An improved bacterial single-cell RNA-seq reveals biofilm heterogeneity
Curation statements for this article:-
Curated by eLife

eLife Assessment
This work introduces an important new method for depleting ribosomal RNA from bacterial single-cell RNA sequencing libraries, demonstrating its applicability for studying heterogeneity in microbial biofilms. The findings provide convincing evidence for a distinct subpopulation of cells at the biofilm base that upregulates PdeI expression. Future studies exploring the functional relationship between PdeI and c-di-GMP levels, along with the roles of co-expressed genes within the same cluster, could further enhance the depth and impact of these conclusions.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
In contrast to mammalian cells, bacterial cells lack mRNA polyadenylated tails, presenting a hurdle in isolating mRNA amidst the prevalent rRNA during single-cell RNA-seq. This study introduces a novel method, ribosomal RNA-derived cDNA depletion (RiboD), seamlessly integrated into the PETRI-seq technique, yielding RiboD-PETRI. This innovative approach offers a cost-effective, equipment-free, and high-throughput solution for bacterial single-cell RNA sequencing (scRNA-seq). By efficiently eliminating rRNA reads and substantially enhancing mRNA detection rates (up to 92%), our method enables precise exploration of bacterial population heterogeneity. Applying RiboD-PETRI to investigate biofilm heterogeneity, distinctive subpopulations marked by unique genes within biofilms were successfully identified. Notably, PdeI, a marker for the cell-surface attachment subpopulation, was observed to elevate cyclic diguanylate (c-di-GMP) levels, promoting persister cell formation. Thus, we address a persistent challenge in bacterial single-cell RNA-seq regarding rRNA abundance, exemplifying the utility of this method in exploring biofilm heterogeneity. Our method effectively tackles a long-standing issue in bacterial scRNA-seq: the overwhelming abundance of rRNA. This advancement significantly enhances our ability to investigate the intricate heterogeneity within biofilms at unprecedented resolution.
Article activity feed
-

-
-
-

eLife Assessment
This work introduces an important new method for depleting ribosomal RNA from bacterial single-cell RNA sequencing libraries, demonstrating its applicability for studying heterogeneity in microbial biofilms. The findings provide convincing evidence for a distinct subpopulation of cells at the biofilm base that upregulates PdeI expression. Future studies exploring the functional relationship between PdeI and c-di-GMP levels, along with the roles of co-expressed genes within the same cluster, could further enhance the depth and impact of these conclusions.
-
Reviewer #1 (Public review):
Summary:
In this manuscript, Yan and colleagues introduce a modification to the previously published PETRI-seq bacterial single cell protocol to include a ribosomal depletion step based on a DNA probe set that selectively hybridizes with ribosome-derived (rRNA) cDNA fragments. They show that their modification of the PETRI-seq protocol increases the fraction of informative non-rRNA reads from ~4-10% to 54-92%. The authors apply their protocol to investigating heterogeneity in a biofilm model of E. coli, and convincingly show how their technology can detect minority subpopulations within a complex community.
Strengths:
The method the authors propose is a straightforward and inexpensive modification of an established split-pool single cell RNA-seq protocol that greatly increases its utility, and should be of …
Reviewer #1 (Public review):
Summary:
In this manuscript, Yan and colleagues introduce a modification to the previously published PETRI-seq bacterial single cell protocol to include a ribosomal depletion step based on a DNA probe set that selectively hybridizes with ribosome-derived (rRNA) cDNA fragments. They show that their modification of the PETRI-seq protocol increases the fraction of informative non-rRNA reads from ~4-10% to 54-92%. The authors apply their protocol to investigating heterogeneity in a biofilm model of E. coli, and convincingly show how their technology can detect minority subpopulations within a complex community.
Strengths:
The method the authors propose is a straightforward and inexpensive modification of an established split-pool single cell RNA-seq protocol that greatly increases its utility, and should be of interest to a wide community working in the field of bacterial single cell RNA-seq.
Comments on revised version:
The reviewers have responded thoughtfully and comprehensively to all of my comments. I believe the details of the protocol are now much easier to understand, and the text and methods have been significantly clarified. I have no further comments.
-
Reviewer #2 (Public review):
Summary:
This work introduces a new method of depleting the ribosomal reads from the single-cell RNA sequencing library prepared with one of the prokaryotic scRNA-seq techniques, PETRI-seq. The advance is very useful since it allows broader access to the technology by lowering the cost of sequencing. It also allows more transcript recovery with fewer sequencing reads. The authors demonstrate the utility and performance of the method for three different model species and find a subpopulation of cells in the E.coli biofilm that express a protein, PdeI, which causes elevated c-di-GMP levels. These cells were shown to be in a state that promotes persister formation in response to ampicillin treatment.
Strengths:
The introduced rRNA depletion method is highly efficient, with the depletion for E.coli resulting in …
Reviewer #2 (Public review):
Summary:
This work introduces a new method of depleting the ribosomal reads from the single-cell RNA sequencing library prepared with one of the prokaryotic scRNA-seq techniques, PETRI-seq. The advance is very useful since it allows broader access to the technology by lowering the cost of sequencing. It also allows more transcript recovery with fewer sequencing reads. The authors demonstrate the utility and performance of the method for three different model species and find a subpopulation of cells in the E.coli biofilm that express a protein, PdeI, which causes elevated c-di-GMP levels. These cells were shown to be in a state that promotes persister formation in response to ampicillin treatment.
Strengths:
The introduced rRNA depletion method is highly efficient, with the depletion for E.coli resulting in over 90% of reads containing mRNA. The method is ready to use with existing PETRI-seq libraries which is a large advantage, given that no other rRNA depletion methods were published for split-pool bacterial scRNA-seq methods. Therefore, the value of the method for the field is high. There is also evidence that a small number of cells at the bottom of a static biofilm express PdeI which is causing the elevated c-di-GMP levels that are associated with persister formation. This finding highlights the potentially complex role of PdeI in regulation of c-di-GMP levels and persister formation in microbial biofilms.
Comments on revised version:
The authors edited the manuscript thoroughly in response to the comments, including both performing new experiments and showing more data and information. Most of the major points raised between both reviewers were addressed. The authors explained the seeming contradiction between c-di-GMP levels and PdeI expression.
-
Author response:
The following is the authors’ response to the previous reviews.
eLife Assessment
This work presents an important method for depleting ribosomal RNA from bacterial single-cell RNA sequencing libraries, enabling the study of cellular heterogeneity within microbial biofilms. The approach convincingly identifies a small subpopulation of cells at the biofilm's base with upregulated PdeI expression, offering invaluable insights into the biology of bacterial biofilms and the formation of persister cells. Further integrated analysis of gene interactions within these datasets could deepen our understanding of biofilm dynamics and resilience.
Thank you for your valuable feedback and for recognizing the importance of our method for depleting ribosomal RNA from bacterial single-cell RNA sequencing libraries. We are pleased that …
Author response:
The following is the authors’ response to the previous reviews.
eLife Assessment
This work presents an important method for depleting ribosomal RNA from bacterial single-cell RNA sequencing libraries, enabling the study of cellular heterogeneity within microbial biofilms. The approach convincingly identifies a small subpopulation of cells at the biofilm's base with upregulated PdeI expression, offering invaluable insights into the biology of bacterial biofilms and the formation of persister cells. Further integrated analysis of gene interactions within these datasets could deepen our understanding of biofilm dynamics and resilience.
Thank you for your valuable feedback and for recognizing the importance of our method for depleting ribosomal RNA from bacterial single-cell RNA sequencing libraries. We are pleased that our approach has convincingly identified a small subpopulation of cells at the base of the biofilm with upregulated PdeI expression, providing significant insights into the biology of bacterial biofilms and the formation of persister cells.
We acknowledge your suggestion for a more comprehensive analysis of multiple genes and their interactions. While we conducted a broad analysis across the transcriptome, our decision to focus on the heterogeneously expressed gene PdeI was primarily informed by its critical role in biofilm biology. In addition to PdeI, we investigated other marker genes and noted that lptE and sstT exhibited potential associations with persister cells. However, our interaction analysis revealed that LptE and SstT did not demonstrate significant relationships with c-di-GMP and PdeI based on current knowledge. This insight led us to concentrate on PdeI, given its direct relevance to biofilm formation and its close connection to the c-di-GMP signaling pathway.
We fully agree that other marker genes may also have important regulatory roles in different aspects of biofilm dynamics. Thus, we plan to explore the expression patterns and potential functions of these genes in our future research. Specifically, we intend to conduct more extensive gene network analyses to uncover the complex regulatory mechanisms involved in biofilm formation and resilience.
Public Reviews:
Reviewer #1 (Public review):
Summary:
In this manuscript, Yan and colleagues introduce a modification to the previously published PETRI-seq bacterial single cell protocol to include a ribosomal depletion step based on a DNA probe set that selectively hybridizes with ribosome-derived (rRNA) cDNA fragments. They show that their modification of the PETRI-seq protocol increases the fraction of informative non-rRNA reads from ~4-10% to 54-92%. The authors apply their protocol to investigating heterogeneity in a biofilm model of E. coli, and convincingly show how their technology can detect minority subpopulations within a complex community.
Strengths:
The method the authors propose is a straightforward and inexpensive modification of an established split-pool single cell RNA-seq protocol that greatly increases its utility, and should be of interest to a wide community working in the field of bacterial single cell RNA-seq.
We sincerely thank the reviewer for their thoughtful and positive evaluation of our work. We appreciate the recognition of our modification to the PETRI-seq bacterial single-cell RNA sequencing protocol by incorporating a ribosomal depletion step. The significant increase in the fraction of informative non-rRNA reads, as noted in the reviewer’s summary, underscores the effectiveness of our method in enhancing the utility of the PETRI-seq approach. We are also encouraged by the reviewer's acknowledgment of our ability to detect minority subpopulations within complex biofilm communities. Our team is committed to further validating and optimizing this method, and we believe that RiboD-PETRI will contribute meaningfully to the field of bacterial single-cell transcriptomics. We hope this innovative approach will facilitate new discoveries in microbial ecology and biofilm research.
Reviewer #2 (Public review):
Summary:
This work introduces a new method of depleting the ribosomal reads from the single-cell RNA sequencing library prepared with one of the prokaryotic scRNA-seq techniques, PETRI-seq. The advance is very useful since it allows broader access to the technology by lowering the cost of sequencing. It also allows more transcript recovery with fewer sequencing reads. The authors demonstrate the utility and performance of the method for three different model species and find a subpopulation of cells in the E.coli biofilm that express a protein, PdeI, which causes elevated c-di-GMP levels. These cells were shown to be in a state that promotes persister formation in response to ampicillin treatment.
Strengths:
The introduced rRNA depletion method is highly efficient, with the depletion for E.coli resulting in over 90% of reads containing mRNA. The method is ready to use with existing PETRI-seq libraries which is a large advantage, given that no other rRNA depletion methods were published for split-pool bacterial scRNA-seq methods. Therefore, the value of the method for the field is high. There is also evidence that a small number of cells at the bottom of a static biofilm express PdeI which is causing the elevated c-di-GMP levels that are associated with persister formation. This finding highlights the potentially complex role of PdeI in regulation of c-di-GMP levels and persister formation in microbial biofilms.
Weaknesses:
Given many current methods that also introduce different techniques for ribosomal RNA depletion in bacterial single-cell RNA sequencing, it is unclear what is the place and role of RiboD-PETRI. The efficiency of rRNA depletion varies greatly between species for the majority of the available methods, so it is not easy to select the best fitting technique for a specific application.
Thank you for your insightful comments regarding the place and role of RiboD-PETRI in the landscape of ribosomal RNA depletion techniques for bacterial single-cell RNA sequencing. We appreciate the opportunity to address your concerns and clarify the significance of our method.
We acknowledge that the field of rRNA depletion in bacterial single-cell RNA sequencing is diverse, with many methods offering different approaches. We also recognize the challenge of selecting the best technique for a specific application, given the variability in rRNA depletion efficiency across species for many available methods. In light of these considerations, we believe RiboD-PETRI occupies a distinct and valuable niche in this landscape due to following reasons: 1) Low-input compatibility: Our method is specifically tailored for the low-input requirements of single-cell RNA sequencing, maintaining high efficiency even with limited starting material. This makes RiboD-PETRI particularly suitable for single-cell studies where sample quantity is often a limiting factor. 2) Equipment-free protocol: One of the unique advantages of RiboD-PETRI is that it can be conducted in any lab without the need for specialized equipment. This accessibility ensures that a wide range of researchers can implement our method, regardless of their laboratory setup. 3) Broad species coverage: Through comprehensive probe design targeting highly conserved regions of bacterial rRNA, RiboD-PETRI offers a robust solution for samples involving multiple bacterial species or complex microbial communities. This approach aims to provide consistent performance across diverse taxa, addressing the variability issue you mentioned. 4) Versatility and compatibility: RiboD-PETRI is designed to be compatible with various downstream single-cell RNA sequencing protocols, enhancing its utility in different experimental setups and research contexts.
