Sir2 and Fun30 regulate ribosomal DNA replication timing via MCM helicase positioning and nucleosome occupancy
Curation statements for this article:-
Curated by eLife

eLife Assessment
This valuable study is a detailed investigation of how chromatin structure influences replication origin function in yeast ribosomal DNA, with a focus on the role of the histone deacetylase Sir2 and the chromatin remodeler Fun30. The paper shows that Sir2 does not affect origin licensing but rather affects local transcription and nucleosome positioning which correlates with increased origin firing. Overall, the evidence is convincing and the model is plausible.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The association between late replication timing and low transcription rates in eukaryotic heterochromatin is well known, yet the specific mechanisms underlying this link remain uncertain. In Saccharomyces cerevisiae , the histone deacetylase Sir2 is required for both transcriptional silencing and late replication at the repetitive ribosomal DNA (rDNA) arrays. We have previously reported that in the absence of SIR2 , a de-repressed RNA PolII repositions MCM replicative helicases from their loading site at the ribosomal origin, where they abut well-positioned, high-occupancy nucleosomes, to an adjacent region with lower nucleosome occupancy. By developing a method that can distinguish activation of closely spaced MCM complexes, here we show that the displaced MCMs at rDNA origins have increased firing propensity compared to the nondisplaced MCMs. Furthermore, we found that both activation of the repositioned MCMs and low occupancy of the adjacent nucleosomes critically depend on the chromatin remodeling activity of FUN30 . Our study elucidates the mechanism by which Sir2 delays replication timing, and it demonstrates, for the first time, that activation of a specific replication origin in vivo relies on the nucleosome context shaped by a single chromatin remodeler.
Article activity feed
-

-
-
-

eLife Assessment
This valuable study is a detailed investigation of how chromatin structure influences replication origin function in yeast ribosomal DNA, with a focus on the role of the histone deacetylase Sir2 and the chromatin remodeler Fun30. The paper shows that Sir2 does not affect origin licensing but rather affects local transcription and nucleosome positioning which correlates with increased origin firing. Overall, the evidence is convincing and the model is plausible.
-
Reviewer #1 (Public review):
This paper presents a mechanistic study of rDNA origin regulation in yeast by SIR2. Each of the ~180 tandemly repeated rDNA gene copies contains a potential replication origin. Early-efficient initiation of these origins is suppressed by Sir2, reducing competition with origins distributed throughout the genome for rate-limiting initiation factors. Previous studies by these authors showed that SIR2 deletion advances replication timing of rDNA origins by a complex mechanism of transcriptional de-repression of a local PolII promoter causing licensed origin proteins (MCMcomplexes) to re-localize (slide along the DNA) to a different (and altered) chromatin environment. In this study, they identify a chromatin remodeler, FUN30, that suppresses the sir2∆ effect, and remarkably, results in a contraction of the rDNA …
Reviewer #1 (Public review):
This paper presents a mechanistic study of rDNA origin regulation in yeast by SIR2. Each of the ~180 tandemly repeated rDNA gene copies contains a potential replication origin. Early-efficient initiation of these origins is suppressed by Sir2, reducing competition with origins distributed throughout the genome for rate-limiting initiation factors. Previous studies by these authors showed that SIR2 deletion advances replication timing of rDNA origins by a complex mechanism of transcriptional de-repression of a local PolII promoter causing licensed origin proteins (MCMcomplexes) to re-localize (slide along the DNA) to a different (and altered) chromatin environment. In this study, they identify a chromatin remodeler, FUN30, that suppresses the sir2∆ effect, and remarkably, results in a contraction of the rDNA to about one-quarter it's normal length/number of repeats, implicating replication defects of the rDNA. Through examination of replication timing, MCM occupancy and nucleosome occupancy on the chromatin in sir2, fun30, and double mutants, they propose a model where nucleosome position relative to the licensed origin (MCM complexes) intrinsically determines origin timing/efficiency. While their interpretations of the data are largely reasonable and can be interpreted to support their model, a key weakness is the connection between Mcm ChEC signal disappearance and origin firing. While the cyclical chromatin association-dissociation of MCM proteins with potential origin sequences may be generally interpreted as licensing followed by firing, dissociation may also result from passive replication and as shown here, displacement by transcription and/or chromatin remodeling. Moreover, linking its disappearance from chromatin in the ChEC method with such precise resolution needs to be validated against an independent method to determine the initiation site(s). Differences in rDNA copy number and relative transcription levels also are not directly accounted for, obscuring a clearer interpretation of the results. Nevertheless, this paper makes a valuable advance with the finding of Fun30 involvement, which substantially reduces rDNA repeat number in sir2∆ background. The model they develop is compelling and I am inclined to agree, but I think the evidence on this specific point is purely correlative and a better method is needed to address the initiation site question. The authors deserve credit for their efforts to elucidate our obscure understanding of the intricacies of chromatin regulation.
Overall, the paper is improved by providing additional data and improved analysis. The paper nicely characterizes the effect of Fun30. The model is reasonable but remains lacking in precise details of mechanism.
-
Reviewer #2 (Public review):
Summary:
In this manuscript, the authors follow up on their previous work showing that in the absence of the Sir2 deacetylase the MCM replicative helicase at the rDNA spacer region is repositioned to a region of low nucleosome occupancy. Here they show that the repositioned displaced MCMs have increased firing propensity relative to non-displaced MCMs. In addition, they show that activation of the repositioned MCMs and low nucleosome occupancy in the adjacent region depend on the chromatin remodeling activity of Fun30.
Strengths:
The paper provides new information on the role of a conserved chromatin remodeling protein in regulation of origin firing and in addition provides evidence that not all loaded MCMs fire and that origin firing is regulated at a step downstream of MCM loading.
Comments on revisions:
Th…
Reviewer #2 (Public review):
Summary:
In this manuscript, the authors follow up on their previous work showing that in the absence of the Sir2 deacetylase the MCM replicative helicase at the rDNA spacer region is repositioned to a region of low nucleosome occupancy. Here they show that the repositioned displaced MCMs have increased firing propensity relative to non-displaced MCMs. In addition, they show that activation of the repositioned MCMs and low nucleosome occupancy in the adjacent region depend on the chromatin remodeling activity of Fun30.
Strengths:
The paper provides new information on the role of a conserved chromatin remodeling protein in regulation of origin firing and in addition provides evidence that not all loaded MCMs fire and that origin firing is regulated at a step downstream of MCM loading.
Comments on revisions:
The authors have addressed my concerns with the addition of new experiments and analysis.
