (see below). However, we were unable to reproducibly get the same results in subsequent experiments after we purchased a new stock of this drug. To solve this problem, we tried compstatin from a different company as well as increasing the concentration, but none of our troubleshooting efforts yielded the same results that we had originally observed. Obviously, this is incredibly disappointing to us. So, although these results initially repeated, we did not feel it was ethical to publish this data. (In the figure below, wildtype and mutant embryos were treated with 1% DMSO or 50 µM compstatin in 1% DMSO from 24 hpf to 55 hpf. The number of SCs was quantified with a Sox10 antibody and confocal imaging at 55 hpf).
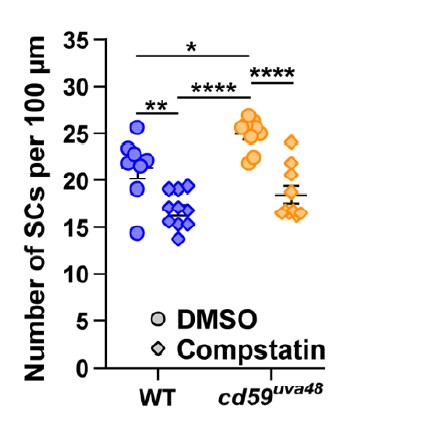
Given these technical limitations, we ultimately decided to include the dexamethasone experiments because they were reproducible. Considering the broader effects of dexamethasone on the immune system, we have softened our claims to include inflammation as well as complement activation. That said, we hope future studies will be able to use this model to gather more information on the specific pathways that are regulating Cd59-dependent SC proliferation.
Page 49, lines 437-439: Here the authors claim that their data "demonstrates that developmental inflammation aids in normal SC proliferation and that this process is amplified when cd59 is mutated." The data presented in Figure 6C-D and commented by the authors on page 49, lines 435-437, show however that "Dex treatment in cd59uva48 mutant embryos restored SC numbers to wildtype levels, whereas wildtype SCs were not significantly affected by Dex application". Dex (dexamethasone) was used here to inhibit inflammation and associated complement activation. Therefore, these data do not show that developmental inflammation aids in normal SC proliferation, but rather that it has no influence.
Thank you for your comment. When compared alone, there are significantly fewer SCs in dexamethasone-treated wildtype larvae compared to DMSO-treated wildtype larvae. We have updated the figure and text to better highlight this relationship (please see Figure 7A, C and line 457). We also quantified EdU incorporation into SCs treated with dexamethasone. Here we also observed a decrease in EdU-positive SCs in wildtype larvae treated with dexamethasone, supporting our observation that developmental inflammation is contributing to normal SC proliferation (please see Figure 7B, D).
Dexamethasone treatment: The authors claim that dexamethasone treatment, by decreasing inflammation and associated complement activation, leads to a decrease of SC proliferation in the cd59 mutant. To support this, there is only Figure 7-Figure Supplement 1 showing a decreased SC number in the mutant treated by dexamethasone as compared to vehicle-treated mutant. To strengthen this point, the authors also need to specifically quantify proliferation by EdU incorporation, as they did in Figure 4, and also cell death.
Thank you for your comment. We have added quantification of EdU incorporation after dex treatment (please see Figure 7B, D). Dr. Feltri, the Reviewing Editor, told us that measuring apoptosis after treatment was not necessary for the revision.
In addition, the mechanistic hypothesis of increased proliferation in cd59 mutant is that cd59 interferes with the activation of the complement and complement-induced pore formation in the plasma membrane. However, dexamethasone is not a specific inhibitor of the complement. Therefore, its potential effect on SC proliferation could be due to other effects than complement inactivation. It is unclear why the authors did not use an inhibitor of the complement that is more specific than dexamethasone.
Thank you for your comment. Please see our previous response to this comment.