In conclusion, RiboD-PETRI's unique combination of low-input compatibility, equipment-free protocol, broad species coverage, and versatility positions it as a robust and accessible option in the landscape of rRNA depletion methods for bacterial single-cell RNA sequencing. We are committed to further validating and improving our method to ensure its valuable contribution to the field and to provide researchers with a reliable tool for their diverse experimental needs.
Despite transcriptome-wide coverage, the authors focused on the role of a single heterogeneously expressed gene, PdeI. A more integrated analysis of multiple genes and\or interactions between them using these data could reveal more insights into the biofilm biology.
Thank you for your valuable feedback. We understand your suggestion for a more comprehensive analysis of multiple genes and their interactions. While we indeed conducted a broad analysis across the transcriptome, our decision to focus on the heterogeneously expressed gene PdeI was primarily based on its crucial role in biofilm biology. Beyond PdeI, we also conducted overexpression experiments on several other marker genes and examined their phenotypes. Notably, the lptE and sstT genes showed potential associations with persister cells. We performed an interaction analysis, which revealed that LptE and SstT did not show significant relationships with c-di-GMP and PdeI based on current knowledge. This finding led us to concentrate our attention on PdeI. Given PdeI's direct relevance to biofilm formation and its close connection to the c-di-GMP signaling pathway, we believed that an in-depth study of PdeI was most likely to reveal key biological mechanisms.
We fully agree with your point that other marker genes may play regulatory roles in different aspects. The expression patterns and potential functions of these genes will be an important direction in our future research. In our future work, we plan to conduct more extensive gene network analyses to uncover the complex regulatory mechanisms of biofilm formation.
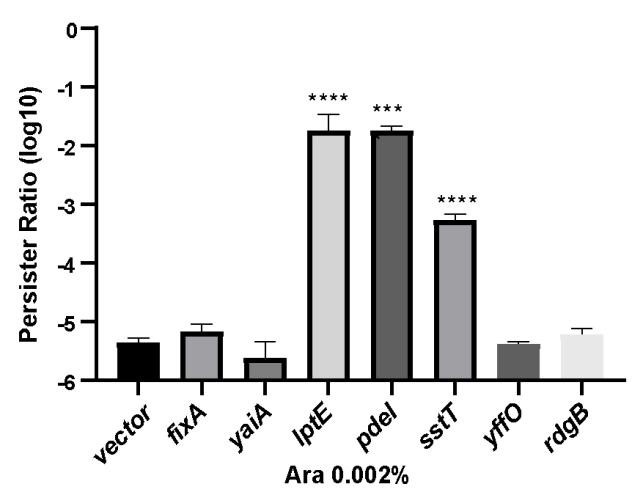
Author response image 1.
The proportion of persister cells in the partially maker genes and empty vector control groups. Following induction of expression with 0.002% arabinose for 2 hours, a persister counting assay was conducted on the strains using 150 μg/ml ampicillin.
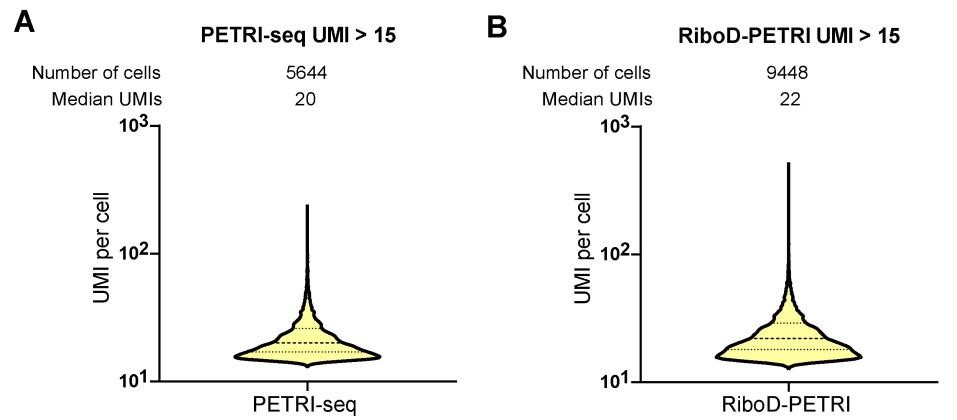
The authors should also present the UMIs capture metrics for RiboD-PETRI method for all cells passing initial quality filter (>=15 UMIs/cell) both in the text and in the figures. Selection of the top few cells with higher UMI count may introduce biological biases in the analysis (the top 5% of cells could represent a distinct subpopulation with very high gene expression due to a biological process). For single-cell RNA sequencing, showing the statistics for a 'top' group of cells creates confusion and inflates the perceived resolution, especially when used to compare to other methods (e.g. the parent method PETRI-seq itself).
Thank you for your valuable feedback regarding the presentation of UMI capture metrics for the RiboD-PETRI method. We appreciate your concern about potential biological biases and the importance of comprehensive data representation in single-cell RNA sequencing analysis. We have now included the UMI capture metrics for all cells passing the initial quality filter (≥15 UMIs/cell) for the RiboD-PETRI method. This information has been added to both the main text and the relevant figures, providing a more complete picture of our method's performance across the entire range of captured cells. These revisions strengthen our manuscript and provide readers with a more complete understanding of the RiboD-PETRI method in the context of single-cell RNA sequencing.
Recommendations for the authors:
Reviewer #1 (Recommendations for the authors):
The reviewers have responded thoughtfully and comprehensively to all of my comments. I believe the details of the protocol are now much easier to understand, and the text and methods have been significantly clarified. I have no further comments.
Reviewer #2 (Recommendations for the authors):
The authors edited the manuscript thoroughly in response to the comments, including both performing new experiments and showing more data and information. Most of the major points raised between both reviewers were addressed. The authors explained the seeming contradiction between c-di-GMP levels and PdeI expression. Despite these improvements, a few issues remain:
- Despite now depositing the data and analysis files to GEO, the access is embargoed and the reviewer token was not provided to evaluate the shared data and accessory files.
Please note that although the data and analysis files have been deposited to GEO, access is currently embargoed. To evaluate the shared data and accessory files, you will need a reviewer token, which appears to have not been provided.
To gain access, please follow these steps:
Visit the GEO accession page at: https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE260458
In the designated field, enter the reviewer token: ehipgqiohhcvjev
- Despite now discussing performance metrics for RiboD-PETRI method for all cells passing initial quality filter (>=15 UMIs/cell) in the text, the authors continued to also include the statistics for top 1000 cells, 5,000 cells and so on. Critically, Figure 2A-B is still showing the UMI and gene distributions per cell only for these select groups of cells. The intent to focus on these metrics is not quite clear, as selection of the top few cells with higher UMI count may introduce biological biases in the analysis (what if the top 5% of cells are unusual because they represent a distinct subpopulation with very high gene expression due to a biological process). I understand the desire to demonstrate the performance of the method by highlighting a few select 'best' cells, however, for single-cell RNA sequencing showing the statistics for a 'top' group of cells is not appropriate and creates confusion, especially when used to compare to other methods (e.g. the parent method PETRI-seq itself).
We appreciate your insightful feedback regarding our presentation of the RiboD-PETRI method's performance metrics. We acknowledge the concerns you've raised and agree that our current approach requires refinement. We have revised our analysis to prominently feature metrics for all cells that pass the initial quality filter (≥15 UMIs/cell) (Fig. 2A, Fig. 3A, Supplementary Fig. 1A, B and Supplementary Fig. 2A, G). This approach provides a more representative view of the method's performance across the entire dataset, avoiding potential biases introduced by focusing solely on top-performing cells.
We recognize that selecting only the top cells based on UMI counts can indeed introduce biological biases, as these cells may represent distinct subpopulations with unique biological processes rather than typical cellular states. To address this, we have clearly stated the potential for bias when highlighting select 'best' cells. We also provided context for why these high-performing cells are shown, explaining that they demonstrate the upper limits of the method's capabilities (lines 139). In addition, when comparing RiboD-PETRI to other methods, including the parent PETRI-seq, we ensured that comparisons are made using consistent criteria across all methods.
By implementing these changes, we aim to provide a more accurate, unbiased, and comprehensive representation of the RiboD-PETRI method's performance while maintaining scientific rigor and transparency. We appreciate your critical feedback, as it helps us improve the quality and reliability of our research presentation.
- Line 151 " The findings reveal that our sequencing saturation is 100% (Fig. S1B, C)" - I suggest the authors revisit this calculation as this parameter is typically very challenging to get above 95-96%. The sequencing saturation should be calculated from the statistics of alignment themselves, i.e. the parameter calculated by Cell Ranger as described here https://kb.10xgenomics.com/hc/en-us/articles/115003646912-How-is-sequencing-saturation-calculated :
"The web_summary.html output from cellranger count includes a metric called "Sequencing Saturation". This metric quantifies the fraction of reads originating from an already-observed UMI. More specifically, this is the fraction of confidently mapped, valid cell-barcode, valid UMI reads that are non-unique (match an existing cell-barcode, UMI, gene combination).
The formula for calculating this metric is as follows:
Sequencing Saturation = 1 - (n_deduped_reads / n_reads)
where
n_deduped_reads = Number of unique (valid cell-barcode, valid UMI, gene) combinations among confidently mapped reads.
n_reads = Total number of confidently mapped, valid cell-barcode, valid UMI reads.
Note that the numerator of the fraction is n_deduped_reads, not the non-unique reads that are mentioned in the definition. n_deduped_reads is a degree of uniqueness, not a degree of duplication/saturation. Therefore we take the complement of (n_deduped_reads / n_reads) to measure saturation."
We appreciate your insightful comment regarding our sequencing saturation calculation. The sequencing saturation algorithm we initially employed was based on the methodology used in the BacDrop study (PMID: PMC10014032, https://pmc.ncbi.nlm.nih.gov/articles/PMC10014032/).
We acknowledge the importance of using standardized and widely accepted methods for calculating sequencing saturation. As per your suggestion, we have recalculated our sequencing saturation using the method described by 10x Genomics. Given the differences between RiboD-PETRI and 10x Genomics datasets, we have adapted the calculation as follows:
· n_deduped_reads: We used the number of UMIs as a measure of unique reads.
· n_reads: We used the total number of confidently mapped reads.
After applying this adapted calculation method, we found that our sequencing saturation ranges from 92.16% to 93.51%. This range aligns more closely with typical expectations for sequencing saturation in single-cell RNA sequencing experiments, suggesting that we have captured a substantial portion of the transcript diversity in our samples. We also updated Figure S1 to reflect these recalculated sequencing saturation values. We will also provide a detailed description of our calculation method in the methods section to ensure transparency and reproducibility. It's important to note that this saturation calculation method was originally designed for 10× Genomics data. While we've adapted it for our study, we acknowledge that its applicability to our specific experimental setup may be limited.
We thank you for bringing this important point to our attention. This recalculation not only improves the accuracy of our reported results but also aligns our methodology more closely with established standards in the field. We believe these revisions strengthen the overall quality and reliability of our study.
- Further, this calculated saturation should be taken into account when comparing the performance of the method in terms of retrieving diverse transcripts from cells. I.e., if the RiboD-Petri dataset was subsampled to the same saturation as the original PETRI-seq dataset was obtained with, would the median UMIs/cell for all cells above filter be comparable? In other words, does rRNA depletion just decreases the cost to sequence to saturation, or does it provide UMI capture benefits at a comparable saturation?
We appreciate your insightful question regarding the comparison of method performance in terms of transcript retrieval diversity and the impact of saturation. To address your concerns, we conducted an additional analysis comparing the RiboD-PETRI and original PETRI-seq datasets at equivalent saturation levels besides our original analysis with equivalent sequencing depth.
With equivalent sequencing depth, RiboD-PETRI demonstrates a significantly enhanced Unique Molecular Identifier (UMI) counts detection rate compared to PETRI-seq alone (Fig. 1C). This method recovered approximately 20175 cells (92.6% recovery rate) with ≥ 15 UMIs per cell with a median UMI count of 42 per cell, which was significantly higher than PETRI-seq's recovery rate of 17.9% with a median UMI count of 20 per cell (Figure S1A, B), indicating the number of detected mRNA per cell increased prominently.
When we subsampled the RiboD-PETRI dataset to match the saturation level of the original PETRI-seq dataset (i.e., equalizing the n_deduped_reads/n_reads ratio), we found that the median UMIs/cell for all cells above the filter threshold was higher in the RiboD-PETRI dataset compared to the original PETRI-seq (as shown in Author response image 2). This observation can be primarily attributed to the introduction of the rRNA depletion step in the RiboD-PETRI method. Our analysis suggests that rRNA depletion not only reduces the cost of sequencing to saturation but also provides additional benefits in UMI capture efficiency at comparable saturation levels.The rRNA depletion step effectively reduces the proportion of rRNA-derived reads in the sequencing output. Consequently, at equivalent saturation levels, this leads to a relative increase in the number of n_deduped_reads corresponding to mRNA transcripts. This shift in read composition enhances the capture of informative UMIs, resulting in improved transcript diversity and detection.
