Protective mitochondrial fission induced by stress-responsive protein GJA1-20k
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
This study identifies a cardioprotective factor, GJA1-20k (a truncated form of Cx43), which appears to confer protection against ischemia/reperfusion (I/R) injury via promotion of mitochondrial fission. This finding is particularly interesting given that hyperfission is generally thought of as an index of toxicity in I/R or hypoxic injury. I/R lesion size in a GJA1 heterozygous mutant mouse is strikingly exacerbated compared to control animals, providing strong in vivo evidence supporting a role for this factor in protection from I/R. However, while the findings are interesting and novel, key results require additional experimental support, including to address the lack of key control data, and a significant revision will be necessary to address these issues.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #3 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The Connexin43 gap junction gene GJA1 has one coding exon, but its mRNA undergoes internal translation to generate N-terminal truncated isoforms of Connexin43 with the predominant isoform being only 20 kDa in size (GJA1-20k). Endogenous GJA1-20k protein is not membrane bound and has been found to increase in response to ischemic stress, localize to mitochondria, and mimic ischemic preconditioning protection in the heart. However, it is not known how GJA1-20k benefits mitochondria to provide this protection. Here, using human cells and mice, we identify that GJA1-20k polymerizes actin around mitochondria which induces focal constriction sites. Mitochondrial fission events occur within about 45 s of GJA1-20k recruitment of actin. Interestingly, GJA1-20k mediated fission is independent of canonical Dynamin-Related Protein 1 (DRP1). We find that GJA1-20k-induced smaller mitochondria have decreased reactive oxygen species (ROS) generation and, in hearts, provide potent protection against ischemia-reperfusion injury. The results indicate that stress responsive internally translated GJA1-20k stabilizes polymerized actin filaments to stimulate non-canonical mitochondrial fission which limits ischemic-reperfusion induced myocardial infarction.
Article activity feed
-

-

Author Response:
Reviewer #3:
Weaknesses:
Previously it was suggested that mitochondrial biogenesis was increased with increased levels of GJA1-20k. Is this a difference in the cellular model (HEK) and do the changes in cell culture accurately recapitulate the changes seen in animals?
The Reviewer is correct that GJA1-20k did not alter the mitochondrial biogenesis in HEK293 cells (Figure 1–figure supplement 2) whereas AAV9-transduced adult cardiomyocytes showed increased mitochondrial DNA copy number (Figure 1–figure supplement 2C), consistent with our previous study (Basheer et al., JCI insight, 2018). We expect that increased mitochondrial biogenesis is a function of chronic GJA1-20k overexpression in vivo, and thus a separate phenomenon from the acute mitochondrial fission which occurs within one minute of GJA1-20k accumulation …
Author Response:
Reviewer #3:
Weaknesses:
Previously it was suggested that mitochondrial biogenesis was increased with increased levels of GJA1-20k. Is this a difference in the cellular model (HEK) and do the changes in cell culture accurately recapitulate the changes seen in animals?
The Reviewer is correct that GJA1-20k did not alter the mitochondrial biogenesis in HEK293 cells (Figure 1–figure supplement 2) whereas AAV9-transduced adult cardiomyocytes showed increased mitochondrial DNA copy number (Figure 1–figure supplement 2C), consistent with our previous study (Basheer et al., JCI insight, 2018). We expect that increased mitochondrial biogenesis is a function of chronic GJA1-20k overexpression in vivo, and thus a separate phenomenon from the acute mitochondrial fission which occurs within one minute of GJA1-20k accumulation around a mitochondrion (Figure 4). The HEK cell line, in which overexpressed GJA1-20k is present for a much shorter time, does not induce mitochondrial biogenesis (Figure 1–figure supplement 2), and thus is an excellent cellular model in which we can study GJA1-20k induced fission.