-
Reviewer #3 (Public review):
Summary:
Heterochromatin is characterized by low transcription activity and late replication timing, both dependent on the NAD-dependent protein deacetylase Sir2, the founding member of the sirtuins. This manuscript addresses the mechanism by which Sir2 delays replication timing at the rDNA in budding yeast. Previous work from the same laboratory (Foss et al. PLoS Genetics 15, e1008138) showed that Sir2 represses transcription-dependent displacement of the Mcm helicase in the rDNA. In this manuscript, the authors show convincingly that the repositioned Mcms fire earlier and that this early firing partly depends on the ATPase activity of the nucleosome remodeler Fun30. Using read-depth analysis of sorted G1/S cells, fun30 was the only chromatin remodeler mutant that somewhat delayed replication timing in sir2 …
Reviewer #3 (Public review):
Summary:
Heterochromatin is characterized by low transcription activity and late replication timing, both dependent on the NAD-dependent protein deacetylase Sir2, the founding member of the sirtuins. This manuscript addresses the mechanism by which Sir2 delays replication timing at the rDNA in budding yeast. Previous work from the same laboratory (Foss et al. PLoS Genetics 15, e1008138) showed that Sir2 represses transcription-dependent displacement of the Mcm helicase in the rDNA. In this manuscript, the authors show convincingly that the repositioned Mcms fire earlier and that this early firing partly depends on the ATPase activity of the nucleosome remodeler Fun30. Using read-depth analysis of sorted G1/S cells, fun30 was the only chromatin remodeler mutant that somewhat delayed replication timing in sir2 mutants, while nhp10, chd1, isw1, htl1, swr1, isw2, and irc5 had no effect. The conclusion was corroborated with orthogonal assays including two-dimensional gel electrophoresis and analysis of EdU incorporation at early origins. Using an insightful analysis with an Mcm-MNase fusion (Mcm-ChEC), the authors show that the repositioned Mcms in sir2 mutants fire earlier than the Mcm at the normal position in wild type. This early firing at the repositioned Mcms is partially suppressed by Fun30. In addition, the authors show Fun30 affects nucleosome occupancy at the sites of the repositioned Mcm, providing a plausible mechanism for the effect of Fun30 on Mcm firing at that position. However, the results from the MNAse-seq and ChEC-seq assays are not fully congruent for the fun30 single mutant. Overall, the results support the conclusions providing a much better mechanistic understanding how Sir2 affects replication timing at rDNA,
Strengths:
(1) The data clearly show that the repositioned Mcm helicase fires earlier than the Mcm in the wild type position.
(2) The study identifies a specific role for Fun30 in replication timing and an effect on nucleosome occupancy around the newly positioned Mcm helicase in sir2 cells.
Comments on revisions:
In the previous revision the authors addressed my concerns and improved the manuscript and the presentation of the data. All my recommendations were implemented.
-
Author response:
The following is the authors’ response to the previous reviews.
eLife Assessment
This valuable study is a detailed investigation of how chromatin structure influences replication origin function in yeast ribosomal DNA, with a focus on the role of the histone deacetylase Sir2 and the chromatin remodeler Fun30. Convincing evidence shows that Sir2 does not affect origin licensing but rather affects local transcription and nucleosome positioning which correlates with increased origin firing. Overall, the evidence is solid and the model plausible. However, the methods employed do not rigorously establish a key aspect of the mechanism where initiation precisely occurs or rigorously exclude alternative models and the effect of Sir2 on transcription is not re-examined in the fun30 context.
Clarification on Sir2 Effect on …
Author response:
The following is the authors’ response to the previous reviews.
eLife Assessment
This valuable study is a detailed investigation of how chromatin structure influences replication origin function in yeast ribosomal DNA, with a focus on the role of the histone deacetylase Sir2 and the chromatin remodeler Fun30. Convincing evidence shows that Sir2 does not affect origin licensing but rather affects local transcription and nucleosome positioning which correlates with increased origin firing. Overall, the evidence is solid and the model plausible. However, the methods employed do not rigorously establish a key aspect of the mechanism where initiation precisely occurs or rigorously exclude alternative models and the effect of Sir2 on transcription is not re-examined in the fun30 context.
Clarification on Sir2 Effect on Transcription in the fun30 Context
We appreciate the reviewers’ thorough assessment but would like to clarify that the effect of Sir2 on transcription in the fun30 context was addressed in both the original and revised manuscripts. However, we recognize that the presentation of the qPCR results may have been unclear, as we initially plotted absolute transcript levels without normalizing for rDNA array size differences among the genotypes. We have now corrected this.
After normalizing for copy number variations, the qPCR data show that the sir2 fun30 double mutant results in a ~40-fold increase in C-pro transcription relative to WT, compared to a 4-fold and 19-fold increase in fun30 and sir2 single mutants, respectively (Figure 5, figure supplement 6). These results have been discussed in the manuscript result section, where we note that "C-pro RNA levels were approximately twice as high in sir2 fun30 compared to sir2 cells when adjusted for rDNA size differences." This observation is critical for addressing both alternative models of MCM disappearance and for pinpointing transcription initiation sites, as detailed in the following sections.
Public Reviews:
Reviewer #1 (Public review):
Summary:
This paper presents a mechanistic study of rDNA origin regulation in yeast by SIR2. Each of the ~180 tandemly repeated rDNA gene copies contains a potential replication origin. Earlyefficient initiation of these origins is suppressed by Sir2, reducing competition with origins distributed throughout the genome for rate-limiting initiation factors. Previous studies by these authors showed that SIR2 deletion advances replication timing of rDNA origins by a complex mechanism of transcriptional de-repression of a local PolII promoter causing licensed origin proteins (MCMcomplexes) to re-localize (slide along the DNA) to a different (and altered) chromatin environment. In this study, they identify a chromatin remodeler, FUN30, that suppresses the sir2∆ effect, and remarkably, results in a contraction of the rDNA to about onequarter it's normal length/number of repeats, implicating replication defects of the rDNA. Through examination of replication timing, MCM occupancy and nucleosome occupancy on the chromatin in sir2, fun30, and double mutants, they propose a model where nucleosome position relative to the licensed origin (MCM complexes) intrinsically determines origin timing/efficiency. While their interpretations of the data are largely reasonable and can be interpreted to support their model, a key weakness is the connection between Mcm ChEC signal disappearance and origin firing. While the cyclical chromatin association-dissociation of MCM proteins with potential origin sequences may be generally interpreted as licensing followed by firing, dissociation may also result from passive replication and as shown here, displacement by transcription and/or chromatin remodeling. Moreover, linking its disappearance from chromatin in the ChEC method with such precise resolution needs to be validated against an independent method to determine the initiation site(s). Differences in rDNA copy number and relative transcription levels also are not directly accounted for, obscuring a clearer interpretation of the results. Nevertheless, this paper makes a valuable advance with the finding of Fun30 involvement, which substantially reduces rDNA repeat number in sir2∆ background. The model they develop is compelling and I am inclined to agree, but I think the evidence on this specific point is purely correlative and a better method is needed to address the initiation site question. The authors deserve credit for their efforts to elucidate our obscure understanding of the intricacies of chromatin regulation. At a minimum, I suggest their conclusions on these points of concern should be softened and caveats discussed. Statistical analysis is lacking for some claims.
Strengths are the identification of FUN30 as suppressor, examination of specific mutants of FUN30 to distinguish likely functional involvement. Use of multiple methods to analyze replication and protein occupancies on chromatin. Development of a coherent model.
Weaknesses are failure to address copy number as a variable; insufficient validation of ChEC method relationship to exact initiation locus; lack of statistical analysis in some cases.
Review of revised version and response letter:
In the response, the authors make some improvements by better quantifying 2D gels, adding some missing statistical analyses, analyzing the effect of fun30 on rDNA replication in strains with reduced rDNA copy number, and using ChIP-seq of MCMs to support the ChEC-seq data. However, these additions do not address the main issue that is at the heart of their model: where initiation precisely occurs and whether the location is altered in the mutant(s). Thus, mechanistic insight is limited.