In conclusion, our findings indicate that the rRNA depletion step in RiboD-PETRI offers dual advantages: it decreases the cost to sequence to saturation and provides enhanced UMI capture benefits at comparable saturation levels, ultimately leading to more efficient and informative single-cell transcriptome profiling.
Author response image 2.
At almost the same sequencing saturation (64% and 67%), the number of cells exceeding the screening criteria (≥15 UMIs ) and the median number of UMIs in cells in Ribod-PETRI and PETRI-seq data of exponential period E. coli (3h).
- smRandom-seq and BaSSSh-seq need to also be discussed since these newer methods are also demonstrating rRNA depletion techniques. (https://doi.org/10.1038/s41467-023-40137-9 and https://doi.org/10.1101/2024.06.28.601229)
Thank you for your valuable feedback. We appreciate the opportunity to discuss our method, RiboD-PETRI, in the context of other recent advances in bacterial RNA sequencing techniques, particularly smRandom-seq and BaSSSh-seq.
RiboD-PETRI employs a Ribosomal RNA-derived cDNA Depletion (RiboD) protocol. This method uses probe primers that span all regions of the bacterial rRNA sequence, with the 3'-end complementary to rRNA-derived cDNA and the 5'-end complementary to a biotin-labeled universal primer. After hybridization, Streptavidin magnetic beads are used to eliminate the hybridized rRNA-derived cDNA, leaving mRNA-derived cDNA in the supernatant. smRandom-seq utilizes a CRISPR-based rRNA depletion technique. This method is designed for high-throughput single-microbe RNA sequencing and has been shown to reduce the rRNA proportion from 83% to 32%, effectively increasing the mRNA proportion four times (from 16% to 63%). While specific details about BaSSSh-seq's rRNA depletion technique are not provided in the available information, it is described as employing a rational probe design for efficient rRNA depletion. This technique aims to minimize the loss of mRNA during the depletion process, ensuring a more accurate representation of the transcriptome.
RiboD-PETRI demonstrates significant enhancement in rRNA-derived cDNA depletion across both gram-negative and gram-positive bacterial species. It increases the mRNA ratio from 8.2% to 81% for E. coli in exponential phase, from 10% to 92% for S. aureus in stationary phase, and from 3.9% to 54% for C. crescentus in exponential phase. smRandom-seq shows high species specificity (99%), a minor doublet rate (1.6%), and a reduced rRNA percentage (32%). These metrics indicate its efficiency in single-microbe RNA sequencing. While specific performance metrics for BaSSSh-seq are not provided in the available information, its rational probe design approach suggests a focus on maintaining mRNA integrity during the depletion process.
RiboD-PETRI is described as a cost-effective ($0.0049 per cell), equipment-free, and high-throughput solution for bacterial scRNA-seq. This makes it an attractive option for researchers with budget constraints. While specific cost information is not provided, the efficiency of smRandom-seq is noted to be affected by the overwhelming quantity of rRNAs (>80% of mapped reads). The CRISPR-based depletion technique likely adds to the complexity and cost of the method. Cost and accessibility information for BaSSSh-seq is not provided in the available data, making a direct comparison difficult.
All three methods represent significant advancements in bacterial RNA sequencing, each offering unique approaches to the challenge of rRNA depletion. RiboD-PETRI stands out for its cost-effectiveness and demonstrated success in complex systems like biofilms. Its ability to significantly increase mRNA ratios across different bacterial species and growth phases is particularly noteworthy. smRandom-seq's CRISPR-based approach offers high specificity and efficiency, which could be advantageous in certain research contexts, particularly where single-microbe resolution is crucial. However, the complexity of the CRISPR system might impact its accessibility and cost-effectiveness. BaSSSh-seq's focus on minimizing mRNA loss during depletion could be beneficial for studies requiring highly accurate transcriptome representations, although more detailed performance data would be needed for a comprehensive comparison. The choice between these methods would depend on specific research needs. RiboD-PETRI's cost-effectiveness and proven application in biofilm studies make it particularly suitable for complex bacterial community analyses. smRandom-seq might be preferred for studies requiring high-throughput single-cell resolution. BaSSSh-seq could be the method of choice when preserving the integrity of the mRNA profile is paramount.
In conclusion, while all three methods offer valuable solutions for rRNA depletion in bacterial RNA sequencing, RiboD-PETRI's combination of efficiency, cost-effectiveness, and demonstrated application in complex biological systems positions it as a highly competitive option in the field of bacterial transcriptomics.
We have revised our discussion in the manuscript according to the above analysis (lines 116-119)
- Ctrl and Delta-Delta abbreviations are used in main text but not defined there (lines 107-110).
Thank you for your valuable feedback. We have now defined the abbreviations "Ctrl" and "Delta-Delta" in the main text for clarity.
- The utility of Figs 2E and 3E is questionable - the same information can be conveyed in text.
Thank you for your thoughtful observation regarding Figures 2E and 3E. We appreciate your feedback and would like to address the concerns you've raised.
While we acknowledge that some of the information in these figures could be conveyed textually, we believe that their visual representation offers several advantages. Figures 2E and 3E provide a comprehensive visual overview of the pathway enrichment analysis for marker genes, which may be more easily digestible than a textual description. This analysis was conducted in response to another reviewer's request, demonstrating our commitment to addressing diverse perspectives in our research.
These figures allow for a systematic interpretation of gene expression data, revealing complex interactions between genes and their involvement in biological pathways that might be less apparent in a text-only format. Visual representations can make complex data more accessible to readers with different learning styles or those who prefer graphical summaries. Additionally, including such figures is consistent with standard practices in our field, facilitating comparison with other studies. We believe that the pathway enrichment analysis results presented in these figures provide valuable insights that merit inclusion as visual elements. However, we are open to discussing alternative ways to present this information if you have specific suggestions for improvement.
-
-
eLife Assessment
This work presents an important method for depleting ribosomal RNA from bacterial single-cell RNA sequencing libraries, enabling the study of cellular heterogeneity within microbial biofilms. The approach convincingly identifies a small subpopulation of cells at the biofilm's base with upregulated PdeI expression, offering invaluable insights into the biology of bacterial biofilms and the formation of persister cells. Further integrated analysis of gene interactions within these datasets could deepen our understanding of biofilm dynamics and resilience.
-
Reviewer #1 (Public review):
Summary:
In this manuscript, Yan and colleagues introduce a modification to the previously published PETRI-seq bacterial single cell protocol to include a ribosomal depletion step based on a DNA probe set that selectively hybridizes with ribosome-derived (rRNA) cDNA fragments. They show that their modification of the PETRI-seq protocol increases the fraction of informative non-rRNA reads from ~4-10% to 54-92%. The authors apply their protocol to investigating heterogeneity in a biofilm model of E. coli, and convincingly show how their technology can detect minority subpopulations within a complex community.
Strengths:
The method the authors propose is a straightforward and inexpensive modification of an established split-pool single cell RNA-seq protocol that greatly increases its utility, and should be of …
Reviewer #1 (Public review):
Summary:
In this manuscript, Yan and colleagues introduce a modification to the previously published PETRI-seq bacterial single cell protocol to include a ribosomal depletion step based on a DNA probe set that selectively hybridizes with ribosome-derived (rRNA) cDNA fragments. They show that their modification of the PETRI-seq protocol increases the fraction of informative non-rRNA reads from ~4-10% to 54-92%. The authors apply their protocol to investigating heterogeneity in a biofilm model of E. coli, and convincingly show how their technology can detect minority subpopulations within a complex community.
Strengths:
The method the authors propose is a straightforward and inexpensive modification of an established split-pool single cell RNA-seq protocol that greatly increases its utility, and should be of interest to a wide community working in the field of bacterial single cell RNA-seq.
-
Reviewer #2 (Public review):
Summary:
This work introduces a new method of depleting the ribosomal reads from the single-cell RNA sequencing library prepared with one of the prokaryotic scRNA-seq techniques, PETRI-seq. The advance is very useful since it allows broader access to the technology by lowering the cost of sequencing. It also allows more transcript recovery with fewer sequencing reads. The authors demonstrate the utility and performance of the method for three different model species and find a subpopulation of cells in the E.coli biofilm that express a protein, PdeI, which causes elevated c-di-GMP levels. These cells were shown to be in a state that promotes persister formation in response to ampicillin treatment.
Strengths:
The introduced rRNA depletion method is highly efficient, with the depletion for E.coli resulting in …
Reviewer #2 (Public review):
Summary:
This work introduces a new method of depleting the ribosomal reads from the single-cell RNA sequencing library prepared with one of the prokaryotic scRNA-seq techniques, PETRI-seq. The advance is very useful since it allows broader access to the technology by lowering the cost of sequencing. It also allows more transcript recovery with fewer sequencing reads. The authors demonstrate the utility and performance of the method for three different model species and find a subpopulation of cells in the E.coli biofilm that express a protein, PdeI, which causes elevated c-di-GMP levels. These cells were shown to be in a state that promotes persister formation in response to ampicillin treatment.
Strengths:
The introduced rRNA depletion method is highly efficient, with the depletion for E.coli resulting in over 90% of reads containing mRNA. The method is ready to use with existing PETRI-seq libraries which is a large advantage, given that no other rRNA depletion methods were published for split-pool bacterial scRNA-seq methods. Therefore, the value of the method for the field is high. There is also evidence that a small number of cells at the bottom of a static biofilm express PdeI which is causing the elevated c-di-GMP levels that are associated with persister formation. This finding highlights the potentially complex role of PdeI in regulation of c-di-GMP levels and persister formation in microbial biofilms.
Weaknesses:
Given many current methods that also introduce different techniques for ribosomal RNA depletion in bacterial single-cell RNA sequencing, it is unclear what is the place and role of RiboD-PETRI. The efficiency of rRNA depletion varies greatly between species for the majority of the available methods, so it is not easy to select the best fitting technique for a specific application.
Despite transcriptome-wide coverage, the authors focused on the role of a single heterogeneously expressed gene, PdeI. A more integrated analysis of multiple genes and\or interactions between them using these data could reveal more insights into the biofilm biology.
The authors should also present the UMIs capture metrics for RiboD-PETRI method for all cells passing initial quality filter (>=15 UMIs/cell) both in the text and in the figures. Selection of the top few cells with higher UMI count may introduce biological biases in the analysis (the top 5% of cells could represent a distinct subpopulation with very high gene expression due to a biological process). For single-cell RNA sequencing, showing the statistics for a 'top' group of cells creates confusion and inflates the perceived resolution, especially when used to compare to other methods (e.g. the parent method PETRI-seq itself).
-
Author response:
The following is the authors’ response to the original reviews.
eLife Assessment
The work introduces a valuable new method for depleting the ribosomal RNA from bacterial single-cell RNA sequencing libraries and shows that this method is applicable to studying the heterogeneity in microbial biofilms. The evidence for a small subpopulation of cells at the bottom of the biofilm which upregulates PdeI expression is solid. However, more investigation into the unresolved functional relationship between PdeI and c-di-GMP levels with the help of other genes co-expressed in the same cluster would have made the conclusions more significant.
Many thanks for eLife’s assessment of our manuscript and the constructive feedback. We are encouraged by the recognition of our bacterial single-cell RNA-seq methodology as valuable and its …
Author response:
The following is the authors’ response to the original reviews.
eLife Assessment
The work introduces a valuable new method for depleting the ribosomal RNA from bacterial single-cell RNA sequencing libraries and shows that this method is applicable to studying the heterogeneity in microbial biofilms. The evidence for a small subpopulation of cells at the bottom of the biofilm which upregulates PdeI expression is solid. However, more investigation into the unresolved functional relationship between PdeI and c-di-GMP levels with the help of other genes co-expressed in the same cluster would have made the conclusions more significant.
Many thanks for eLife’s assessment of our manuscript and the constructive feedback. We are encouraged by the recognition of our bacterial single-cell RNA-seq methodology as valuable and its efficacy in studying bacterial population heterogeneity. We appreciate the suggestion for additional investigation into the functional relationship between PdeI and c-di-GMP levels. We concur that such an exploration could substantially enhance the impact of our conclusions. To address this, we have implemented the following revisions: We have expanded our data analysis to identify and characterize genes co-expressed with PdeI within the same cellular cluster (Fig. 3F, G, Response Fig. 10); We conducted additional experiments to validate the functional relationships between PdeI and c-di-GMP, followed by detailed phenotypic analyses (Response Fig. 9B). Our analysis reveals that while other marker genes in this cluster are co-expressed, they do not significantly impact biofilm formation or directly relate to c-di-GMP or PdeI. We believe these revisions have substantially enhanced the comprehensiveness and context of our manuscript, thereby reinforcing the significance of our discoveries related to microbial biofilms. The expanded investigation provides a more thorough understanding of the PdeI-associated subpopulation and its role in biofilm formation, addressing the concerns raised in the initial assessment.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
In this manuscript, Yan and colleagues introduce a modification to the previously published PETRI-seq bacterial single-cell protocol to include a ribosomal depletion step based on a DNA probe set that selectively hybridizes with ribosome-derived (rRNA) cDNA fragments. They show that their modification of the PETRI-seq protocol increases the fraction of informative non-rRNA reads from ~4-10% to 54-92%. The authors apply their protocol to investigating heterogeneity in a biofilm model of E. coli, and convincingly show how their technology can detect minority subpopulations within a complex community.