Page 54, lines 456-457: The following statement "Collectively, these data demonstrate that inflammation-induced SC proliferation contributes to perturbed myelin and node of Ranvier development." is not accurate since these data remain correlative. Indeed, there is in this study nothing showing that increased SC proliferation between 48 and 55 hpf leads to perturbed myelin and node of Ranvier development. In addition, the term "inflammation" is not precise enough here. What the authors attempt to show is an increase of complement activation due to the absence of cd59 expression in SCs. The authors did not try to induce inflammation in wild type animals to see whether this induces proliferation and perturbed myelin and node of Ranvier development. They also did not try to directly knock down C8/C9 in cd59 mutants to see whether they would rescue the phenotype of the cd59 mutant, at least to some extent. In addition, their statement mentioned above needs to be more precise by stating that their findings apply to cd59 mutants and not to wild type animals.
Thank you for your comment. Please see our previous responses to these comments.
Reviewer #3 (Public Review):
Wiltbank and colleagues explore the function of CD59 in developmental Schwann cell myelination. Using previously published transcriptomics data sets they arrive at CD59 as a differentially expressed gene in myelinating glia. In addition, patients with pathogenic variants have neuropathy. The authors construct a transgenic zebrafish reporter line for cd59. Surprisingly, it labels a very, very small percentage of Schwann cells (less than 10% throughout development). The authors then construct several loss-of-function mutants for cd59. They report these mutants have increased numbers of Schwann cells, but nerves are smaller and EM shows they have reduced the number of myelin wraps. Consistent with impaired myelination they also observe fewer nodes of Ranvier. The authors suggest loss of cd59 results in increased MAC deposition on myelinating Schwann cells. Remarkably, using an inhibitor of inflammation (dexamethasone), the authors show that they can normalize/rescue the main phenotypes: 1) normalize the number of SCs, 2, dramatically improve myelination to normal nerve volumes, and 3) rescue node of Ranvier formation. This last experiment that rescues the phenotype is really terrific. The experiments are mostly very well done and the story is both interesting and conceptually novel. Nevertheless, there are a few points that I think the authors could address:
- It is very surprising that the cd59 reporter line only showed expression in a small subset (10% or less) of Schwann cells. How do the authors explain the widespread effects? Similarly, the authors make a point of stating that motor Schwann cells did not express cd59. Did myelinated motor axons show the same phenotype - reduced myelination, impaired node formation? How can the expression of cd59 in only 10% of cells cause widespread effects throughout the nerve? How can it limit overproliferation if 9/10 cells don't even express it?
Thank you for the question. It is really interesting that a small subset of cells can have such a big impact on nerve development. One of our current hypotheses is that overproliferation of SCs has led to activation of contact-inhibition pathways, which in turn are negatively regulating myelination. We expand further on this hypothesis in our discussion (please see line 536). We also suggest questions addressing glial cell heterogeneity to explore in the future (please see line 603).
In regards to the motor nerves, we quantified the number of Sox10-positive cells (SCs and MEP glia) on motor nerves at 72 hpf and showed that there was no overproliferation of these cell types (please see Figure 4I, J). We have not observed any issues with motor nerve myelination, which is what we would expect if motor SC proliferation was unaffected. That said, these differences between motor and pLLN SCs are really interesting because it opens up discussion for glial cell heterogeneity between nerve types (e.g. sensory versus motor nerves). We see similar evidence of this in satellite glia that populate the cochlear spiral ganglion versus those that surround the dorsal root ganglia (see Tasdemir-Yilmaz et al. 2021 or Wiltbank and Kucenas 2021), so it makes sense that there could be some SC diversity between nerve types as well. We expand further on these ideas in our discussion (please see line 603).
- It is surprising to me that there is a significant increase in SC proliferation, but no change in the length of myelin sheaths. Does this mean there are more SCs that remain unmyelinated and undifferentiated?
Thank you for your comment. We were also surprised. With our current tools, we are unable to determine the fate of these extra SCs but hope that future studies will be able to clarify this question. We have added discussion around this topic to the text (please see line 554).