We discuss the issue regarding the initiation site below.
Under the section "Addressing Alternative Explanations", the authors claim that processes like transcription and passive replication cannot affect the displaced complex specifically. Why? They are not on same DNA (as mentioned in the Fig 1 legend).
Premature origin activation, not transcription, drives the disappearance of repositioned MCM complexes in sir2 mutants in HU.
Indeed, the reviewer is correct in suggesting that C-pro transcription confined to rDNA units with repositioned MCM complexes could selectively displace those complexes, potentially explaining the selective disappearance of displaced MCMs in sir2 cells. However, our analysis of C-pro transcription and MCM occupancy in G1 versus HU across the genotypes allows us to rule out this possibility.
We show that the fraction of repositioned MCMs in G1 cells is proportional to the level of C-pro transcription (WT < fun30 << sir2 < sir2 fun30), consistent with the involvement of transcription in the repositioning process during MCM loading in G1. Accordingly, with approximately twice the transcription in sir2 fun30 compared to sir2, we observe more repositioned MCMs in sir2 fun30 cells than in sir2 cells in G1 (Fig 5C).
However, if the disappearance of repositioned MCMs in HU were solely due to C-pro transcription rather than origin activation, we would expect the repositioned MCMs to disappear more quickly in sir2 fun30 cells. Contrary to this expectation, our data show that repositioned MCM complexes are more stable in sir2 fun30 mutants compared to sir2 mutants, indicating that transcription is not the primary factor in the disappearance of displaced MCM complexes in HU; rather, rDNA origin activation appears to be the key factor.
Replication initiation site in sir2. Using multiple independent approaches, including 2D gels, ChIP-seq, and EdU incorporation, we have demonstrated that rDNA origins fire prematurely in sir2 mutants, a conclusion that the reviewer does not contest. Once an origin fires, the MCM signal disappears from the site of its initial deposition, as expected, and this is confirmed in our MCM ChIP and HU ChEC data, both at rDNA origins and across the genome.
Given that the majority of MCM complexes in sir2 mutants are repositioned, it is expected that these repositioned complexes disappear following premature origin activation. With less than half of the licensed origins (or <30% of total rDNA copies) retaining MCM at non-repositioned sites in sir2 mutants, if only these non-repositioned complexes were firing, and the repositioned MCM complexes were disappearing via mechanisms other than replication initiation (e.g., transcription), rDNA replication in sir2 mutants would be severely compromised rather than accelerated. Given this, and the strong experimental evidence that repositioned MCM complexes fire prematurely, continued focus on alternative explanations for MCM complex disappearance seems unwarranted.
We present this analysis in the results section as follows:
“Finally, although deletion of FUN30 could suppress replication initiation at the rDNA either by inhibiting the firing of the active, repositioned MCM complex or by preventing MCM repositioning to the "active location" in the first place, our results suggest that suppression occurs through the former mechanism. Consistent with previous reports that fun30 mutants are deficient in transcriptional silencing (Neves-Costa et al. 2009), C-pro RNA levels were approximately twice as high in sir2 fun30 cells compared to sir2 cells when adjusted for rDNA size (Figure 5—figure supplement 6).
Moreover, deletion of FUN30 shifts the distribution toward the repositioned MCM location over the non-repositioned one in G1 cells (Figure 5C), aligning with the increased C-pro transcription observed in fun30 mutants. This shift is evident in both sir2 and SIR2 cells. Despite the increased transcription-mediated repositioning in sir2 fun30 cells compared to sir2 cells during G1, repositioned MCM persists longer in sir2 fun30 cells than in sir2 cells after release into HU. Additionally, sir2 fun30 mutants exhibit reduced MCM accumulation at the RFB compared to sir2 mutants after release into HU, supporting the conclusion that MCM disappearance in HU reflects origin activation rather than transcription-mediated displacement.”
The model in Fig 7 implies that initiation sites are different in WT versus the mutants and this determines their timing/efficiency. But they also suggest that the same site might be used with different efficiencies in this response. I agree that both are possibilities and are not resolved.
Adjustment of the model to account for repositioned MCMs in WT cells In Figure 5—figure supplement 5, we demonstrate that even in WT cells, a small fraction of repositioned MCMs (~5%) can be detected, and that these repositioned MCM complexes disappear prematurely. However, because this represents a very small fraction of MCMs in WT cells, we initially did not include it in our overall model in Figure 7. In light of the reviewer's comment, we have now revised the model to incorporate this detail.
Supporting their model requires better resolution to determine the actual replication initiation site. While this may be challenging, it should be feasible with methods to map nascent strands like DNAscent, or Okazaki fragment mapping.
The initiation site in sir2 mutants has been thoroughly analyzed and supported by extensive experimental data, as discussed above. While high-resolution techniques such as DNAscent or Okazaki fragment mapping could potentially offer another layer of validation, the likelihood of obtaining finer detail that would change the conclusions is minimal. The methods we employed provide sufficient resolution to pinpoint the initiation site, and our results align consistently with established replication models.
Further experimentation would not only be redundant but also unlikely to provide new insights beyond revalidation. Given the strength of our current data, we believe the conclusions regarding replication initiation are robust and well-supported, making additional experiments unnecessary at this stage. Our priority is to focus on advancing other aspects of the research that require deeper exploration.
The 2D gel analysis of strains with reduced rDNA copy numbers adequately addresses the copy number variable with regard to the replication effect.
Overall, the paper is improved by providing additional data and improved analysis. The paper nicely characterizes the effect of Fun30. The model is reasonable but remains lacking in precise details of mechanism.
Reviewer #2 (Public review):
Summary:
In this manuscript, the authors follow up on their previous work showing that in the absence of the Sir2 deacetylase the MCM replicative helicase at the rDNA spacer region is repositioned to a region of low nucleosome occupancy. Here they show that the repositioned displaced MCMs have increased firing propensity relative to non-displaced MCMs. In addition, they show that activation of the repositioned MCMs and low nucleosome occupancy in the adjacent region depend on the chromatin remodeling activity of Fun30.
Strengths:
The paper provides new information on the role of a conserved chromatin remodeling protein in regulation of origin firing and in addition provides evidence that not all loaded MCMs fire and that origin firing is regulated at a step downstream of MCM loading.
Weaknesses:
The relationship between the authors results and prior work on the role of Sir2 (and Fob1) in regulation of rDNA recombination and copy number maintenance is not explored, making it difficult to place the results in a broader context. Sir2 has previously been shown to be recruited by Fob1, which is also required for DSB formation and recombination-mediated changes in rDNA copy number. Are the changes that the authors observe specifically in fun30 sir2 cells related to this pathway? Is Fob1 required for the reduced rDNA copy number in fun30 sir2 double mutant cells?