Strengths:
The method the authors propose is a straightforward and inexpensive modification of an established split-pool single-cell RNA-seq protocol that greatly increases its utility, and should be of interest to a wide community working in the field of bacterial single-cell RNA-seq.
Weaknesses:
The manuscript is written in a very compressed style and many technical details of the evaluations conducted are unclear and processed data has not been made available for evaluation, limiting the ability of the reader to independently judge the merits of the method.
Thank you for your thoughtful and constructive review of our manuscript. We appreciate your recognition of the strengths of our work and the potential impact of our modified PETRI-seq protocol on the field of bacterial single-cell RNA-seq. We are grateful for the opportunity to address your concerns and improve the clarity and accessibility of our manuscript.
We acknowledge your feedback regarding the compressed writing style and lack of technical details, which are constrained by the requirements of the Short Report format in eLife. We have addressed these issues in our revised manuscript as follows:
(1) Expanded methodology section: We have provided a more comprehensive description of our experimental procedures, including detailed protocols for the ribosomal depletion step (lines 435-453) and data analysis pipeline (lines 471-528). This will enable readers to better understand and potentially replicate our methods.
(2) Clarification of technical evaluations: We have elaborated on the specifics of our evaluations, including the criteria used for assessing the efficiency of ribosomal depletion (lines 99-120), and the methods employed for identifying and characterizing subpopulations (lines 155-159, 161-163 and 163-167).
(3) Data availability: We apologize for the oversight in not making our processed data readily available. We have deposited all relevant datasets, including raw and source data, in appropriate public repositories (GEO: GSE260458) and provide clear instructions for accessing this data in the revised manuscript.
(4) Supplementary information: To maintain the concise nature of the main text while providing necessary details, we have included additional supplementary information. This will cover extended methodology (lines 311-318, 321-323, 327-340, 450-453, 533, and 578-589), detailed statistical analyses (lines 492-493, 499-501 and 509-528), and comprehensive data tables to support our findings.
We believe these changes significantly improved the clarity and reproducibility of our work, allowing readers to better evaluate the merits of our method.
Reviewer #2 (Public Review):
Summary:
This work introduces a new method of depleting the ribosomal reads from the single-cell RNA sequencing library prepared with one of the prokaryotic scRNA-seq techniques, PETRI-seq. The advance is very useful since it allows broader access to the technology by lowering the cost of sequencing. It also allows more transcript recovery with fewer sequencing reads. The authors demonstrate the utility and performance of the method for three different model species and find a subpopulation of cells in the E.coli biofilm that express a protein, PdeI, which causes elevated c-di-GMP levels. These cells were shown to be in a state that promotes persister formation in response to ampicillin treatment.
Strengths:
The introduced rRNA depletion method is highly efficient, with the depletion for E.coli resulting in over 90% of reads containing mRNA. The method is ready to use with existing PETRI-seq libraries which is a large advantage, given that no other rRNA depletion methods were published for split-pool bacterial scRNA-seq methods. Therefore, the value of the method for the field is high. There is also evidence that a small number of cells at the bottom of a static biofilm express PdeI which is causing the elevated c-di-GMP levels that are associated with persister formation. Given that PdeI is a phosphodiesterase, which is supposed to promote hydrolysis of c-di-GMP, this finding is unexpected.
Weaknesses:
With the descriptions and writing of the manuscript, it is hard to place the findings about the PdeI into existing context (i.e. it is well known that c-di-GMP is involved in biofilm development and is heterogeneously distributed in several species' biofilms; it is also known that E.coli diesterases regulate this second messenger, i.e. https://journals.asm.org/doi/full/10.1128/jb.00604-15).
There is also no explanation for the apparently contradictory upregulation of c-di-GMP in cells expressing higher PdeI levels. Perhaps the examination of the rest of the genes in cluster 2 of the biofilm sample could be useful to explain the observed association.
Thank you for your thoughtful and constructive review of our manuscript. We are pleased that the reviewer recognizes the value and efficiency of our rRNA depletion method for PETRI-seq, as well as its potential impact on the field. We would like to address the points raised by the reviewer and provide additional context and clarification regarding the function of PdeI in c-di-GMP regulation.
We acknowledge that c-di-GMP’s role in biofilm development and its heterogeneous distribution in bacterial biofilms are well studied. We appreciate the reviewer's observation regarding the seemingly contradictory relationship between increased PdeI expression and elevated c-di-GMP levels. This is indeed an intriguing finding that warrants further explanation.
PdeI is predicted to function as a phosphodiesterase involved in c-di-GMP degradation, based on sequence analysis demonstrating the presence of an intact EAL domain, which is known for this function. However, it is important to note that PdeI also harbors a divergent GGDEF domain, typically associated with c-di-GMP synthesis. This dual-domain structure indicates that PdeI may play complex regulatory roles. Previous studies have shown that knocking out the major phosphodiesterase PdeH in E. coli results in the accumulation of c-di-GMP. Moreover, introducing a point mutation (G412S) in PdeI's divergent GGDEF domain within this PdeH knockout background led to decreased c-di-GMP levels2. This finding implies that the wild-type GGDEF domain in PdeI contributes to maintaining or increasing cellular c-di-GMP levels.
Importantly, our single-cell experiments demonstrated a positive correlation between PdeI expression levels and c-di-GMP levels (Figure 4D). In this revision, we also constructed a PdeI(G412S)-BFP mutation strain. Notably, our observations of this strain revealed that c-di-GMP levels remained constant despite an increase in BFP fluorescence, which serves as a proxy for PdeI(G412S) expression levels (Figure 4D). This experimental evidence, coupled with domain analyses, suggests that PdeI may also contribute to c-di-GMP synthesis, rebutting the notion that it acts solely as a phosphodiesterase. HPLC LC-MS/MS analysis further confirmed that the overexpression of PdeI, induced by arabinose, resulted in increased c-di-GMP levels (Fig. 4E) . These findings strongly suggest that PdeI plays a pivotal role in upregulating c-di-GMP levels.
Our further analysis indicated that PdeI contains a CHASE (cyclases/histidine kinase-associated sensory) domain. Combined with our experimental results showing that PdeI is a membrane-associated protein, we hypothesize that PdeI acts as a sensor, integrating environmental signals with c-di-GMP production under complex regulatory mechanisms.
We understand your interest in the other genes present in cluster 2 of the biofilm and their potential relationship to PdeI and c-di-GMP. Upon careful analysis, we have determined that the other marker genes in this cluster do not significantly impact biofilm formation, nor have we identified any direct relationship between these genes, c-di-GMP, or PdeI. Our focus on PdeI within this cluster is justified by its unique and significant role in c-di-GMP regulation and biofilm formation, as demonstrated by our experimental results. While other genes in this cluster may be co-expressed, their functions appear unrelated to the PdeI-c-di-GMP pathway we are investigating. Therefore, we opted not to elaborate on these genes in our main discussion, as they do not contribute directly to our understanding of the PdeI-c-di-GMP association. However, we can include a brief mention of these genes in the manuscript, indicating their lack of relevance to the PdeI-c-di-GMP pathway. This addition will provide a more comprehensive view of the cluster's composition while maintaining our focus on the key findings related to PdeI and c-di-GMP.
We have also included the aforementioned explanations and supporting experimental data within the manuscript to clarify this important point (lines 193-217). Thank you for highlighting this apparent contradiction, allowing us to provide a more detailed explanation of our findings.
Recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
Overall, I found the main text of the manuscript well written and easy to understand, though too compressed in parts to fully understand the details of the work presented, some examples are outlined below. The materials and methods appeared to be less carefully compiled and could use some careful proof-reading for spelling (e.g. repeated use of "minuts" for minutes, "datas" for data) and grammar and sentence fragments (e.g. "For exponential period E. coli data." Line 333). In general, the meaning is still clear enough to be understood. I also was unable to find figure captions for the supplementary figures, making these difficult to understand.
We appreciate your careful review, which has helped us improve the clarity and quality of our manuscript. We acknowledge that some parts of the main text may have been overly compressed due to Short Report format in eLife. We have thoroughly reviewed the manuscript and expanded on key areas to provide more comprehensive explanations. We have carefully revised the Materials and Methods section to address the following: Corrected all spelling and grammatical error, including "minuts" to "minutes" and "datas" to "data". Corrected grammatical issues and sentence fragments throughout the section. We sincerely apologize for the omission of captions for the supplementary figures. We have now added detailed captions for all supplementary figures to ensure they are easily understandable. We believe these revisions address your concerns and enhance the overall readability and comprehension of our work.
General comments:
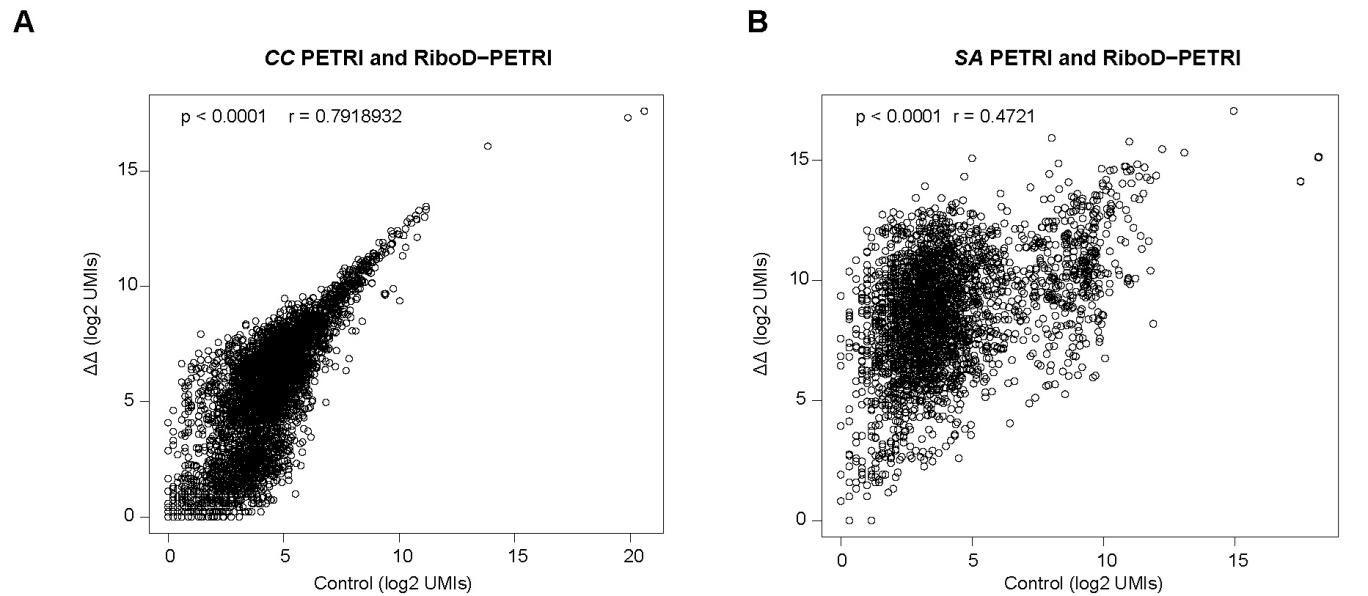
(1) To evaluate the performance of RiboD-PETRI, it would be helpful to have more details in general, particularly to do with the development of the sequencing protocol and the statistics shown. Some examples: How many reads were sequenced in each experiment? Of these, how many are mapped to the bacterial genome? How many reads were recovered per cell? Have the authors performed some kind of subsampling analysis to determine if their sequencing has saturated the detection of expressed genes? The authors show e.g. correlations between classic PETRI-seq and RiboD-PETRI for E. coli in Figure 1, but also have similar data for C. crescentus and S. aureus - do these data behave similarly? These are just a few examples, but I'm sure the authors have asked themselves many similar questions while developing this project; more details, hard numbers, and comparisons would be very much appreciated.
Thank you for your valuable feedback. To address your concerns, we have added a table in the supplementary material that clarifies the details of sequencing.
The correlation values of PETRI-seq and RiboD-PETRI data in C. crescentus are relatively good. However, the correlation values between PETRI-seq and RiboD-PETRI data in SA data are relatively less high. The reason is that the sequencing depths of RiboD-PETRI and PETRI-seq are different, resulting in much higher gene expression in the RiboD-PETRI sequencing results than in PETRI-seq, and the calculated correlation coefficient is only about 0.47. This indicates that there is some positive correlation between the two sets of data, but it is not particularly strong. This indicates that there is a certain positive correlation between these two sets of data, but it is not particularly strong. However, we have counted the expression of 2763 genes in total, and even though the calculated correlation coefficient is relatively low, it still shows that there is some consistency between the two groups of samples.
Author response image 1.