The revised manuscript has been modified to include the above new data (Figure 1–figure supplement 2) and discussion:
—Results section (lines 121 – 129): Previously we reported that GJA1-20k is involved in mitochondrial biogenesis (Basheer, Fu et al. 2018). Consistent with our previous study, AAV9-transduced adult cardiomyocytes showed increased mitochondrial DNA copy number and GJA1-20k deficient mice (Gja1M213L/M213L) had decreased copy number. However, exogenous GJA1-20k did not alter the mitochondrial biogenesis in HEK293 cells. Nor did exogenous GJA1-20k affect membrane potential or baseline ATP production (Figure 1–figure supplement 2A–C). In addition to mitochondrial DNA copy number, neither biogenesis nor mitophagy protein markers were altered in either GJA1-20k transfected HEK293 cells or Gja1M213L/M213L mouse hearts (Figure 1–figure supplement 2D – G).
—Discussion section (lines 289 – 292): Yet the presence of GJA1-20k, while inducing mitochondrial fission and smaller mitochondria (Figure 1, 3 and 4), does not either reduce MFN1 or MFN2, activate DRP1, change membrane potential, ATP production, mitochondrial biogenesis, or mitophagy (Figure 2; Figure 1 – figure supplement 2).
Mdivi-1 is not a selective Drp1 inhibitor. It is a Complex I inhibitor, leading to unintended changes in mitochondrial dynamics in response to ETC stress. Rather than Mdivi-1, a dominant negative Drp1 mutant K38A could be overexpressed to see whether this prevents GJA1-20k-mediated fission. If it still goes through, then I agree that Drp1 is not involved at all.
We appreciate Reviewer #3’s thoughtful suggestion and, in this revised manuscript, we studied mitochondrial morphology in the presence of K38A. As seen in Figure 2C and D of the revised manuscript, K38A elongated mitochondria, as expected from inhibited Drp1 mediated fission. However, despite Drp1 inhibition by K38A, in the presence of GJA1-20k, mitochondria remain small, further supporting that GJA1-20k-mediated fission is DRP1-independent.
—Results section (lines 140 – 150): To further investigate whether GJA1-20k induced reduction in mitochondrial size is dependent on DRP1, we analyzed mitochondrial morphology after inhibiting DRP1 by performing siRNA- mediated DRP1 knock-down (Figure 2—figure supplement 1A–C) or transfecting DRP1 dominant negative mutant (K38A), all with or without GJA1-20k transfection. With either method of DRP1 inhibition, the average area of individual mitochondria increased, consistent with inhibiting canonical fission (Figure 2C, D). In addition, K38A has more pronounced DRP1 inhibition which resulted in greater mitochondrial enlargement than siDRP1 (Figure 2C, D; Figure 2—figure supplement 1F). However, GJA1-20k acts epistatically to DRP1 loss or interference and prevents DRP1-mediated mitochondrial enlargement (Figure 2C–F; Figure 2— figure supplement 1B, C), indicating GJA1-20k can act at or downstream of DRP1.
For the kinetics studies (see Fig 4), I think it is important to measure the timing of the actin recruitment and eventual fission when Drp1 is knocked down and/or when a DN mutant (K38A) is involved. Again, I do not trust the chemical inhibitor (Mdivi-1) data since this does not inhibit Drp1 activity.
We would like to thank Reviewer #3 for suggesting we use an additional method of inhibiting Drp1. We analyzed real time actin dynamics under direct DRP1 knock-down. As seen in Mdivi-1 treatment, GJA1- 20k accumulated and then actin assembled around mitochondria and induced fission under DRP1 knockdown (Figure 4 and Video 1 of revised manuscript). The kinetic parameters of fission were also similar between Drp1 knockdown and Mdivi-1 treatment. The original Figure 4 and Video 1 and 2 have been moved to Figure 4–figure supplement 1 and Video 2 and 3, respectively, in order to accommodate the new Drp1 knockdown data (Figure 4 and Video 1).