Reviewer #3 (Public review):
Summary:
Heterochromatin is characterized by low transcription activity and late replication timing, both dependent on the NAD-dependent protein deacetylase Sir2, the founding member of the sirtuins. This manuscript addresses the mechanism by which Sir2 delays replication timing at the rDNA in budding yeast. Previous work from the same laboratory (Foss et al. PLoS Genetics 15, e1008138) showed that Sir2 represses transcription-dependent displacement of the Mcm helicase in the rDNA. In this manuscript, the authors show convincingly that the repositioned Mcms fire earlier and that this early firing partly depends on the ATPase activity of the nucleosome remodeler Fun30. Using read-depth analysis of sorted G1/S cells, fun30 was the only chromatin remodeler mutant that somewhat delayed replication timing in sir2 mutants, while nhp10, chd1, isw1, htl1, swr1, isw2, and irc5 had no effect. The conclusion was corroborated with orthogonal assays including two-dimensional gel electrophoresis and analysis of EdU incorporation at early origins. Using an insightful analysis with an Mcm-MNase fusion (Mcm-ChEC), the authors
show that the repositioned Mcms in sir2 mutants fire earlier than the Mcm at the normal position in wild type. This early firing at the repositioned Mcms is partially suppressed by Fun30. In addition, the authors show Fun30 affects nucleosome occupancy at the sites of the repositioned Mcm, providing a plausible mechanism for the effect of Fun30 on Mcm firing at that position. However, the results from the MNAse-seq and ChEC-seq assays are not fully congruent for the fun30 single mutant. Overall, the results support the conclusions providing a much better mechanistic understanding how Sir2 affects replication timing at rDNA,
Strengths
(1) The data clearly show that the repositioned Mcm helicase fires earlier than the Mcm in the wild type position.
(2) The study identifies a specific role for Fun30 in replication timing and an effect on nucleosome occupancy around the newly positioned Mcm helicase in sir2 cells.
Weaknesses
(1) It is unclear which strains were used in each experiment.
(2) The relevance of the fun30 phospho-site mutant (S20AS28A) is unclear.
(3) For some experiments (Figs. 3, 4, 6) it is unclear whether the data are reproducible and the differences significant. Information about the number of independent experiments and quantitation is lacking. This affects the interpretation, as fun30 seems to affect the +3 nucleosome much more than let on in the description.
Recommendations for the authors:
Reviewer #2 (Recommendations for the authors):
The authors have addressed my concerns by the addition of new experiments and analysis.
One point remains unclear regarding additional support for the Mcm-ChEC results using ChIP experiments to verify whether MCM redistributes in sir2D cells. In their rebuttal, the authors state that, "New supporting based evidence: ChIP at rDNA Origins. Our ChIP analysis also shows that the disappearance of the MCM signal at rDNA origins in sir2Δ cells released into HU is accompanied by signal accumulation at the replication fork barrier (RFB), indicative of stalled replication forks at this location (Figure 5 figure supplement 3)...." The ChIP data in Figure 5 supplement 3 show accumulation of the Mcm2 ChIP signal to the left of the RFB in sir2D cells but it doesn't look like there is any decrease in the MCM signal in sir2D relative to wild-type cells for the peak C-Pro. There is a new MCM peak suggesting perhaps a new MCM loading event.
Figure 5 figure supplement 3 shows the relative abundance of the MCM ChIP signal across the ~2 kb rDNA region, spanning from the MCM loading site at the rDNA origin (on the left) to the replication fork barrier (RFB) on the right. The MCM-ChIP data are normalized to the highest signal within this rDNA region rather than across the entire genome, meaning that only the relative abundance of MCM within this region is represented, and not comparisons between different conditions. We have now presented the results with the same axes for both alpha factor and HU.
In wild-type (WT) cells, the MCM signal remains primarily at the initial loading site. However, in sir2 mutants, a significant portion of the MCM signal shifts rightward, consistent with rDNA origin activation and the movement of MCM along with the progressing replication fork. While some replication forks stall at the RFB, others are positioned between the MCM loading site and the RFB. The additional MCM peak observed does not represent a new MCM loading event, as the experiment was conducted during S-phase, when new MCM loading is not possible.
Reviewer #3 (Recommendations for the authors):
In this revision the authors addressed my concerns and improved the manuscript and the presentation of the data. All my recommendations were implemented.
-
-
eLife Assessment
This valuable study is a detailed investigation of how chromatin structure influences replication origin function in yeast ribosomal DNA, with a focus on the role of the histone deacetylase Sir2 and the chromatin remodeler Fun30. Convincing evidence shows that Sir2 does not affect origin licensing but rather affects local transcription and nucleosome positioning which correlates with increased origin firing. Overall, the evidence is solid and the model plausible. However, the methods employed do not rigorously establish a key aspect of the mechanism where initiation precisely occurs or rigorously exclude alternative models and the effect of Sir2 on transcription is not re-examined in the fun30 context.
-
Reviewer #1 (Public review):
Summary:
This paper presents a mechanistic study of rDNA origin regulation in yeast by SIR2. Each of the ~180 tandemly repeated rDNA gene copies contains a potential replication origin. Early-efficient initiation of these origins is suppressed by Sir2, reducing competition with origins distributed throughout the genome for rate-limiting initiation factors. Previous studies by these authors showed that SIR2 deletion advances replication timing of rDNA origins by a complex mechanism of transcriptional de-repression of a local PolII promoter causing licensed origin proteins (MCMcomplexes) to re-localize (slide along the DNA) to a different (and altered) chromatin environment. In this study, they identify a chromatin remodeler, FUN30, that suppresses the sir2∆ effect, and remarkably, results in a contraction of …
Reviewer #1 (Public review):
Summary:
This paper presents a mechanistic study of rDNA origin regulation in yeast by SIR2. Each of the ~180 tandemly repeated rDNA gene copies contains a potential replication origin. Early-efficient initiation of these origins is suppressed by Sir2, reducing competition with origins distributed throughout the genome for rate-limiting initiation factors. Previous studies by these authors showed that SIR2 deletion advances replication timing of rDNA origins by a complex mechanism of transcriptional de-repression of a local PolII promoter causing licensed origin proteins (MCMcomplexes) to re-localize (slide along the DNA) to a different (and altered) chromatin environment. In this study, they identify a chromatin remodeler, FUN30, that suppresses the sir2∆ effect, and remarkably, results in a contraction of the rDNA to about one-quarter it's normal length/number of repeats, implicating replication defects of the rDNA. Through examination of replication timing, MCM occupancy and nucleosome occupancy on the chromatin in sir2, fun30, and double mutants, they propose a model where nucleosome position relative to the licensed origin (MCM complexes) intrinsically determines origin timing/efficiency. While their interpretations of the data are largely reasonable and can be interpreted to support their model, a key weakness is the connection between Mcm ChEC signal disappearance and origin firing. While the cyclical chromatin association-dissociation of MCM proteins with potential origin sequences may be generally interpreted as licensing followed by firing, dissociation may also result from passive replication and as shown here, displacement by transcription and/or chromatin remodeling. Moreover, linking its disappearance from chromatin in the ChEC method with such precise resolution needs to be validated against an independent method to determine the initiation site(s). Differences in rDNA copy number and relative transcription levels also are not directly accounted for, obscuring a clearer interpretation of the results. Nevertheless, this paper makes a valuable advance with the finding of Fun30 involvement, which substantially reduces rDNA repeat number in sir2∆ background. The model they develop is compelling and I am inclined to agree, but I think the evidence on this specific point is purely correlative and a better method is needed to address the initiation site question. The authors deserve credit for their efforts to elucidate our obscure understanding of the intricacies of chromatin regulation. At a minimum, I suggest their conclusions on these points of concern should be softened and caveats discussed. Statistical analysis is lacking for some claims.