Assessment of the effect of rRNA depletion on transcriptional profiles of (A) C. crescentus (CC) and (B) S. aureus (SA) . The Pearson correlation coefficient (r) of UMI counts per gene (log2 UMIs) between RiboD-PETRI and PETRI-seq was calculated for 4097 genes (A) and 2763 genes (B). The "ΔΔ" label represents the RiboD-PETRI protocol; The "Ctrl" label represents the classic PETRI-seq protocol we performed. Each point represents a gene.
(2) Additionally, I think it is critical that the authors provide processed read counts per cell and gene in their supplementary information to allow others to investigate the performance of their method without going back to raw FASTQ files, as this can represent a significant hurdle for reanalysis.
Thank you for your suggestion. However, it's important to clarify that reads and UMIs (Unique Molecular Identifiers) are distinct concepts in single-cell RNA sequencing. Reads can be influenced by PCR amplification during library construction, making their quantity less stable. In contrast, UMIs serve as a more reliable indicator of the number of mRNA molecules detected after PCR amplification. Throughout our study, we primarily utilized UMI counts for quantification. To address your concern about data accessibility, we have included the UMI counts per cell and gene in our supplementary materials provided above (Table S7-15. Some of the files are too large in memory and are therefore stored in GEO: GSE260458). This approach provides a more accurate representation of gene expression levels and allows for robust reanalysis without the need to process raw FASTQ files.
(3) Finally, the authors should also discuss other approaches to ribosomal depletion in bacterial scRNA-seq. One of the figures appears to contain such a comparison, but it is never mentioned in the text that I can find, and one could read this manuscript and come away believing this is the first attempt to deplete rRNA from bacterial scRNA-seq.
We have addressed this concern by including a comparison of different methods for depleting rRNA from bacterial scRNA-seq in Table S4 and make a short text comparison as follows: “Additionally, we compared our findings with other reported methods (Fig. 1B; Table S4). The original PETRI-seq protocol, which does not include an rRNA depletion step, exhibited an mRNA detection rate of approximately 5%. The MicroSPLiT-seq method, which utilizes Poly A Polymerase for mRNA enrichment, achieved a detection rate of 7%. Similarly, M3-seq and BacDrop-seq, which employ RNase H to digest rRNA post-DNA probe hybridization in cells, reported mRNA detection rates of 65% and 61%, respectively. MATQ-DASH, which utilizes Cas9-mediated targeted rRNA depletion, yielded a detection rate of 30%. Among these, RiboD-PETRI demonstrated superior performance in mRNA detection while requiring the least sequencing depth.” We have added this content in the main text (lines 110-120), specifically in relation to Figure 1B and Table S4. This addition provides context for our method and clarifies its position among existing techniques.
Detailed comments:
Line 78: the authors describe the multiplet frequency, but it is not clear to me how this was determined, for which experiments, or where in the SI I should look to see this. Often this is done by mixing cultures of two distinct bacteria, but I see no evidence of this key experiment in the manuscript.
The multiplet frequency we discuss in the manuscript is not determined through experimental mixing of distinct bacterial cultures.The PETRI-seq and mirco-SPLIT articles have also done experiments mixing the two libraries to determine the single-cell rate, and both gave good results. Our technique is derived from these two articles (mainly PETRI-seq), and the biggest difference is the difference in the later RiboD part, so we did not do this experiment separately. So the multiple frequencies here are theoretical predictions based on our sequencing results, calculated using a Poisson distribution. We have made this distinction clearer in our manuscript (lines 93-97). The method is available in Materials and Methods section (lines 520-528). The data is available in Table S2. To elaborate:
To assess the efficiency of single-cell capture in RiboD-PETRI, we calculated the multiplet frequency using a Poisson distribution based on our sequencing results
(1) Definition: In our study, multiplet frequency is defined as the probability of a non-empty barcode corresponding to more than one cell.
(2) Calculation Method: We use a Poisson distribution-based approach to calculate the predicted multiplet frequency. The process involves several steps:
We first calculate the proportion of barcodes corresponding to zero cells:
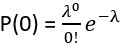 . Then, we calculate the proportion corresponding to one cell:
. Then, we calculate the proportion corresponding to one cell: 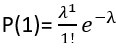 . We derive the proportion for more than zero cells: P(≥1) = 1 - P(0). And for more than one cell: P(≥2) = 1 - P(1) - P(0). Finally, the multiplet frequency is calculated as:
. We derive the proportion for more than zero cells: P(≥1) = 1 - P(0). And for more than one cell: P(≥2) = 1 - P(1) - P(0). Finally, the multiplet frequency is calculated as: 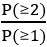
(3) Parameter λ: This is the ratio of the number of cells to the total number of possible barcode combinations. For instance, when detecting 10,000 cells,
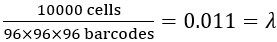 .
.Line 94: the concept of "percentage of gene expression" is never clearly defined. Does this mean the authors detect 99.86% of genes expressed in some cells? How is "expressed" defined - is this just detecting a single UMI?
The term "percentage gene expression" refers to the proportion of genes in the bacterial strain that were detected as expressed in the sequenced cell population. Specifically, in this context, it means that 99.86% of all genes in the bacterial strain were detected as expressed in at least one cell in our sequencing results. To define "expressed" more clearly: a gene is considered expressed if at least one UMI (Unique Molecular Identifier) detected in a cell in the population. This definition allows for the detection of even low-level gene expression. To enhance clarity in the manuscript, we have rephrased the sentence as “transcriptome-wide gene coverage across the cell population”.
Line 98: The authors discuss the number of recovered UMIs throughout this paragraph, but there is no clear discussion of the number of detected expressed genes per cell. Could the authors include a discussion of this as well, as this is another important measure of sensitivity?
We appreciate your suggestion to include a discussion on the number of detected expressed genes per cell, as this is indeed another important measure of sensitivity. We would like to clarify that we have actually included statistics on the number of genes detected across all cells in the main text of our paper. This information is presented as percentages. However, we understand that you may be looking for a more detailed representation, similar to the UMI statistics we provided. To address this, we have now added a new analysis showing the number of genes detected per cell (lines 132-133, 138-139, 144-145 and 184-186, Fig. 2B, 3B and S2B). This additional result complements our existing UMI data and provides a more comprehensive view of the sensitivity of our method. We have included this new gene-per-cell statistical graph in the supplementary materials.
Figure 1B: I presume ctrl and delta delta represent the classic PETRI-seq and RiboD protocols, respectively, but this is not specified. This should be clarified in the figure caption, or the names changed.
We appreciate you bringing this to our attention. We acknowledge that the labeling in the figure could have been clearer. We have now clarified this information in the figure caption. To provide more specificity: The "ΔΔ" label represents the RiboD-PETRI protocol; The "Ctrl" label represents the classic PETRI-seq protocol we performed. We have updated the figure caption to include these details, which should help readers better understand the protocols being compared in the figure.
Line 104: the authors claim "This performance surpassed other reported bacterial scRNA-seq methods" with a long number of references to other methods. "Performance" is not clearly defined, and it is unclear what the exact claim being made is. The authors should clarify what they're claiming, and further discuss the other methods and comparisons they have made with them in a thorough and fair fashion.
We appreciate your request for clarification, and we acknowledge that our definition of "performance" should have been more explicit. We would like to clarify that in this context, we define performance primarily in terms of the proportion of mRNA captured. Our improved method demonstrates a significantly higher rate of rRNA removal compared to other bacterial single-cell library construction methods. This results in a higher proportion of mRNA in our sequencing data, which we consider a key performance metric for single-cell RNA sequencing in bacteria. Additionally, when compared to our previous method, PETRI-seq, our improved approach not only enhances rRNA removal but also reduces library construction costs. This dual improvement in both data quality and cost-effectiveness is what we intended to convey with our performance claim.
We recognize that a more thorough and fair discussion of other methods and their comparisons would be beneficial. We have summarized the comparison in Table S4 and make a short text discussion in the main text (lines 106-120). This addition provides context for our method and clarifies its position among existing techniques.
Figure 1D: Do the authors have any explanation for the relatively lower performance of their C. crescentus depletion?
We appreciate your attention to detail and the opportunity to address this point. The lower efficiency of rRNA removal in C. crescentus compared to other species can be attributed to inherent differences between species. It's important to note that a single method for rRNA depletion may not be universally effective across all bacterial species due to variations in their genetic makeup and rRNA structures. Different bacterial species can have unique rRNA sequences, secondary structures, or associated proteins that may affect the efficiency of our depletion method. This species-specific variation highlights the challenges in developing a one-size-fits-all approach for bacterial rRNA depletion. While our method has shown high efficiency across several species, the results with C. crescentus underscore the need for continued refinement and possibly species-specific optimizations in rRNA depletion techniques. We thank you for bringing attention to this point, as it provides valuable insight into the complexities of bacterial rRNA depletion and areas for future improvement in our method.
Line 118: The authors claim RiboD-PETRI has a "consistent ability to unveil within-population heterogeneity", however the preceding paragraph shows it detects potential heterogeneity, but provides no evidence this inferred heterogeneity reflects the reality of gene expression in individual cells.
We appreciate your careful reading and the opportunity to clarify this point. We acknowledge that our wording may have been too assertive given the evidence presented. We acknowledge that the subpopulations of cells identified in other species have not undergone experimental verification. Our intention in presenting these results was to demonstrate RiboD-PETRI's capability to detect “potential” heterogeneity consistently across different bacterial species, showcasing the method's sensitivity and potential utility in exploring within-population diversity. However, we agree that without further experimental validation, we cannot definitively claim that these detected differences represent true biological heterogeneity in all cases. We have revised this section to reflect the current state of our findings more accurately, emphasizing that while RiboD-PETRI consistently detects potential heterogeneity across species, further experimental validation would be required to confirm the biological significance of the observations (lines 169-171).
Figure 1 H&I: I'm not entirely sure what I am meant to see in these figures, presumably some evidence for heterogeneity in gene expression. Are there better visualizations that could be used to communicate this?
We appreciate your suggestion for improving the visualization of gene expression heterogeneity. We have explored alternative visualization methods in the revised manuscript. Specifically, for the expression levels of marker genes shown in Figure 1H (which is Figure 2D now), we have created violin plots (Supplementary Fig. 4). These plots offer a more comprehensive view of the distribution of expression levels across different cell populations, making it easier to discern heterogeneity. However, due to the number of marker genes and the resulting volume of data, these violin plots are quite extensive and would occupy a significant amount of space. Given the space constraints of the main figure, we propose to include these violin plots as a Fig. S4 immediately following Figure 1 H&I (which is Figure 2D&E now). This arrangement will allow readers to access more detailed information about these marker genes while maintaining the concise style of the main figure.
Regarding the pathway enrichment figure (Figure 2E), we have also considered your suggestion for improvement. We attempted to use a dot plot to display the KEGG pathway enrichment of the genes. However, our analysis revealed that the genes were only enriched in a single pathway. As a result, the visual representation using a dot plot still did not produce a particularly aesthetically pleasing or informative figure.
Line 124: The authors state no significant batch effect was observed, but in the methods on line 344 they specify batch effects were removed using Harmony. It's unclear what exactly S2 is showing without a figure caption, but the authors should clarify this discrepancy.
We apologize for any confusion caused by the lack of a clear figure caption for Figure S2 (which is Figure S3D now). To address your concern, in addition to adding figure captions for supplementary figure, we would also like to provide more context about the batch effect analysis. In Supplementary Fig. S3, Panel C represents the results without using Harmony for batch effect removal, while Panel D shows the results after applying Harmony. In both panels A and B, the distribution of samples one and two do not show substantial differences. Based on this observation, we concluded that there was no significant batch effect between the two samples. However, we acknowledge that even subtle batch effects could potentially influence downstream analyses. Therefore, out of an abundance of caution and to ensure the highest quality of our results, we decided to apply Harmony to remove any potential minor batch effects. This approach aligns with best practices in single-cell analysis, where even small technical variations are often accounted for to enhance the robustness of the results.
To improve clarity, we have revised our manuscript to better explain this nuanced approach: 1. We have updated the statement to reflect that while no major batch effect was observed, we applied batch correction as a precautionary measure (lines 181-182). 2. We have added a detailed caption to Figure S3, explaining the comparison between non-corrected and batch-corrected data. 3. We have modified the methods section to clarify that Harmony was applied as a precautionary step, despite the absence of obvious batch effects (lines 492-493).
Figure 2D: I found this panel fairly uninformative, is there a better way to communicate this finding?
Thank you for your feedback regarding Figure 2D. We have explored alternative ways to present this information, using a dot plot to display the enrichment pathways, as this is often an effective method for visualizing such data. Meanwhile, we also provided a more detailed textual description of the enrichment results in the main text, highlighting the most significant findings.
Figure 2I: the figure itself and caption say GFP, but in the text and elsewhere the authors say this is a BFP fusion.