- The results showing deposition of the MAC (via C5b-9+C5b-8 immunostaining) are not convincing. The overall background level of immunostaining is dramatically increased. This result is central to the overall story in the paper. What controls were performed to confirm this doesn't simply reflect an overall higher background artifact during immunostaining?
Thank you for your comment. We have added our antibody controls to the supplemental figures (please see Figure 6 – figure supplement 1A) demonstrating that we can increase MAC deposition by inducing complement activation (either through heat-related damage or DNase-elicited DNA damage). We also do not observe signal when the primary antibody is not present. Based on our controls, we do not think the extra MAC labeling is background. Rather, we believe that MAC deposition has increased globally in the cd59 mutant embryos. This is not surprising given that complement activation leads to a positive feedback loop of more complement and immune activation, which is likely occurring in the cd59 mutants.
To help clarify the MAC data, we have also added Imaris renderings of the MACs that are bound to the SC membranes, demonstrating that there are more MACs embedded in the cd59 mutant SC membranes compared to wildtype SCs (see Figure 6B).
- Can the authors speculate on a mechanism for how promoting more MAC results in increased proliferation?
Thank you for your question. We have added discussion around this topic to the text (please see line 585).
-
Evaluation Summary:
This paper by Wiltbank et al. "Cd59 and inflammation orchestrate Schwann cell development" investigates the function of the small GPI-anchored protein Cd59, a protein known to suppress complement mediated inflammation, in Schwann cell development and myelination, using zebrafish as a model system. This paper will be of broad interest to developmental biologists, glial biologists and immunologists, as it suggests the interesting and novel findings that Cd59 regulates Schwann cell development, mainly by modulating Schwann cell proliferation.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. The reviewers remained anonymous to the authors.)
-
Reviewer #1 (Public Review):
This study focuses on elucidating the function of CD59, a small GPI-anchored glycoprotein, in Schwann cell development. Patients with CD59 deficiency suffer from neurological dysfunctions, but the link between CD59 deficiency and the development of neurological dysfunctions remains unclear. To clarify this link, the authors used zebrafish as an animal model. They generated cd59 mutant zebrafish and studied their Schwann cell development. The authors started this study by showing CD59 expression data from different sources in the Schwann cell and oligodendrocyte lineages in zebrafish and mice. They continued by demonstrating that CD59 is expressed only by a subset of developing Schwann cells, which is very interesting conceptually for the identification of different Schwann cell populations and their specific …
Reviewer #1 (Public Review):
This study focuses on elucidating the function of CD59, a small GPI-anchored glycoprotein, in Schwann cell development. Patients with CD59 deficiency suffer from neurological dysfunctions, but the link between CD59 deficiency and the development of neurological dysfunctions remains unclear. To clarify this link, the authors used zebrafish as an animal model. They generated cd59 mutant zebrafish and studied their Schwann cell development. The authors started this study by showing CD59 expression data from different sources in the Schwann cell and oligodendrocyte lineages in zebrafish and mice. They continued by demonstrating that CD59 is expressed only by a subset of developing Schwann cells, which is very interesting conceptually for the identification of different Schwann cell populations and their specific functions and also for the potential development of future techniques targeting specific Schwann cell populations. However, since the authors focused in the following parts of the article on Schwann cell development, it is unclear why they have included data on oligodendrocytes at the start of the manuscript.
In this study, the authors show that cd59 ablation in zebrafish leads to increased Schwann cell proliferation between 48 and 55 hpf (hours post fertilization), which is quite convincing. However, they claim that this transient increase in proliferation leads to impaired myelination and node of Ranvier formation. Unfortunately, these findings remain correlative and it appears unclear why an increased number of Schwann cells that stop proliferating at the same time-point as wild type Schwann cells would impair myelination and node of Ranvier formation. This phenotype is attributed by the authors to increased proliferation of Schwann cells between 48 and 55 hpf, which seems rather unlikely or not supported by the data currently presented. The hypomyelination phenotype is rather mild, while the impairment of node of Ranvier formation seems quite strong - however, the data currently presented is not very convincing and needs improvement. The data showing an increase of complement activation in cd59 mutants is also not very convincing and should be improved. In addition, the link between increased complement activation and increased proliferation remains to be proven in the context of this study, and the choice of dexamethasone as an inhibitor of complement activation does not appear to be the best choice since it is not specific to the complement.