Strengths are the identification of FUN30 as suppressor, examination of specific mutants of FUN30 to distinguish likely functional involvement. Use of multiple methods to analyze replication and protein occupancies on chromatin. Development of a coherent model.
Weaknesses are failure to address copy number as a variable; insufficient validation of ChEC method relationship to exact initiation locus; lack of statistical analysis in some cases.
Review of revised version and response letter:
In the response, the authors make some improvements by better quantifying 2D gels, adding some missing statistical analyses, analyzing the effect of fun30 on rDNA replication in strains with reduced rDNA copy number, and using ChIP-seq of MCMs to support the ChEC-seq data. However, these additions do not address the main issue that is at the heart of their model: where initiation precisely occurs and whether the location is altered in the mutant(s). Thus, mechanistic insight is limited.
Under the section "Addressing Alternative Explanations", the authors claim that processes like transcription and passive replication cannot affect the displaced complex specifically. Why? They are not on same DNA (as mentioned in the Fig 1 legend).
The model in Fig 7 implies that initiation sites are different in WT versus the mutants and this determines their timing/efficiency. But they also suggest that the same site might be used with different efficiencies in this response. I agree that both are possibilities and are not resolved.
Supporting their model requires better resolution to determine the actual replication initiation site. While this may be challenging, it should be feasible with methods to map nascent strands like DNAscent, or Okazaki fragment mapping.
The 2D gel analysis of strains with reduced rDNA copy numbers adequately addresses the copy number variable with regard to the replication effect.
Overall, the paper is improved by providing additional data and improved analysis. The paper nicely characterizes the effect of Fun30. The model is reasonable but remains lacking in precise details of mechanism.
-
Reviewer #2 (Public review):
Summary:
In this manuscript, the authors follow up on their previous work showing that in the absence of the Sir2 deacetylase the MCM replicative helicase at the rDNA spacer region is repositioned to a region of low nucleosome occupancy. Here they show that the repositioned displaced MCMs have increased firing propensity relative to non-displaced MCMs. In addition, they show that activation of the repositioned MCMs and low nucleosome occupancy in the adjacent region depend on the chromatin remodeling activity of Fun30.
Strengths:
The paper provides new information on the role of a conserved chromatin remodeling protein in regulation of origin firing and in addition provides evidence that not all loaded MCMs fire and that origin firing is regulated at a step downstream of MCM loading.
Weaknesses:
The …
Reviewer #2 (Public review):
Summary:
In this manuscript, the authors follow up on their previous work showing that in the absence of the Sir2 deacetylase the MCM replicative helicase at the rDNA spacer region is repositioned to a region of low nucleosome occupancy. Here they show that the repositioned displaced MCMs have increased firing propensity relative to non-displaced MCMs. In addition, they show that activation of the repositioned MCMs and low nucleosome occupancy in the adjacent region depend on the chromatin remodeling activity of Fun30.
Strengths:
The paper provides new information on the role of a conserved chromatin remodeling protein in regulation of origin firing and in addition provides evidence that not all loaded MCMs fire and that origin firing is regulated at a step downstream of MCM loading.
Weaknesses:
The relationship between the authors results and prior work on the role of Sir2 (and Fob1) in regulation of rDNA recombination and copy number maintenance is not explored, making it difficult to place the results in a broader context. Sir2 has previously been shown to be recruited by Fob1, which is also required for DSB formation and recombination-mediated changes in rDNA copy number. Are the changes that the authors observe specifically in fun30 sir2 cells related to this pathway? Is Fob1 required for the reduced rDNA copy number in fun30 sir2 double mutant cells?
-
Reviewer #3 (Public review):
Summary:
Heterochromatin is characterized by low transcription activity and late replication timing, both dependent on the NAD-dependent protein deacetylase Sir2, the founding member of the sirtuins. This manuscript addresses the mechanism by which Sir2 delays replication timing at the rDNA in budding yeast. Previous work from the same laboratory (Foss et al. PLoS Genetics 15, e1008138) showed that Sir2 represses transcription-dependent displacement of the Mcm helicase in the rDNA. In this manuscript, the authors show convincingly that the repositioned Mcms fire earlier and that this early firing partly depends on the ATPase activity of the nucleosome remodeler Fun30. Using read-depth analysis of sorted G1/S cells, fun30 was the only chromatin remodeler mutant that somewhat delayed replication timing in sir2 …
Reviewer #3 (Public review):
Summary:
Heterochromatin is characterized by low transcription activity and late replication timing, both dependent on the NAD-dependent protein deacetylase Sir2, the founding member of the sirtuins. This manuscript addresses the mechanism by which Sir2 delays replication timing at the rDNA in budding yeast. Previous work from the same laboratory (Foss et al. PLoS Genetics 15, e1008138) showed that Sir2 represses transcription-dependent displacement of the Mcm helicase in the rDNA. In this manuscript, the authors show convincingly that the repositioned Mcms fire earlier and that this early firing partly depends on the ATPase activity of the nucleosome remodeler Fun30. Using read-depth analysis of sorted G1/S cells, fun30 was the only chromatin remodeler mutant that somewhat delayed replication timing in sir2 mutants, while nhp10, chd1, isw1, htl1, swr1, isw2, and irc5 had no effect. The conclusion was corroborated with orthogonal assays including two-dimensional gel electrophoresis and analysis of EdU incorporation at early origins. Using an insightful analysis with an Mcm-MNase fusion (Mcm-ChEC), the authors show that the repositioned Mcms in sir2 mutants fire earlier than the Mcm at the normal position in wild type. This early firing at the repositioned Mcms is partially suppressed by Fun30. In addition, the authors show Fun30 affects nucleosome occupancy at the sites of the repositioned Mcm, providing a plausible mechanism for the effect of Fun30 on Mcm firing at that position. However, the results from the MNAse-seq and ChEC-seq assays are not fully congruent for the fun30 single mutant. Overall, the results support the conclusions providing a much better mechanistic understanding how Sir2 affects replication timing at rDNA,
Strengths
(1) The data clearly show that the repositioned Mcm helicase fires earlier than the Mcm in the wild type position.
(2) The study identifies a specific role for Fun30 in replication timing and an effect on nucleosome occupancy around the newly positioned Mcm helicase in sir2 cells.Weaknesses
(1) It is unclear which strains were used in each experiment.
(2) The relevance of the fun30 phospho-site mutant (S20AS28A) is unclear.
(3) For some experiments (Figs. 3, 4, 6) it is unclear whether the data are reproducible and the differences significant. Information about the number of independent experiments and quantitation is lacking. This affects the interpretation, as fun30 seems to affect the +3 nucleosome much more than let on in the description. -
Author response:
The following is the authors’ response to the original reviews.
eLife Assessment
This study is a detailed investigation of how chromatin structure influences replication origin function in yeast ribosomal DNA, with focus on the role of the histone deacetylase Sir2 and the chromatin remodeler Fun30. Convincing evidence shows that Sir2 does not affect origin licensing but rather affects local transcription and nucleosome positioning which correlates with increased origin firing. However, the evidence remains incomplete as the methods employed do not rigorously establish a key aspect of the mechanism, fully address some alternative models, or sufficiently relate to prior results. Overall, this is a valuable advance for the field that could be improved to establish a more robust paradigm.