We appreciate your careful review of our manuscript and figures. We apologize for any confusion this may have caused. To clarify: Both GFP (Green Fluorescent Protein) and BFP (Blue Fluorescent Protein) were indeed used in our experiments, but for different purposes: 1. GFP was used for imaging to observe location of PdeI in bacteria and persister cell growth, which is shown in Figure 4C and 4K. 2. BFP was used for cell sorting, imaging of location in biofilm, and detecting the proportion of persister cells which shown in Figure 4D, 4F-J. To address this inconsistency and improve clarity, we will make the following corrections: 1. We have reviewed the main text to ensure that references to GFP and BFP are accurate and consistent with their respective uses in our experiments. 2. We have added a note in the figure caption for Figure 4C to explicitly state that this particular image shows GFP fluorescence for location of PdeI. 3. In the methods section, we have provided a clear explanation of how both fluorescent proteins were used in different aspects of our study (lines 326-340).
Line 156: The authors compare prices between RiboD and PETRI-seq. It would be helpful to provide a full cost breakdown, e.g. in supplementary information, as it is unclear exactly how the authors came to these numbers or where the major savings are (presumably in sequencing depth?)
We appreciate your suggestion to provide a more detailed cost breakdown, and we agree that this would enhance the transparency and reproducibility of our cost analysis. In response to your feedback, we have prepared a comprehensive cost breakdown that includes all materials and reagents used in the library preparation process. Additionally, we've factored in the sequencing depth (50G) and the unit price for sequencing (25¥/G). These calculations allow us to determine the cost per cell after sequencing. As you correctly surmised, a significant portion of the cost reduction is indeed related to sequencing depth. However, there are also savings in the library preparation steps that contribute to the overall cost-effectiveness of our method. We propose to include this detailed cost breakdown as a supplementary table (Table S6) in our paper. This table will provide a clear, itemized list of all expenses involved, including: 1. Reagents and materials for library preparation 2. Sequencing costs (depth and price per G) 3. Calculated cost per cell.
Line 291: The design and production of the depletion probes are not clearly explained. How did the authors design them? How were they synthesized? Also, it appears the authors have separate probe sets for E. coli, C. crescentus, and S. aureus - this should be clarified, possibly in the main text.
Thank you for your important questions regarding the design and production of our depletion probes. We included the detailed probe information in Supplementary Table S1, however, we didn’t clarify the information in the main text due to the constrains of the requirements of the Short Report format in eLife. We appreciate the opportunity to provide clarifications.
The core principle behind our probe design is that the probe sequences are reverse complementary to the r-cDNA sequences. This design allows for specific recognition of r-cDNA. The probes are then bound to magnetic beads, allowing the r-cDNA-probe-bead complexes to be separated from the rest of the library. To address your specific questions: 1. Probe Design: We designed separate probe sets for E. coli, C. crescentus, and S. aureus. Each set was specifically constructed to be reverse complementary to the r-cDNA sequences of its respective bacterial species. This species-specific approach ensures high efficiency and specificity in rRNA depletion for each organism. The hybrid DNA complex wasthen removed by Streptavidin magnetic beads. 2. Probe Synthesis: The probes were synthesized based on these design principles. 3. Species-Specific Probe Sets: You are correct in noting that we used separate probe sets for each bacterial species. We have clarified this important point in the main text to ensure readers understand the specificity of our approach. To further illustrate this process, we have created a schematic diagram showing the principle of rRNA removal and clarified the design principle in figure legend, which we have included in the figure legend of Fig. 1A.
Line 362: I didn't see a description of the construction of the PdeI-BFP strain, I assume this would be important for anyone interested in the specific work on PdeI.
Thank you for your astute observation regarding the construction of the PdeI-BFP strain. We appreciate the opportunity to provide this important information. The PdeI-BFP strain was constructed as follows: 1. We cloned the pdeI gene along with its native promoter region (250bp) into a pBAD vector. 2. The original promoter region of the pBAD vector was removed to avoid any potential interference. 3. This construction enables the expression of the PdeI-BFP fusion protein to be regulated by the native promoter of pdeI, thus maintaining its physiological control mechanisms. 4. The BFP coding sequence was fused to the pdeI gene to create the PdeI-BFP fusion construct. We have added a detailed description of the PdeI-BFP strain construction to our methods section (lines 327-334).
Reviewer #2 (Recommendations For The Authors):
(1) General remarks:
Reconsider using 'advanced' in the title. It is highly generic and misleading. Perhaps 'cost-efficient' would be a more precise substitute.
Thank you for your valuable suggestion. After careful consideration, we have decided to use "improved" in the title. Firstly, our method presents an efficient solution to a persistent challenge in bacterial single-cell RNA sequencing, specifically addressing rRNA abundance. Secondly, it facilitates precise exploration of bacterial population heterogeneity. We believe our method encompasses more than just cost-effectiveness, justifying the use of the term "advanced."
Consider expanding the introduction. The introduction does not explain the setup of the biological question or basic details such as the organism(s) for which the technique has been developed, or which species biofilms were studied.
Thank you for your valuable feedback regarding our introduction. We acknowledge our compressed writing style due to constrains of the requirements of the Short Report format in eLife. We appreciate opportunity to expand this crucial section of our manuscript, which will undoubtedly improve the clarity and impact of our manuscript's introduction.
We revised our introduction (lines 53-80) according to following principles:
(1) Initial Biological Question: We explained the initial biological question that motivated our research—understanding the heterogeneity in E. coli biofilms—to provide essential context for our technological development.
(2) Limitations of Existing Techniques: We briefly described the limitations of current single-cell sequencing techniques for bacteria, particularly regarding their application in biofilm studies.
(3) Introduction of Improved Technique: We introduced our improved technique, initially developed for E. coli.
(4) Research Evolution: We highlighted how our research has evolved, demonstrating that our technique is applicable not only to E. coli but also to Gram-positive bacteria and other Gram-negative species, showcasing the broad applicability of our method.
(5) Specific Organisms Studied: We provided examples of the specific organisms we studied, encompassing both Gram-positive and Gram-negative bacteria.
(6) Potential Implications: Finally, we outlined the potential implications of our technique for studying bacterial heterogeneity across various species and contexts, extending beyond biofilms.
(2) Writing remarks:
43-45 Reword: "Thus, we address a persistent challenge in bacterial single-cell RNA-seq regarding rRNA abundance, exemplifying the utility of this method in exploring biofilm heterogeneity.".
Thank you for highlighting this sentence and requesting a rewording. I appreciate the opportunity to improve the clarity and impact of our statement. We have reworded the sentence as: "Our method effectively tackles a long-standing issue in bacterial single-cell RNA-seq: the overwhelming abundance of rRNA. This advancement significantly enhances our ability to investigate the intricate heterogeneity within biofilms at unprecedented resolution." (lines 47-50)
49 "Biofilms, comprising approximately 80% of chronic and recurrent microbial infections in the human body..." - probably meant 'contribute to'.
Thank you for catching this imprecision in our statement. We have reworded the sentence as: "Biofilms contribute to approximately 80% of chronic and recurrent microbial infections in the human body..."
54-55 Please expand on "this".
Thank you for your request to expand on the use of "this" in the sentence. You're right that more clarity would be beneficial here. We have revised and expanded this section in lines 54-69.
81-84 Unclear why these species samples were either at exponential or stationary phases. The growth stage can influence the proportion of rRNA and other transcripts in the population.
Thank you for raising this important point about the growth phases of the bacterial samples used in our study. We appreciate the opportunity to clarify our experimental design. To evaluate the performance of RiboD-PETRI, we designed a comprehensive assessment of rRNA depletion efficiency under diverse physiological conditions, specifically contrasting exponential and stationary phases. This approach allows us to understand how these different growth states impact rRNA depletion efficacy. Additionally, we included a variety of bacterial species, encompassing both gram-negative and gram-positive organisms, to ensure that our findings are broadly applicable across different types of bacteria. By incorporating these variables, we aim to provide insights into the robustness and reliability of the RiboD-PETRI method in various biological contexts. We have included this rationale in our result section (lines 99-106), providing readers with a clear understanding of our experimental design choices.
86 "compared TO PETRI-seq " (typo).
We have corrected this typo in our manuscript.
94 "gene expression collectively" rephrase. Probably this means coverage of the entire gene set across all cells. Same for downstream usage of the phrase.
Thank you for pointing out this ambiguity in our phrasing. Your interpretation of our intended meaning is accurate. We have rephrased the sentence as “transcriptome-wide gene coverage across the cell population”.
97 What were the median UMIs for the 30,000 cell library {greater than or equal to}15 UMIs? Same question for the other datasets. This would reflect a more comparable statistic with previous studies than the top 3% of the cells for example, since the distributions of the single-cell UMIs typically have a long tail.
Thank you for this insightful question and for pointing out the importance of providing more comparable statistics. We agree that median values offer a more robust measure of central tendency, especially for datasets with long-tailed distributions, which are common in single-cell studies. The suggestion to include median Unique Molecular Identifier (UMI) counts would indeed provide a more comparable statistic with previous studies. We have analyzed the median UMIs for our libraries as follows and revised our manuscript according to the analysis (lines 126-130, 133-136, 139-142 and 175-180).
(1) Median UMI count in Exponential Phase E. coli:
Total: 102 UMIs per cell
Top 1,000 cells: 462 UMIs per cell
Top 5,000 cells: 259 UMIs per cell
Top 10,000 cells: 193 UMIs per cell
(2) Median UMI count in Stationary Phase S. aureus:
Total: 142 UMIs per cell
Top 1,000 cells: 378 UMIs per cell
Top 5,000 cells: 207 UMIs per cell
Top 8,000 cells: 167 UMIs per cell
(3) Median UMI count in Exponential Phase C. crescentus:
Total: 182 UMIs per cell
Top 1,000 cells: 2,190 UMIs per cell
Top 5,000 cells: 662 UMIs per cell
Top 10,000 cells: 225 UMIs per cell
(4) Median UMI count in Static E. coli Biofilm:
Total of Replicate 1: 34 UMIs per cell
Total of Replicate 2: 52 UMIs per cell
Top 1,621 cells of Replicate 1: 283 UMIs per cell
Top 3,999 cells of Replicate 2: 239 UMIs per cell
104-105 The performance metric should again be the median UMIs of the majority of the cells passing the filter (15 mRNA UMIs is reasonable). The top 3-5% are always much higher in resolution because of the heavy tail of the single-cell UMI distribution. It is unclear if the performance surpasses the other methods using the comparable metric. Recommend removing this line.
We appreciate your suggestion regarding the use of median UMIs as a more appropriate performance metric, and we agree that comparing the top 3-5% of cells can be misleading due to the heavy tail of the single-cell UMI distribution. We have removed the line in question (104-105) that compares our method's performance based on the top 3-5% of cells in the revised manuscript. Instead, we focused on presenting the median UMI counts for cells passing the filter (≥15 mRNA UMIs) as the primary performance metric. This will provide a more representative and comparable measure of our method's performance. We have also revised the surrounding text to reflect this change, ensuring that our claims about performance are based on these more robust statistics (lines 126-130, 133-136, 139-142 and 175-180).
106-108 The sequencing saturation of the libraries (in %), and downsampling analysis should be added to illustrate this point.
Thank you for your valuable suggestion. Your recommendation to add sequencing saturation and downsampling analysis is highly valuable and will help better illustrate our point. Based on your feedback, we have revised our manuscript by adding the following content:
To provide a thorough evaluation of our sequencing depth and library quality, we performed sequencing saturation analysis on our sequencing samples. The findings reveal that our sequencing saturation is 100% (Fig. 8A & B), indicating that our sequencing depth is sufficient to capture the diversity of most transcripts. To further illustrate the impact of our downstream analysis on the datasets, we have demonstrated the data distribution before and after applying our filtering criteria (Fig. S1B & C). These figures effectively visualized the influence of our filtering process on the data quality and distribution. After filtering, we can have a more refined dataset with reduced noise and outliers, which enhances the reliability of our downstream analyses.
We have also ensured that a detailed description of the sequencing saturation method is included in the manuscript to provide readers with a comprehensive understanding of our methodology. We appreciate your feedback and believe these additions significantly improve our work.
122: Please provide more details about the biofilm setup, including the media used. I did not find them in the methods.
We appreciate your attention to detail, and we agree that this information is crucial for the reproducibility of our experiments. We propose to add the following information to our methods section (lines 311-318):
"For the biofilm setup, bacterial cultures were grown overnight. The next day, we diluted the culture 1:100 in a petri dish. We added 2ml of LB medium to the dish. If the bacteria contain a plasmid, the appropriate antibiotic needs to be added to LB. The petri dish was then incubated statically in a growth chamber for 24 hours. After incubation, we performed imaging directly under the microscope. The petri dishes used were glass-bottom dishes from Biosharp (catalog number BS-20-GJM), allowing for direct microscopic imaging without the need for cover slips or slides. This setup allowed us to grow and image the biofilms in situ, providing a more accurate representation of their natural structure and composition."
125: "sequenced 1,563 reads" missing "with"
Thank you for correcting our grammar. We have revisd the phrase as “sequenced with 1,563 reads”.
126: "283/239 UMIs per cell" unclear. 283 and 239 UMIs per cell per replicate, respectively?
Thank you for correcting our grammar. We have revised the phrase as “283 and 239 UMIs per cell per replicate, respectively” (lines 184).
Figure 1D: Please indicate where the comparison datasets are from.