The revised manuscript has been modified to include the above new data (Figure 4; Video 1):
—Results section (lines 198 – 219): Simultaneous use of fluorescently labelled actin, GJA1-20k, and mitochondria in live cells permit real time imaging of mitochondrial fission events at actin assembly sites. As seen in Video 1 and Figure 4B, GJA1-20k recruits actin to mitochondria, which results in fission. In Video 1, the actin network can be seen to develop around mitochondria and, coinciding with GJA1-20k intensity, forms an increasingly tight band across a mitochondrion which, within one minute, results in mitochondrial fission. The imaging in the bottom row of Figure 4B, and in the right column of Video 1 were obtained by multiplying GJA1-20k signal with actin signal, highlighting the locations at which GJA1-20k and actin are coincident. The respective line-scan profiles in Figure 4C indicate that mitochondrial fission occurs at points where the product of GJA1-20k and actin is the highest. Following accumulation of GJA1-20k and actin (red lines) at these points, a drop in mitochondrial signal (blue lines) is apparent when fission occurs. Fission (low point of blue lines) occurs approximately 45 seconds after co-accumulation of GJA1-20k and actin (high point of red lines, Figure 4C). Time to fission was computed from the time of peak GJA1-20k and actin intensity product, to the time of mitochondrial signal being reduced to background (Figure 4D–F). Statistically, this time to fission occurred at a median of 45 seconds, with a standard deviation of 11 seconds (Figure 4G). Note, the real time imaging shown in Video 1, and Figure 4 were performed under siDRP1. Therefore, the mitochondrial fission induced by cooperation between GJA1-20k and actin can be independent of canonical DRP1-mediated fission. To rule out inadvertent bias by siRNA, we used pharmacologic Mdivi-1 to inhibit DRP1 and, similar to the use of DRP1 siRNA, actin formed around mitochondria at GJA1-20k sites (Figure 4—figure supplement 1A–D) and fission occurred within a similar timescale (Video 2 and 3; Figure 4— figure supplement 1E–H).
The assessment of the impact of ischemic stress with the heterozygous animal (M213L/WT) is hard to interpret. How reduced is the expression of GJA1-20k in these animals and how is mitochondrial function impacted based on Seahorse analysis? The mitochondrial morphology is not altered in these animals, so would mitochondrial function be largely unchanged as well? It is not clear how much GJA1-20k is needed to observe changes in mitochondrial shape and function, and comparisons with the homozygous mutant (M213L/M213L) are not the same, making it difficult to resolve the interpretation of these data.
We appreciate Reviewer #3’s thoughtful and valuable comments. We previously reported that the heterozygous mutant (M213L/WT) expresses approximately half of GJA1-20k compared to WT (Figure 1 in Xiao and Shimura et al., J Clin Invest, 2020). Unfortunately, homozygous mutants die before adulthood, preventing effective comparison of GJA1-20k content on mitochondrial function in adult cardiomyocytes. To compare the impact of the amount of endogenous GJA1-20k on mitochondrial function, we added seahorse data from heterozygous neonatal CMs (Figure 5 C, D) and compared these data to seahorse data from neonatal cardiomyocytes from both wildtype and homozygous mutants. Even though there was no significant difference in mitochondrial size between WT and M213L/WT (Figure 5I, J; Figure 5–figure supplement 1A, B) under basal conditions, the seahorse OCR levels from M213L/WT myocytes is in between that of WT and homozygous (M213L/M213L) (Figure 5 C, D; Figure 5–figure supplement 1C) cardiomyocytes. Since GJA1-20k is a stress responsive peptide which increases under ischemic stress, in the present manuscript, we should like to emphasize that even a partial (50%) decrease in GJA1-20k expression induces mitochondrial fragility to oxidative stress. As shown in new Figure 5 I – L of the revised manuscript, the heterozygous mutant (M213L/WT) has more elongated mitochondria and a high distribution of damaged mitochondria post-I/R compared to WT, consistent with TTC staining, even with no change in mitochondrial size under basal conditions.