We have added extensive new …
Author response:
The following is the authors’ response to the original reviews.
eLife Assessment
This study is a detailed investigation of how chromatin structure influences replication origin function in yeast ribosomal DNA, with focus on the role of the histone deacetylase Sir2 and the chromatin remodeler Fun30. Convincing evidence shows that Sir2 does not affect origin licensing but rather affects local transcription and nucleosome positioning which correlates with increased origin firing. However, the evidence remains incomplete as the methods employed do not rigorously establish a key aspect of the mechanism, fully address some alternative models, or sufficiently relate to prior results. Overall, this is a valuable advance for the field that could be improved to establish a more robust paradigm.
We have added extensive new results to the manuscript that, we believe, address all three criticisms above, namely that the methods employed do not (1) rigorously establish a key aspect of the mechanism; (2) fully address some alternative models; or (3) sufficiently relate to prior results.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
This paper presents a mechanistic study of rDNA origin regulation in yeast by SIR2. Each of the ~180 tandemly repeated rDNA gene copies contains a potential replication origin. Earlyefficient initiation of these origins is suppressed by Sir2, reducing competition with origins distributed throughout the genome for rate-limiting initiation factors. Previous studies by these authors showed that SIR2 deletion advances replication timing of rDNA origins by a complex mechanism of transcriptional de-repression of a local PolII promoter causing licensed origin proteins (MCMcomplexes) to re-localize (slide along the DNA) to a different (and altered) chromatin environment. In this study, they identify a chromatin remodeler, FUN30, that suppresses the sir2∆ effect, and remarkably, results in a contraction of the rDNA to about onequarter it's normal length/number of repeats, implicating replication defects of the rDNA. Through examination of replication timing, MCM occupancy and nucleosome occupancy on the chromatin in sir2, fun30, and double mutants, they propose a model where nucleosome position relative to the licensed origin (MCM complexes) intrinsically determines origin timing/efficiency. While their interpretations of the data are largely reasonable and can be interpreted to support their model, a key weakness is the connection between Mcm ChEC signal disappearance and origin firing.
Criticism: The reviewer expressed concern about the connection between Mcm ChEC signal disappearance and origin firing.
To further support our claim that the disappearance of the MCM signal in our ChEC datasets reflects origin firing, we now present additional data using the well-established method of MCM Chromatin IP (ChIP).
(1) New Supporting Evidence: ChIP at genome-wide origins. In Figure 5 figure supplement 2, we demonstrate that the Mcm2 ChIP signal in cells released into hydroxyurea (HU) is significantly reduced at early origins compared to late origins, which mirrors the pattern observed with the MCM2 ChEC signal. This reduction in the ChIP signal at early origins supports the interpretation that the MCM signal disappearance is associated with origin firing.
(2) New supporting based evidence: ChIP at rDNA Origins. Our ChIP analysis also shows that the disappearance of the MCM signal at rDNA origins in sir2Δ cells released into HU is accompanied by signal accumulation at the replication fork barrier (RFB), indicative of stalled replication forks at this location (Figure 5 figure supplement 3). This pattern is consistent with the initiation of replication at these origins and fork stalling at the RFB.
(3) New supporting evidence: 2D gels with quantification. Furthermore, additional 2D gel electrophoresis results provide ample independent evidence of rDNA origin firing in HU in sir2Δ mutants and suppression of origin firing in sir2 fun30 cells. These new data include 1) quantification of 2D gels in Figure 4D and 2) new 2D gels presented in Figure 4C as described below in greater detail. Collectively, these results demonstrate that rDNA origins fire prematurely in HU in sir2 cells and that firing is suppressed by FUN30 deletion. These additional data reinforce our model and support the association between MCM signal disappearance and replication initiation.
While the cyclical chromatin association-dissociation of MCM proteins with potential origin sequences may be generally interpreted as licensing followed by firing, dissociation may also result from passive replication and as shown here, displacement by transcription and/or chromatin remodeling.
The reviewer raised a concern that the cyclical chromatin association-dissociation of MCM proteins could be interpreted as licensing followed by firing, but might also result from passive replication or displacement by transcription and chromatin remodeling.
Addressing Alternative Explanations:
(1) Selective Disappearance of MCM Complexes: While transcription and passive replication can indeed cause the MCM-ChEC signal to disappear, these processes cannot selectively cause the disappearance of the displaced MCM complex without also affecting the non-displaced MCM complex. Specifically, RNA polymerase transcribing C-pro would first need to dislodge the normally positioned MCM complex before reaching the displaced complex, which is not observed in our data.
(2) Role of FUN30 Deletion: FUN30 deletion results in increased C-pro transcription and reduced disappearance of the displaced MCM complex. This observation supports our model, as transcription alone would not selectively affect the displaced MCM complex while leaving the normally positioned MCM complex unaffected.
(3) Licensing Restrictions: It is crucial to note that continuous replenishment of displaced MCMs with newly loaded MCMs is not possible in our experimental conditions, as the cells are in S phase and licensing is restricted to G1. This temporal restriction further supports our interpretation that the disappearance of the MCM signal reflects origin firing rather than alternative processes.
In summary, while alternative explanations such as transcription and passive replication could potentially account for MCM signal disappearance, our data indicate that these processes cannot selectively affect the displaced MCM complex without impacting the non-displaced complex. The selective disappearance observed in our experiments, along with the effects of FUN30 deletion and the temporal constraints on MCM loading, strongly support our interpretation that the disappearance of the MCM signal reflects origin firing.
Moreover, linking its disappearance from chromatin in the ChEC method with such precise resolution needs to be validated against an independent method to determine the initiation site(s). Differences in rDNA copy number and relative transcription levels also are not directly accounted for, obscuring a clearer interpretation of the results.
The reviewer raised concerns about the need to validate the disappearance of MCM from chromatin observed using the ChEC method against an independent method to determine initiation sites. Additionally, they pointed out that differences in rDNA copy number and relative transcription levels are not directly accounted for, which may obscure the interpretation of the results.
(1) Reduced rDNA Copy Number promotes Early Replication: Copy number reduction of the magnitude caused by deletion of both SIR2 and FUN30 is not expected to suppress early rDNA replication in sir2, but rather to exacerbate it. Specifically, deletion of SIR2 and FUN30 causes the rDNA to shrink to approximately 35 copies. Kwan et al., 2023 (PMID: 36842087) have shown that a reduction in rDNA copy number to 35 copies results in a dramatic acceleration of rDNA replication in a SIR2+ strain. Therefore, the effect of rDNA size on replication timing reinforces our conclusion that deletion of FUN30 suppresses rDNA replication.