We appreciate your question regarding the source of the comparison datasets in Figure 1D. All data presented in Figure 1D are from our own sequencing experiments. We did not use data from other publications for this comparison. Specifically, we performed sequencing on E. coli cells in the exponential growth phase using three different library preparation methods: RiboD-PETRI, PETRI-seq, and RNA-seq. The data shown in Figure 1D represent a comparison of UMIs and/or reads correlations obtained from these three methods. All sequencing results have been uploaded to the Gene Expression Omnibus (GEO) database. The accession number is GSE260458. We have updated the figure legend for Figure 1D to clearly state that all datasets are from our own experiments, specifying the different methods used.
Figure 1I, 2D: Unable to interpret the color block in the data.
We apologize for any confusion regarding the interpretation of the color blocks in Figures 1I and 2D (which are Figure 2E, 3E now). The color blocks in these figures represent the p-values of the data points. The color scale ranges from red to blue. Red colors indicate smaller p-values, suggesting higher statistical significance and more reliable results. Blue colors indicate larger p-values, suggesting lower statistical significance and less reliable results. We have updated the figure legends for both Figure 2E and Figure 3E to include this explanation of the color scale. Additionally, we have added a color legend to each figure to make the interpretation more intuitive for readers.
Figure1H and 2C: Gene names should be provided where possible. The locus tags are highly annotation-dependent and hard to interpret. Also, a larger size figure should be helpful. The clusters 2 and 3 in 2C are the most important, yet because they have few cells, very hard to see in this panel.
We appreciate your suggestions for improving the clarity and interpretability of Figures 1H and 2C (which is Figure 2D, 3D now). We have replaced the locus tags with gene names where possible in both figures. We have increased the size of both figures to improve visibility and readability. We have also made Clusters 2 and 3 in Figure 3D more prominent in the revised figure. Despite their smaller cell count, we recognize their importance and have adjusted the visualization to ensure they are clearly visible. We believe these modifications will significantly enhance the clarity and informativeness of Figures 2D and 3D.
(3) Questions to consider further expanding on, by more analyses or experiments and in the discussion:
What are the explanations for the apparently contradictory upregulation of c-di-GMP in cells expressing higher PdeI levels? How could a phosphodiesterase lead to increased c-di-GMP levels?
We appreciate the reviewer's observation regarding the seemingly contradictory relationship between increased PdeI expression and elevated c-di-GMP levels. This is indeed an intriguing finding that warrants further explanation.
PdeI was predicted to be a phosphodiesterase responsible for c-di-GMP degradation. This prediction is based on sequence analysis where PdeI contains an intact EAL domain known for degrading c-di-GMP. However, it is noteworthy that PdeI also contains a divergent GGDEF domain, which is typically associated with c-di-GMP synthesis (Fig S8). This dual-domain architecture suggests that PdeI may engage in complex regulatory roles. Previous studies have shown that the knockout of the major phosphodiesterase PdeH in E. coli leads to the accumulation of c-di-GMP. Further, a point mutation on PdeI's divergent GGDEF domain (G412S) in this PdeH knockout strain resulted in decreased c-di-GMP levels2, implying that the wild-type GGDEF domain in PdeI contributes to the maintenance or increase of c-di-GMP levels in the cell. Importantly, our single-cell experiments showed a positive correlation between PdeI expression levels and c-di-GMP levels (Response Fig. 9B). In this revision, we also constructed PdeI(G412S)-BFP mutation strain. Notably, our observations of this strain revealed that c-di-GMP levels remained constant despite increasing BFP fluorescence, which serves as a proxy for PdeI(G412S) expression levels (Fig. 4D). This experimental evidence, along with domain analysis, suggests that PdeI could contribute to c-di-GMP synthesis, rebutting the notion that it solely functions as a phosphodiesterase. HPLC LC-MS/MS analysis further confirmed that PdeI overexpression, induced by arabinose, led to an upregulation of c-di-GMP levels (Fig. 4E). These results strongly suggest that PdeI plays a significant role in upregulating c-di-GMP levels. Our further analysis revealed that PdeI contains a CHASE (cyclases/histidine kinase-associated sensory) domain. Combined with our experimental results demonstrating that PdeI is a membrane-associated protein, we hypothesize that PdeI functions as a sensor that integrates environmental signals with c-di-GMP production under complex regulatory mechanisms.
We have also included this explanation (lines 193-217) and the supporting experimental data (Fig. 4D & 4J) in our manuscript to clarify this important point. Thank you for highlighting this apparent contradiction, as it has allowed us to provide a more comprehensive explanation of our findings.
What about the rest of the genes in cluster 2 of the biofilm? They should be used to help interpret the association between PdeI and c-di-GMP.
We understand your interest in the other genes present in cluster 2 of the biofilm and their potential relationship to PdeI and c-di-GMP. After careful analysis, we have determined that the other marker genes in this cluster do not have a significant impact on biofilm formation. Furthermore, we have not found any direct relationship between these genes and c-di-GMP or PdeI. Our focus on PdeI in this cluster is due to its unique and significant role in c-di-GMP regulation and biofilm formation, as demonstrated by our experimental results. While the other genes in this cluster may be co-expressed, their functions appear to be unrelated to the PdeI and c-di-GMP pathway we are investigating. We chose not to elaborate on these genes in our main discussion as they do not contribute directly to our understanding of the PdeI and c-di-GMP association. Instead, we could include a brief mention of these genes in the manuscript, noting that they were found to be unrelated to the PdeI-c-di-GMP pathway. This would provide a more comprehensive view of the cluster composition while maintaining focus on the key findings related to PdeI and c-di-GMP.
Author response image 2.
Protein-protein interactions of marker genes in cluster 2 of 24-hour static biofilms of E coli data.
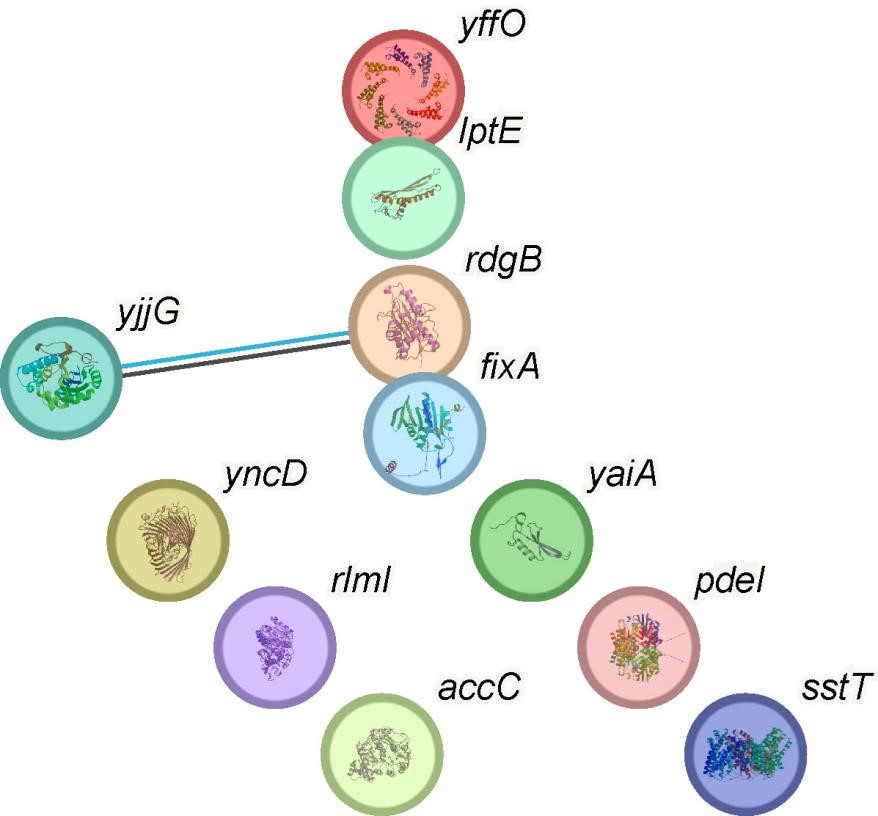
A verification is needed that the protein fusion to PdeI functional/membrane localization is not due to protein interactions with fluorescent protein fusion.
We appreciate your concern regarding the potential impact of the fluorescent protein fusion on the functionality and membrane localization of PdeI. It is crucial to verify that the observed effects are attributable to PdeI itself and not an artifact of its fusion with the fluorescent protein. To address this matter, we have incorporated a control group expressing only the fluorescent protein BFP (without the PdeI fusion) under the same promoter. This experimental design allows us to differentiate between effects caused by PdeI and those potentially arising from the fluorescent protein alone.
Our results revealed the following key observations:
(1) Cellular Localization: The GFP alone exhibited a uniform distribution in the cytoplasm of bacterial cells, whereas the PdeI-GFP fusion protein was specifically localized to the membrane (Fig. 4C).
(2) Localization in the Biofilm Matrix: BFP-positive cells were distributed throughout the entire biofilm community. In contrast, PdeI-BFP positive cells localized at the bottom of the biofilm, where cell-surface adhesion occurs (Fig 4F).
(3) c-di-GMP Levels: Cells with high levels of BFP displayed no increase in c-di-GMP levels. Conversely, cells with high levels of PdeI-BFP exhibited a significant increase in c-di-GMP levels (Fig. 4D).
(4) Persister Cell Ratio: Cells expressing high levels of BFP showed no increase in persister ratios, while cells with elevated levels of PdeI-BFP demonstrated a marked increase in persister ratios (Fig. 4J).
These findings from the control experiments have been included in our manuscript (lines 193-244, Fig. 4C, 4D, 4F, 4G and 4J), providing robust validation of our results concerning the PdeI fusion protein. They confirm that the observed effects are indeed due to PdeI and not merely artifacts of the fluorescent protein fusion.
(!) Vrabioiu, A. M. & Berg, H. C. Signaling events that occur when cells of Escherichia coli encounter a glass surface. Proceedings of the National Academy of Sciences of the United States of America 119, doi:10.1073/pnas.2116830119 (2022). https://doi.org/10.1073/pnas.2116830119
(2)bReinders, A. et al. Expression and Genetic Activation of Cyclic Di-GMP-Specific Phosphodiesterases in Escherichia coli. J Bacteriol 198, 448-462 (2016). https://doi.org:10.1128/JB.00604-15
-
eLife Assessment
This work presents an important method for depleting ribosomal RNA from bacterial single-cell RNA sequencing libraries, enabling the study of cellular heterogeneity within microbial biofilms. The approach convincingly identifies a small subpopulation of cells at the biofilm's base with upregulated PdeI expression, offering invaluable insights into the biology of bacterial biofilms and the formation of persister cells. Further integrated analysis of gene interactions within these datasets could deepen our understanding of biofilm dynamics and resilience.
-
Reviewer #1 (Public review):
Summary:
In this manuscript, Yan and colleagues introduce a modification to the previously published PETRI-seq bacterial single cell protocol to include a ribosomal depletion step based on a DNA probe set that selectively hybridizes with ribosome-derived (rRNA) cDNA fragments. They show that their modification of the PETRI-seq protocol increases the fraction of informative non-rRNA reads from ~4-10% to 54-92%. The authors apply their protocol to investigating heterogeneity in a biofilm model of E. coli, and convincingly show how their technology can detect minority subpopulations within a complex community.
Strengths:
The method the authors propose is a straightforward and inexpensive modification of an established split-pool single cell RNA-seq protocol that greatly increases its utility, and should be of …
Reviewer #1 (Public review):
Summary:
In this manuscript, Yan and colleagues introduce a modification to the previously published PETRI-seq bacterial single cell protocol to include a ribosomal depletion step based on a DNA probe set that selectively hybridizes with ribosome-derived (rRNA) cDNA fragments. They show that their modification of the PETRI-seq protocol increases the fraction of informative non-rRNA reads from ~4-10% to 54-92%. The authors apply their protocol to investigating heterogeneity in a biofilm model of E. coli, and convincingly show how their technology can detect minority subpopulations within a complex community.
Strengths:
The method the authors propose is a straightforward and inexpensive modification of an established split-pool single cell RNA-seq protocol that greatly increases its utility, and should be of interest to a wide community working in the field of bacterial single cell RNA-seq.
-
Reviewer #2 (Public review):
Summary:
This work introduces a new method of depleting the ribosomal reads from the single-cell RNA sequencing library prepared with one of the prokaryotic scRNA-seq techniques, PETRI-seq. The advance is very useful since it allows broader access to the technology by lowering the cost of sequencing. It also allows more transcript recovery with fewer sequencing reads. The authors demonstrate the utility and performance of the method for three different model species and find a subpopulation of cells in the E.coli biofilm that express a protein, PdeI, which causes elevated c-di-GMP levels. These cells were shown to be in a state that promotes persister formation in response to ampicillin treatment.