Page 49, lines 437-439: Here the authors claim that their data "demonstrates that developmental inflammation aids in normal SC proliferation and that this process is amplified when cd59 is mutated." The data presented in Figure 6C-D and commented by the authors on page 49, lines 435-437, show however that "Dex treatment in cd59uva48 mutant embryos restored SC numbers to wildtype levels, whereas wildtype SCs were not significantly affected by Dex application". Dex (dexamethasone) was used here to inhibit inflammation and associated complement activation. Therefore, these data do not show that developmental inflammation aids in normal SC proliferation, but rather that it has no influence.
Dexamethasone treatment: The authors claim that dexamethasone treatment, by decreasing inflammation and associated complement activation, leads to a decrease of SC proliferation in the cd59 mutant. To support this, there is only Figure 7-Figure Supplement 1 showing a decreased SC number in the mutant treated by dexamethasone as compared to vehicle-treated mutant. To strengthen this point, the authors also need to specifically quantify proliferation by EdU incorporation, as they did in Figure 4, and also cell death.
In addition, the mechanistic hypothesis of increased proliferation in cd59 mutant is that cd59 interferes with the activation of the complement and complement-induced pore formation in the plasma membrane. However, dexamethasone is not a specific inhibitor of the complement. Therefore, its potential effect on SC proliferation could be due to other effects than complement inactivation. It is unclear why the authors did not use an inhibitor of the complement that is more specific than dexamethasone.
Page 54, lines 456-457: The following statement "Collectively, these data demonstrate that inflammation-induced SC proliferation contributes to perturbed myelin and node of Ranvier development." is not accurate since these data remain correlative. Indeed, there is in this study nothing showing that increased SC proliferation between 48 and 55 hpf leads to perturbed myelin and node of Ranvier development. In addition, the term "inflammation" is not precise enough here. What the authors attempt to show is an increase of complement activation due to the absence of cd59 expression in SCs. The authors did not try to induce inflammation in wild type animals to see whether this induces proliferation and perturbed myelin and node of Ranvier development. They also did not try to directly knock down C8/C9 in cd59 mutants to see whether they would rescue the phenotype of the cd59 mutant, at least to some extent. In addition, their statement mentioned above needs to be more precise by stating that their findings apply to cd59 mutants and not to wild type animals.
-
Reviewer #2 (Public Review):
Wiltbank et al. investigate the functional relevance of cd59 expression, mainly focusing on the developing peripheral nervous system of zebrafish. By analyzing pre-existing bulk and scRNA-seq datasets they find that cd59 is expressed in both myelinating Schwann cells and oligodendrocytes, the myelinating cells of the peripheral and central nervous system, respectively. Indeed, CD59, a small GPI-anchored glycoprotein, is an abundant myelin protein in the CNS of adult zebrafish according to prior proteome analysis. The authors first generate a zebrafish reporter line to further characterize cd59 expression and then by genome editing (CRISPR/Cas9) generate a loss of function line. By CISH and FISH they validate the RNA-seq data and observe expression of cd59 in a subset of developing myelinating Schwann cells …
Reviewer #2 (Public Review):
Wiltbank et al. investigate the functional relevance of cd59 expression, mainly focusing on the developing peripheral nervous system of zebrafish. By analyzing pre-existing bulk and scRNA-seq datasets they find that cd59 is expressed in both myelinating Schwann cells and oligodendrocytes, the myelinating cells of the peripheral and central nervous system, respectively. Indeed, CD59, a small GPI-anchored glycoprotein, is an abundant myelin protein in the CNS of adult zebrafish according to prior proteome analysis. The authors first generate a zebrafish reporter line to further characterize cd59 expression and then by genome editing (CRISPR/Cas9) generate a loss of function line. By CISH and FISH they validate the RNA-seq data and observe expression of cd59 in a subset of developing myelinating Schwann cells (SCs) in the LLN. They find that CD59 restricts the over-proliferation of Schwann cells and affects the structure of myelin and the nodes of Ranvier. Considering prior knowledge that CD59 plays a role in suppressing the complement system, the authors assessed how the complement reacts in cd59 mutant zebrafish. Strikingly, they found that Schwann cells were not protected from complement attack when CD59 was lacking. Inhibiting inflammation using Dexamethasone not only restored Schwann cell numbers to wildtype levels but also improved Node of Ranvier clustering and myelin volume. Hence, the authors conclude that cd59 expression protects developing Schwann cells via inhibiting developmental inflammation.