(2) New 2D Gels in sir2 and sir2 fun30 strains with equal number of rDNA repeats: To directly address the concern regarding differences in the number of rDNA repeats, we have included new 2D gel analyses in the revised manuscript. By using a fob1
background, we were able to equalize the repeat number between the sir2 and sir2 fun30 strains (Figure 4E). The 2D gels conclusively show that the suppression of rDNA origin firing upon FUN30 deletion is independent of both rDNA size and FOB1.
Nevertheless, this paper makes a valuable advance with the finding of Fun30 involvement, which substantially reduces rDNA repeat number in sir2∆ background. The model they develop is compelling and I am inclined to agree, but I think the evidence on this specific point is purely correlative and a better method is needed to address the initiation site question. The authors deserve credit for their efforts to elucidate our obscure understanding of the intricacies of chromatin regulation. At a minimum, I suggest their conclusions on these points of concern should be softened and caveats discussed. Statistical analysis is lacking for some claims.
Strengths are the identification of FUN30 as suppressor, examination of specific mutants of FUN30 to distinguish likely functional involvement. Use of multiple methods to analyze replication and protein occupancies on chromatin. Development of a coherent model.
Weaknesses are failure to address copy number as a variable; insufficient validation of ChEC method relationship to exact initiation locus; lack of statistical analysis in some cases.
With regard to "insufficient validation of ChEC method relationship to exact initiation locus": The two potential initiation sites that one would monitor (non-displaced and displaced) are separated by less than 150 base pairs, and other techniques simply do not have the resolution necessary to distinguish such differences. Indeed, our new ChIP results presented in Figure 5 figure supplement 3 clearly demonstrate that while the resolution of ChIP is adequate to detect the reduction of MCM signal at the replication initiation site and its relocation to the RFB ( ~2 kb away), it lacks the resolution required to differentiate closely spaced MCM complexes.
Furthermore, as we suggest in the manuscript, our results are consistent with a model in which it is only the displaced MCM complex that is activated, whether in sir2 or WT. If no genotypedependent difference in initiation sites is even expected, it would be hard to interpret even the most precise replication-based assays.
We appreciate the reviewer pointing out that some statistical analyses were lacking: we have added statistical analysis for 2D gels (Figures 4D and 4E), EdU incorporation experiments in Figure 4F and disappearance of MCM ChEC and ChIP signal upon release of cells into HU (Figure 5 supplement 1 and Supplement 2).
Additional background and discussion for public review:
This paper broadly addresses the mechanism(s) that regulate replication origin firing in different chromatin contexts. The rDNA origin is present in each of ~180 tandem repeats of the rDNA sequence, representing a high potential origin density per length of DNA (9.1kb repeat unit). However, the average origin efficiency of rDNA origins is relatively low (~20% in wild-type cells), which reduces the replication load on the overall genome by reducing competition with origins throughout the genome for limiting replication initiation factors. Deletion of histone deacetylase SIR2, which silences PolII transcription within the rDNA, results in increased early activation or the rDNA origins (and reduced rate of overall genome replication). Previous work by the authors showed that MCM complexes loaded onto the rDNA origins (origin licensing) were laterally displaced (sliding) along the rDNA, away from a well-positioned nucleosome on one side. The authors' major hypothesis throughout this work is that the new MCM location(s) are intrinsically more efficient configurations for origin firing. The authors identify a chromatin remodeling enzyme, FUN30, whose deletion appears to suppress the earlier activation of rDNA origins in sir2∆ cells. Indeed, it appears that the reduction of rDNA origin activity in sir2∆ fun30∆ cells is severe enough to results in a substantial reduction in the rDNA array repeat length (number of repeats); the reduced rDNA length presumably facilitates it's more stable replication and maintenance.
Analysis of replication by 2D gels is marginally convincing, using 2D gels for this purpose is very challenging and tricky to quantify.
We address this criticism by carefuly quantifying 2 D gel results using single rARS signal for normalizing bubble arc as discussed below.
The more quantitative analysis by EdU incorporation is more convincing of the suppression of the earlier replication caused by SIR2 deletion.
We have also added quantification of EdU results to strengthen our arguments.
To address the mechanism of suppression, they analyze MCM positioning using ChEC, which in G1 cells shows partial displacement of MCM from normal position A to positions B and C in sir2∆ cells and similar but more complete displacement away from A to positions B and C in sir2fun30 cells. During S-phase in the presence of hydroxyurea, which slows replication progression considerably (and blocks later origin firing) MCM signals redistribute, which is interpreted to represent origin firing and bidirectional movement of MCMs (only one direction is shown), some of which accumulate near the replication fork barrier, consistent with their interpretation. They observe that MCMs displaced (in G1) to sites B or C in sir2∆ cells, disappear more rapidly during S-phase, whereas the similar dynamic is not observed in sir2∆fun30∆. This is the main basis for their conclusion that the B and C sites are more permissive than A. While this may be the simplest interpretation, there are limitations with this assay that undermine a rigorous conclusion (additional points below). The main problem is that we know the MCM complexes are mobile so disappearance may reflect displacement by other means including transcription which is high is the sir2∆ background. Indeed, the double mutant has greater level of transcription per repeat unit which might explain more displaced from A in G1. Thus, displacement might not always represent origin firing. Because the sir2 background profoundly changes transcription, and the double mutant has a much smaller array length associated with higher transcription, how can we rule out greater accessibility at site A, for example in sir2∆, leading to more firing, which is suppressed in sir2 fun30 due to greater MCM displacement away from A?
I think the critical missing data to solidly support their conclusions is a definitive determination of the site(s) of initiation using a more direct method, such as strand specific sequencing of EdU or nascent strand analysis. More direct comparisons of the strains with lower copy number to rule out this facet. As discussed in detail below, copy number reduction is known to suppress at least part of the sir2∆ effect so this looms over the interpretations. I think they are probably correct in their overall model based on the simplest interpretation of the data but I think it remains to be rigorously established. I think they should soften their conclusions in this respect.
Please see discussion below about these issues.
Reviewer #2 (Public Review):
Summary:
In this manuscript, the authors follow up on their previous work showing that in the absence of the Sir2 deacetylase the MCM replicative helicase at the rDNA spacer region is repositioned to a region of low nucleosome occupancy. Here they show that the repositioned displaced MCMs have increased firing propensity relative to non-displaced MCMs. In addition, they show that activation of the repositioned MCMs and low nucleosome occupancy in the adjacent region depend on the chromatin remodeling activity of Fun30.
Strengths:
The paper provides new information on the role of a conserved chromatin remodeling protein in the regulation of origin firing and in addition provides evidence that not all loaded MCMs fire and that origin firing is regulated at a step downstream of MCM loading.
Weaknesses:
The relationship between the author's results and prior work on the role of Sir2 (and Fob1) in regulation of rDNA recombination and copy number maintenance is not explored, making it difficult to place the results in a broader context. Sir2 has previously been shown to be recruited by Fob1, which is also required for DSB formation and recombination-mediated changes in rDNA copy number. Are the changes that the authors observe specifically in fun30 sir2 cells related to this pathway? Is Fob1 required for the reduced rDNA copy number in fun30 sir2 double mutant cells?
We have conducted additional studies in the fob1 background to address how FOB1 and the replication fork barrier (RFB) influence the kinetics of rDNA size reduction upon FUN30 deletion (Figure 2 - figure supplement 2), rDNA replication timing (Figure 2 - figure supplement 3), and rDNA origin firing using 2D gels (Figure 4C).