The revised manuscript has been modified to include the above new data (Figure 5; Figure 5–figure supplement 1) and discussion:
—Results section (lines 227 – 233) Similarly, maximal respiration is increased in neonatal CMs derived from GJA1-20k deficient Gja1M213L/M213L mice and maximal respiration for heterozygous Gja1M213L/WT mice is between that of WT and Gja1M213L/M213L (Figure 5C, D; Figure 5—figure supplement 1A, B). In addition, observing other OCR parameters, we found a decrease in ATP-linked respiration and reserve capacity in Gja1M213L/WT cardiomyocytes, and an increase in proton leak and non-mitochondrial respiration in Gja1M213L/M213L suggesting that there can be compensatory long-term effects of the Gja1 mutation (Figure 5—figure supplement 1C).
—Results section (lines 241 – 250) However, remarkably, reduced GJA1-20k expression results in an almost complete cardiac infarction after I/R injury (Figure 5E, F). Moreover, ROS production after I/R injury was increased in Gja1M213L/WT mice compared to WT post-I/R (Figure 5G, H). There was no significant difference in mitochondria size at the basal condition between WT and Gja1M213L/WT mice adult CMs as with neonatal CMs (Figure 5I, J), whereas the mitochondria size was significantly increased after I/R injury and the heterozygous Gja1M213L/WT mice had larger mitochondria compared to WT mice post-I/R (Figure 5I, J). Interestingly, the area of mitochondrial matrix was also increased, suggesting loss of cristae in Gja1M213L/WT mice heart (Figure 5K, L). These data indicate that even partial deletion of GJA1-20k results in a profoundly impaired response to ischemic stress.
—Discussion section (lines 350 – 357) Because GJA1-20k-induced fission is associated with less ROS production with oxidative stress (Figure 5 – figure supplement 1D, E), the endogenous generation of GJA1-20k and subsequent decreased ROS production could explain a major benefit of pre-conditioning. Of note, genetic GJA1-20k reduction increases infarct size and ROS production post-I/R injury (Figure 5E–H). In addition, the population of damaged mitochondria is significantly increased in heterozygous Gja1M213L/WT mouse heart post-I/R (Figure 5I–L). Therefore, GJA1-20k induced decreases in ROS production could limit the amount of I/R injury induced by myocardial infarction.
It is still unclear to me how GJA1-20k is affecting mitochondrial size and function. Based on previous papers, this peptide localizes to the surface of mitochondria, but it is not clear how, or whether, it directly facilitates actin recruitment. The interplay with the endoplasmic reticulum (ER), which can nucleate actin at sites of mitochondrial fission, was not examined. If actin is driving membrane remodeling, is it mediated by ER crossover at these sites?
We appreciate Reviewer #3’s thoughtful comment and suggestion. Our unpublished data indicate that GJA1-20k has an actin-binding domain, suggesting direct binding and actin dynamics regulation. As shown in Figure 3 in the present study, GJA1-20k recruits actin around mitochondria membrane and their interaction resulted in fission. In addition, as the Reviewer suggested, our preliminary data showed significant increase in ER network in GJA1-20k-transfected cells (Figure below). Therefore, there is the possibility that ER is also involved in GJA1-20k mediated mitochondrial fission, while further research will be required to reveal the detailed mechanisms. In the present manuscript, we would like to focus on the finding that actin is necessary for GJA1-20k-mediated mitochondrial fission but not DRP1.
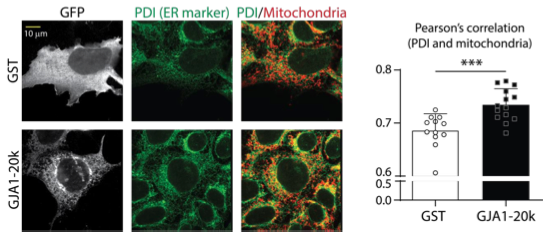
ER network association with mitochondria is increased in GJA1-20k-transfected cells. Left: Representative fixed cell images of HEK293 cells with GFP-tagged GST or GJA1-20k. ER and mitochondria were labeled by Protein disulfide-isomerase (PDI) and Tom20, respectively. Right: The quantification of Pearson’s correlation between PDI and mitochondria. The graph is expressed as mean ± SD. p values were determined by two-tailed Mann-Whitney U-test. ****p < 0.001.*
We have updated the Discussion section to point to this excellent consideration in the future.