Strengths:
The introduced rRNA depletion method is highly efficient, with the depletion for E.coli resulting in …
Reviewer #2 (Public review):
Summary:
This work introduces a new method of depleting the ribosomal reads from the single-cell RNA sequencing library prepared with one of the prokaryotic scRNA-seq techniques, PETRI-seq. The advance is very useful since it allows broader access to the technology by lowering the cost of sequencing. It also allows more transcript recovery with fewer sequencing reads. The authors demonstrate the utility and performance of the method for three different model species and find a subpopulation of cells in the E.coli biofilm that express a protein, PdeI, which causes elevated c-di-GMP levels. These cells were shown to be in a state that promotes persister formation in response to ampicillin treatment.
Strengths:
The introduced rRNA depletion method is highly efficient, with the depletion for E.coli resulting in over 90% of reads containing mRNA. The method is ready to use with existing PETRI-seq libraries which is a large advantage, given that no other rRNA depletion methods were published for split-pool bacterial scRNA-seq methods. Therefore, the value of the method for the field is high. There is also evidence that a small number of cells at the bottom of a static biofilm express PdeI which is causing the elevated c-di-GMP levels that are associated with persister formation. This finding highlights the potentially complex role of PdeI in regulation of c-di-GMP levels and persister formation in microbial biofilms.
Weaknesses:
Given many current methods that also introduce different techniques for ribosomal RNA depletion in bacterial single-cell RNA sequencing, it is unclear what is the place and role of RiboD-PETRI. The efficiency of rRNA depletion varies greatly between species for the majority of the available methods, so it is not easy to select the best fitting technique for a specific application.
Despite transcriptome-wide coverage, the authors focused on the role of a single heterogeneously expressed gene, PdeI. A more integrated analysis of multiple genes and\or interactions between them using these data could reveal more insights into the biofilm biology.
The authors should also present the UMIs capture metrics for RiboD-PETRI method for all cells passing initial quality filter (>=15 UMIs/cell) both in the text and in the figures. Selection of the top few cells with higher UMI count may introduce biological biases in the analysis (the top 5% of cells could represent a distinct subpopulation with very high gene expression due to a biological process). For single-cell RNA sequencing, showing the statistics for a 'top' group of cells creates confusion and inflates the perceived resolution, especially when used to compare to other methods (e.g. the parent method PETRI-seq itself).
-
-

-
Author response:
Public Reviews:
Reviewer #1 (Public Review):
[...] Strengths:
The method the authors propose is a straightforward and inexpensive modification of an established split-pool single-cell RNA-seq protocol that greatly increases its utility, and should be of interest to a wide community working in the field of bacterial single-cell RNA-seq.
Weaknesses:
The manuscript is written in a very compressed style and many technical details of the evaluations conducted are unclear and processed data has not been made available for evaluation, limiting the ability of the reader to independently judge the merits of the method.
Thank you for your thoughtful and constructive review of our manuscript. We appreciate your recognition of the strengths of our work and the potential impact of our modified PETRI-seq protocol on the field of …
Author response:
Public Reviews:
Reviewer #1 (Public Review):
[...] Strengths:
The method the authors propose is a straightforward and inexpensive modification of an established split-pool single-cell RNA-seq protocol that greatly increases its utility, and should be of interest to a wide community working in the field of bacterial single-cell RNA-seq.
Weaknesses:
The manuscript is written in a very compressed style and many technical details of the evaluations conducted are unclear and processed data has not been made available for evaluation, limiting the ability of the reader to independently judge the merits of the method.
Thank you for your thoughtful and constructive review of our manuscript. We appreciate your recognition of the strengths of our work and the potential impact of our modified PETRI-seq protocol on the field of bacterial single-cell RNA-seq. We are grateful for the opportunity to address your concerns and improve the clarity and accessibility of our manuscript.
We acknowledge your feedback regarding the compressed writing style and lack of technical details,which are constrained by the requirements of the Short Report format in eLife. We will addresse these issues in our revised manuscript as follows:
(1) Expanded methodology section: We will provide a more comprehensive description of our experimental procedures, including detailed protocols for the ribosomal depletion step and data analysis pipeline. This will enable readers to better understand and potentially replicate our methods.
(2) Clarification of technical evaluations: We will elaborate on the specifics of our evaluations, including the criteria used for assessing the efficiency of ribosomal depletion and the methods employed for identifying and characterizing subpopulations within the E. coli biofilm model.
(3) Data availability: We apologize for the oversight in not making our processed data readily available. We have deposited all relevant datasets, including raw and source data, in appropriate public repositories (GEO number: GSE260458) and provide clear instructions for accessing this data in the revised manuscript.
(4) Supplementary information: To maintain the concise nature of the main text while providing necessary details, we will inculde additional supplementary information. This will cover extended methodology, detailed statistical analyses, and comprehensive data tables to support our findings.
(5) Discussion of limitations: We will include a more thorough discussion of the potential limitations of our modified protocol and areas for future improvement.
We believe these changes will significantly improve the clarity and reproducibility of our work, allowing readers to better evaluate the merits of our method.
Reviewer #2 (Public Review):
[...] Strengths:
The introduced rRNA depletion method is highly efficient, with the depletion for E.coli resulting in over 90% of reads containing mRNA. The method is ready to use with existing PETRI-seq libraries which is a large advantage, given that no other rRNA depletion methods were published for split-pool bacterial scRNA-seq methods. Therefore, the value of the method for the field is high. There is also evidence that a small number of cells at the bottom of a static biofilm express PdeI which is causing the elevated c-di-GMP levels that are associated with persister formation. Given that PdeI is a phosphodiesterase, which is supposed to promote hydrolysis of c-di-GMP, this finding is unexpected.
Weaknesses:
With the descriptions and writing of the manuscript, it is hard to place the findings about the PdeI into existing context (i.e. it is well known that c-di-GMP is involved in biofilm development and is heterogeneously distributed in several species' biofilms; it is also known that E.coli diesterases regulate this second messenger, i.e. https://journals.asm.org/doi/full/10.1128/jb.00604-15).
There is also no explanation for the apparently contradictory upregulation of c-di-GMP in cells expressing higher PdeI levels. Perhaps the examination of the rest of the genes in cluster 2 of the biofilm sample could be useful to explain the observed association.Thank you for your thoughtful and constructive review of our manuscript. We are pleased that the reviewer recognizes the value and efficiency of our rRNA depletion method for PETRI-seq, as well as its potential impact on the field. We would like to address the points raised by the reviewer and provide additional context and clarification regarding the function of PdeI in c-di-GMP regulation.
We acknowledge that c-di-GMP’s role in biofilm development and its heterogeneous distribution in bacterial biofilms are well studied. We appreciate the reviewer's observation regarding the seemingly contradictory relationship between increased PdeI expression and elevated c-di-GMP levels. This is indeed an intriguing finding that warrants further explanation.
PdeI was predicted to be a phosphodiesterase responsible for c-di-GMP degradation. This prediction is based on sequence analysis where PdeI contains an intact EAL domain known for degrading c-di-GMP. However, it is noteworthy that PdeI also contains a divergent GGDEF domain, which is typically associated with c-di-GMP synthesis. This dual-domain architecture suggests a potential for complex regulatory roles. As reported, the knockout of the major phosphodiesterase PdeH in E. coli leads to the accumulation of c-di-GMP. Further, a point mutation on PdeI's divergent GGDEF domain (G412S) in this PdeH knockout strain resulted in decreased c-di-GMP levels, implying that the wild-type GGDEF domain in PdeI has a role in maintaining or increasing c-di-GMP levels in the cell. Additionally, PdeI contains a CHASE (cyclases/histidine kinase-associated sensory) domain. Combined with our experimental results demonstrating that PdeI is a membrane-associated protein, we predict that PdeI functions as a sensor that integrates environmental signals with c-di-GMP production under complex regulatory mechanisms. The experimental evidence, along with domain analysis, suggests that PdeI could contribute to c-di-GMP synthesis, rebutting the notion that it solely functions as a phosphodiesterase. Furthermore, our single-cell experiments showed a positive correlation between PdeI expression levels and c-di-GMP levels (Fig. 2J). HPLC LC-MS/MS analysis further confirmed that PdeI overexpression (induced by arabinose) upregulated c-di-GMP levels (Fig. 2K). Importantly, in our HPLC LC-MS/MS analysis, we compared the PdeI overexpression strain with the wild-type MG1655 strain, thereby excluding the influence of other genes in cluster 2. In summary, while PdeI is predicted to be a phosphodiesterase based on its sequence and the presence of an EAL domain, the additional presence of a divergent GGDEF domain and experimental evidence suggests that PdeI has a function in upregulating c-di-GMP levels. These findings support the hypothesis that PdeI may have both synthetic and regulatory roles in c-di-GMP metabolism.
-
-
eLife assessment
The work introduces a valuable new method for depleting the ribosomal RNA from bacterial single-cell RNA sequencing libraries and shows that this method is applicable to studying the heterogeneity in microbial biofilms. The evidence for a small subpopulation of cells at the bottom of the biofilm which upregulates PdeI expression is solid. However, more investigation into the unresolved functional relationship between PdeI and c-di-GMP levels with the help of other genes co-expressed in the same cluster would have made the conclusions more significant.
-
Reviewer #1 (Public Review):
Summary:
In this manuscript, Yan and colleagues introduce a modification to the previously published PETRI-seq bacterial single-cell protocol to include a ribosomal depletion step based on a DNA probe set that selectively hybridizes with ribosome-derived (rRNA) cDNA fragments. They show that their modification of the PETRI-seq protocol increases the fraction of informative non-rRNA reads from ~4-10% to 54-92%. The authors apply their protocol to investigating heterogeneity in a biofilm model of E. coli, and convincingly show how their technology can detect minority subpopulations within a complex community.
Strengths:
The method the authors propose is a straightforward and inexpensive modification of an established split-pool single-cell RNA-seq protocol that greatly increases its utility, and should be of …
Reviewer #1 (Public Review):
Summary:
In this manuscript, Yan and colleagues introduce a modification to the previously published PETRI-seq bacterial single-cell protocol to include a ribosomal depletion step based on a DNA probe set that selectively hybridizes with ribosome-derived (rRNA) cDNA fragments. They show that their modification of the PETRI-seq protocol increases the fraction of informative non-rRNA reads from ~4-10% to 54-92%. The authors apply their protocol to investigating heterogeneity in a biofilm model of E. coli, and convincingly show how their technology can detect minority subpopulations within a complex community.
Strengths:
The method the authors propose is a straightforward and inexpensive modification of an established split-pool single-cell RNA-seq protocol that greatly increases its utility, and should be of interest to a wide community working in the field of bacterial single-cell RNA-seq.
Weaknesses:
The manuscript is written in a very compressed style and many technical details of the evaluations conducted are unclear and processed data has not been made available for evaluation, limiting the ability of the reader to independently judge the merits of the method.
-
Reviewer #2 (Public Review):
Summary:
This work introduces a new method of depleting the ribosomal reads from the single-cell RNA sequencing library prepared with one of the prokaryotic scRNA-seq techniques, PETRI-seq. The advance is very useful since it allows broader access to the technology by lowering the cost of sequencing. It also allows more transcript recovery with fewer sequencing reads. The authors demonstrate the utility and performance of the method for three different model species and find a subpopulation of cells in the E.coli biofilm that express a protein, PdeI, which causes elevated c-di-GMP levels. These cells were shown to be in a state that promotes persister formation in response to ampicillin treatment.
Strengths:
The introduced rRNA depletion method is highly efficient, with the depletion for E.coli resulting in …
Reviewer #2 (Public Review):
Summary:
This work introduces a new method of depleting the ribosomal reads from the single-cell RNA sequencing library prepared with one of the prokaryotic scRNA-seq techniques, PETRI-seq. The advance is very useful since it allows broader access to the technology by lowering the cost of sequencing. It also allows more transcript recovery with fewer sequencing reads. The authors demonstrate the utility and performance of the method for three different model species and find a subpopulation of cells in the E.coli biofilm that express a protein, PdeI, which causes elevated c-di-GMP levels. These cells were shown to be in a state that promotes persister formation in response to ampicillin treatment.
Strengths:
The introduced rRNA depletion method is highly efficient, with the depletion for E.coli resulting in over 90% of reads containing mRNA. The method is ready to use with existing PETRI-seq libraries which is a large advantage, given that no other rRNA depletion methods were published for split-pool bacterial scRNA-seq methods. Therefore, the value of the method for the field is high. There is also evidence that a small number of cells at the bottom of a static biofilm express PdeI which is causing the elevated c-di-GMP levels that are associated with persister formation. Given that PdeI is a phosphodiesterase, which is supposed to promote hydrolysis of c-di-GMP, this finding is unexpected.
Weaknesses:
With the descriptions and writing of the manuscript, it is hard to place the findings about the PdeI into existing context (i.e. it is well known that c-di-GMP is involved in biofilm development and is heterogeneously distributed in several species' biofilms; it is also known that E.coli diesterases regulate this second messenger, i.e. https://journals.asm.org/doi/full/10.1128/jb.00604-15).
There is also no explanation for the apparently contradictory upregulation of c-di-GMP in cells expressing higher PdeI levels. Perhaps the examination of the rest of the genes in cluster 2 of the biofilm sample could be useful to explain the observed association. -
-