Generally, the conclusions of the manuscript are well supported by the data. A few minor points regarding phrasing and figure design could be further improved, but overall this is an interesting study that could also become relevant for future therapeutic translation. Together, the study presents a relevant and novel topic and will find an interested readership in the communities working on myelinating cells, the complement system / innate immune system, and their interactions.
-
Reviewer #3 (Public Review):
Wiltbank and colleagues explore the function of CD59 in developmental Schwann cell myelination. Using previously published transcriptomics data sets they arrive at CD59 as a differentially expressed gene in myelinating glia. In addition, patients with pathogenic variants have neuropathy. The authors construct a transgenic zebrafish reporter line for cd59. Surprisingly, it labels a very, very small percentage of Schwann cells (less than 10% throughout development). The authors then construct several loss-of-function mutants for cd59. They report these mutants have increased numbers of Schwann cells, but nerves are smaller and EM shows they have reduced the number of myelin wraps. Consistent with impaired myelination they also observe fewer nodes of Ranvier. The authors suggest loss of cd59 results in …
Reviewer #3 (Public Review):
Wiltbank and colleagues explore the function of CD59 in developmental Schwann cell myelination. Using previously published transcriptomics data sets they arrive at CD59 as a differentially expressed gene in myelinating glia. In addition, patients with pathogenic variants have neuropathy. The authors construct a transgenic zebrafish reporter line for cd59. Surprisingly, it labels a very, very small percentage of Schwann cells (less than 10% throughout development). The authors then construct several loss-of-function mutants for cd59. They report these mutants have increased numbers of Schwann cells, but nerves are smaller and EM shows they have reduced the number of myelin wraps. Consistent with impaired myelination they also observe fewer nodes of Ranvier. The authors suggest loss of cd59 results in increased MAC deposition on myelinating Schwann cells. Remarkably, using an inhibitor of inflammation (dexamethasone), the authors show that they can normalize/rescue the main phenotypes: 1) normalize the number of SCs, 2, dramatically improve myelination to normal nerve volumes, and 3) rescue node of Ranvier formation. This last experiment that rescues the phenotype is really terrific. The experiments are mostly very well done and the story is both interesting and conceptually novel. Nevertheless, there are a few points that I think the authors could address:
1. It is very surprising that the cd59 reporter line only showed expression in a small subset (10% or less) of Schwann cells. How do the authors explain the widespread effects? Similarly, the authors make a point of stating that motor Schwann cells did not express cd59. Did myelinated motor axons show the same phenotype - reduced myelination, impaired node formation? How can the expression of cd59 in only 10% of cells cause widespread effects throughout the nerve? How can it limit overproliferation if 9/10 cells don't even express it?
2. It is surprising to me that there is a significant increase in SC proliferation, but no change in the length of myelin sheaths. Does this mean there are more SCs that remain unmyelinated and undifferentiated?
3. The results showing deposition of the MAC (via C5b-9+C5b-8 immunostaining) are not convincing. The overall background level of immunostaining is dramatically increased. This result is central to the overall story in the paper. What controls were performed to confirm this doesn't simply reflect an overall higher background artifact during immunostaining?
4. Can the authors speculate on a mechanism for how promoting more MAC results in increased proliferation? -