—Discussion section (lines 299 – 302) In addition to actin, the endoplasmic reticulum (ER) membrane can be involved in mitochondrial scission (Friedman, Lackner et al. 2011, Tandler, Hoppel et al. 2018). Future studies should be considered whether GJA1-20k induced actin cytoskeleton arrangements involves ER membrane as well.
-
Reviewer #3 (Public Review):
This manuscript seeks to provide mechanistic insight into the role of GJA1-20k in mitochondrial changes that protect against ischemia-reperfusion damage. In previous studies, this group has shown that GJA1-20k protein increases in response to ischemic stress, localizes to mitochondria, promotes mitochondrial biogenesis, and mimics ischemic preconditioning protection in the heart. These changes did not coincide with changes in mitochondrial dynamics proteins, but the increase in GJA1-20k provides protection through an unknown mechanism. This makes it a potentially attractive therapeutic candidate for protection against ischemia.
The evidence in this manuscript shows that mitochondrial size is affected by GJA1-20k, as over-expression of this fragment reduced mitochondrial area. The authors argue that this …
Reviewer #3 (Public Review):
This manuscript seeks to provide mechanistic insight into the role of GJA1-20k in mitochondrial changes that protect against ischemia-reperfusion damage. In previous studies, this group has shown that GJA1-20k protein increases in response to ischemic stress, localizes to mitochondria, promotes mitochondrial biogenesis, and mimics ischemic preconditioning protection in the heart. These changes did not coincide with changes in mitochondrial dynamics proteins, but the increase in GJA1-20k provides protection through an unknown mechanism. This makes it a potentially attractive therapeutic candidate for protection against ischemia.
The evidence in this manuscript shows that mitochondrial size is affected by GJA1-20k, as over-expression of this fragment reduced mitochondrial area. The authors argue that this change in morphology is independent of Drp1 activity, and actin dynamics drive mitochondrial division. These ultrastructural changes coincide with cytoprotective effects during reperfusion following ischemic events by limiting ROS production.
Strengths:
The data on ultrastructural changes is convincing, and GJA1-20k induced a decrease in mitochondrial size. The imaging looks good and quantification is helpful in the evaluating the impact of these changes.
To complement the use of proposed Drp1 inhibitors, the authors use genetic knock-down (KD) of Drp1, and the KD looks robust. Still see some Drp1 colocalization on the mitochondria in the KD, but the levels are diminished.
The decrease in ROS when HEK cells were treated with H2O2 is convincing. And this coincides with the decreased respiration capacity observed in the Seahorse analysis. This provides some mechanistic insight about a specific change in mitochondrial function that contributes to the protective effects observed.
Weaknesses:
With the introduction of GJA1-20k, there is clearly a difference in mitochondrial size, and total mitochondrial content appears unaltered (i.e. Tom20 does not increase). Previously it was suggested that mitochondrial biogenesis was increased with increased levels of GJA1-20k. Is this a difference in the cellular model (HEK) and do the changes in cell culture accurately recapitulate the changes seen in animals? Having more mitochondrial mass despite decrease in the avg. size of these organelles may represent an important difference.
Mdivi-1 is not a selective Drp1 inhibitor. It is a Complex I inhibitor, leading to unintended changes in mitochondrial dynamics in response to ETC stress. Rather than Mdivi-1, a dominant negative Drp1 mutant K38A could be overexpressed to see whether this prevents GJA1-20k-mediated fission. If it still goes through, then I agree that Drp1 is not involved at all.
For the kinetics studies (see Fig 4), I think it is important to measure the timing of the actin recruitment and eventual fission when Drp1 is knocked down and/or when a DN mutant (K38A) is involved. Again, I do not trust the chemical inhibitor (Mdivi-1) data since this does not inhibit Drp1 activity.
The assessment of the impact of ischemic stress with the heterozygous animal (M213L/WT) is hard to interpret. How reduced is the expression of GJA1-20k in these animals and how is mitochondrial function impacted based on Seahorse analysis? The mitochondrial morphology is not altered in these animals, so would mitochondrial function be largely unchanged as well? It is not clear how much GJA1-20k is needed to observe changes in mitochondrial shape and function, and comparisons with the homozygous mutant (M213L/M213L) are not the same, making it difficult to resolve the interpretation of these data.
It is still unclear to me how GJA1-20k is affecting mitochondrial size and function. Based on previous papers, this peptide localizes to the surface of mitochondria, but it is not clear how, or whether, it directly facilitates actin recruitment. The interplay with the endoplasmic reticulum (ER), which can nucleate actin at sites of mitochondrial fission, was not examined. If actin is driving membrane remodeling, is it mediated by ER crossover at these sites?
-
Reviewer #2 (Public Review):
Shimura et al. have discovered that GJA1-20K may provide protection in ischemic hearts through polymerizing actin around mitochondria and inducing mitochondrial fission. The authors use a series of elegant genetic, chemical, biochemical and cell biology studies including the use of the Gja1 M213L mouse line, which is unable to generate the 20kD Gja1 isoform, in order to determine that the beneficial effects of GJA1-20K. Specifically, the authors discovered that this beneficial effect is due to decreased reactive oxygen species (ROS) generation from smaller mitochondria. The overall work is well done but additional discussion should be provided about the impact of the work, particularly how the work may help realize a goal of therapeutically achieving ischemic preconditioning that has not been achieved in …
Reviewer #2 (Public Review):
Shimura et al. have discovered that GJA1-20K may provide protection in ischemic hearts through polymerizing actin around mitochondria and inducing mitochondrial fission. The authors use a series of elegant genetic, chemical, biochemical and cell biology studies including the use of the Gja1 M213L mouse line, which is unable to generate the 20kD Gja1 isoform, in order to determine that the beneficial effects of GJA1-20K. Specifically, the authors discovered that this beneficial effect is due to decreased reactive oxygen species (ROS) generation from smaller mitochondria. The overall work is well done but additional discussion should be provided about the impact of the work, particularly how the work may help realize a goal of therapeutically achieving ischemic preconditioning that has not been achieved in more than 30 years since ischemic preconditioning was first recognized.
-
Reviewer #1 (Public Review):
Mitochondria hyperfission during ishchemia or hypoxia is generally thought as an index of the severity of cytotoxicity, but this group originally identified Cx43 truncated peptide, GJA1-20K, which induces cardioprotection against ischemia/reperfusion injury in mice through promoting mitochondrial fission. Their proposal that GJA1-20K induces mitochodnrial fission independently of Drp1 activation is interesting, but their indirect results are weak to support it.
-
Evaluation Summary:
This study identifies a cardioprotective factor, GJA1-20k (a truncated form of Cx43), which appears to confer protection against ischemia/reperfusion (I/R) injury via promotion of mitochondrial fission. This finding is particularly interesting given that hyperfission is generally thought of as an index of toxicity in I/R or hypoxic injury. I/R lesion size in a GJA1 heterozygous mutant mouse is strikingly exacerbated compared to control animals, providing strong in vivo evidence supporting a role for this factor in protection from I/R. However, while the findings are interesting and novel, key results require additional experimental support, including to address the lack of key control data, and a significant revision will be necessary to address these issues.
(This preprint has been reviewed by eLife. We include the …
Evaluation Summary:
This study identifies a cardioprotective factor, GJA1-20k (a truncated form of Cx43), which appears to confer protection against ischemia/reperfusion (I/R) injury via promotion of mitochondrial fission. This finding is particularly interesting given that hyperfission is generally thought of as an index of toxicity in I/R or hypoxic injury. I/R lesion size in a GJA1 heterozygous mutant mouse is strikingly exacerbated compared to control animals, providing strong in vivo evidence supporting a role for this factor in protection from I/R. However, while the findings are interesting and novel, key results require additional experimental support, including to address the lack of key control data, and a significant revision will be necessary to address these issues.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #3 agreed to share their name with the authors.)
-