Strains lacking SIR2 exhibit unstable rDNA size, and FOB1 deletion stabilizes rDNA size in a sir2 background (and otherwise). Similarly, we found that FOB1 deletion influences the kinetics of rDNA size reduction in sir2 fun30 cells. Specifically, we were able to generate a fob1 sir2 fun30 strain with more than 150 copies. Nonetheless, and consistent with our model, this strain still exhibited delayed rDNA replication timing (Figure 2 - figure supplement 3), and its rDNA still shrank upon continuous culture (Figure 2 figure supplement 2). These results demonstrate that, although FOB1 affects the kinetics of rDNA size reduction in sir2 fun30 strains, the reduced rDNA array size or delayed replication timing upon FUN30 deletion size does not depend on FOB1.
The use of the fob1 background allowed us to compare the activation of rDNA origins in sir2 and sir2 fun30 strains with equally short rDNA sizes. 2D gels demonstrate robust and reproducible suppression of rDNA origin activity upon deletion of FUN30 in sir2 fob1 strains with 35 rDNA copies (Figure 4C). These results indicate that the main effect we are interested in—FUN30-induced reduction in origin firing—is independent of both FOB1 and rDNA size.
Our additional studies conclusively show that the FUN30-induced reduction in rDNA origin firing is independent of both FOB1 and rDNA size. These findings provide important insights into the mechanisms regulating rDNA copy number maintenance, placing our results within the broader context of existing knowledge on Sir2 and Fob1 functions.
Reviewer #3 (Public Review):
Summary:
Heterochromatin is characterized by low transcription activity and late replication timing, both dependent on the NAD-dependent protein deacetylase Sir2, the founding member of the sirtuins. This manuscript addresses the mechanism by which Sir2 delays replication timing at the rDNA in budding yeast. Previous work from the same laboratory (Foss et al. PLoS Genetics 15, e1008138) showed that Sir2 represses transcription-dependent displacement of the Mcm helicase in the rDNA. In this manuscript, the authors show convincingly that the repositioned Mcms fire earlier and that this early firing partly depends on the ATPase activity of the nucleosome remodeler Fun30. Using read-depth analysis of sorted G1/S cells, fun30 was the only chromatin remodeler mutant that somewhat delayed replication timing in sir2 mutants, while nhp10, chd1, isw1, htl1, swr1, isw2, and irc3 had not effect. The conclusion was corroborated with orthogonal assays including two-dimensional gel electrophoresis and analysis of EdU incorporation at early origins. Using an insightful analysis with an Mcm-MNase fusion (Mcm-ChEC), the authors show that the repositioned Mcms in sir2 mutants fire earlier than the Mcm at the normal position in wild type. This early firing at the repositioned Mcms is partially suppressed by Fun30. In addition, the authors show Fun30 affects nucleosome occupancy at the sites of the repositioned Mcm, providing a plausible mechanism for the effect of Fun30 on Mcm firing at that position. However, the results from the MNAse-seq and ChEC-seq assays are not fully congruent for the fun30 single mutant. Overall, the results support the conclusions providing a much better mechanistic understanding how Sir2 affects replication timing at rDNA,
The observation that the MNase-seq plot in fun30 mutant shows a large signal at the +3 nucleosome and somewhat smaller at position +2, while the ChEC-seq plot exhibits negligible signals, is indeed an important point of consideration. This discrepancy arises because most of the MCM in fun30 mutant remains at its original site where it abuts +1 nucleosome. As a result, the MCM-MNase fusion protein fails to reach and “light up” the +3 nucleosome, which is, nonetheless, well-visualized with exogenous MNase. The paucity of displaced MCMs, which is responsible for cutting +2 nucleosome, explains the discrepancy in the +2 nucleosome signal between exogenous MNase and CheC datasets in the fun30 mutant.
Despite this apparent discrepancy, the overall results support our conclusions and provide a much better mechanistic understanding of how Sir2 affects replication timing at rDNA. The MNaseseq data reflect nucleosome positioning and chromatin structure, while the ChEC-seq data specifically highlights the locations where MCM is bound and active.
Strengths
(1) The data clearly show that the repositioned Mcm helicase fires earlier than the Mcm in the wild type position.
(2) The study identifies a specific role for Fun30 in replication timing and an effect on nucleosome occupancy around the newly positioned Mcm helicase in sir2 cells.
Weaknesses
(1) It is unclear which strains were used in each experiment.
(2) The relevance of the fun30 phospho-site mutant (S20AS28A) is unclear.
We appreciate the reviewer pointing out places in which our manuscript omitted key pieces of information (items 1 and 3), we have included the strain numbers in our revision. With regard to point 2, we had written:
Fun30 is also known to play a role in the DNA damage response; specifically, phosphorylation of Fun30 on S20 and S28 by CDK1 targets Fun30 to sites of DNA damage, where it promotes DNA resection (Chen et al. 2016; Bantele et al. 2017). To determine whether the replication phenotype that we observed might be a consequence of Fun30's role in the DNA damage response, we tested non-phosphorylatable mutants for the ability to suppress early replication of the rDNA in sir2; these mutations had no effect on the replication phenotype (Figure 2B), arguing against a primary role for Fun30 in DNA damage repair that somehow manifests itself in replication.
(3) For some experiments (Figs. 3, 4, 6) it is unclear whether the data are reproducible and the differences significant. Information about the number of independent experiments and quantitation is lacking. This affects the interpretation, as fun30 seems to affect the +3 nucleosome much more than let on in the description.
We have provided replicas and quantitation for the results in these figures.
(Replica ChEC Southern blot with quantification (Figure 3 figure supplement 1), quantification and replicas for 2D gels in Figure 4 and replicas for nucleosome occupancy (Figure 6 supplement 1).
Recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
Fig. 3-Examination of MCM occupancy at the rDNA ARS region using a variation of ChEC.
Presumably these are these G1-arrested cells but does not seem to be stated. Please confirm.
The 2D gels results are not very convincing of their conclusions. We are asked to compare bubble to fork arcs at 30 minutes, but this is not feasible. It is the author's job to quantify the data from multiple replicates, but none is given. After much careful examination, comparing the relative intensities of ascending bubble and Y-arcs, I think I can accept that 4A shows highest early efficiency for sir2 over WT and fun30, which are similar to each other, and lowest for sir2 fun30, at 60 and 90 min.
In the revision we provide a careful quantification of the 2D gels in Figure 4. For assessing rDNA origin activity, we normalized the bubble arc during the HU time course to a single rARS signal, that appears as large 24.4kb Nhe1I fragment originating from the rightmost rDNA repeat (see Figures 4A and 4B). The description of the quantification in the text is provided below.
“Prior to separation on 2D gels, DNA was digested with NheI, which releases a 4.7 kb rARScontaining linear DNA fragment at the internal rDNA repeats (1N) and a much larger, 24.5 kb single-rARS-containing fragment originating from the rightmost repeat. In 2D gels, active origins generate replication bubble arc signals, whereas passive replication of an origin appears as a y-arc. Having a signal emanating from a single ARS-containing fragment simplifies the comparison of rDNA origin activity in strains with different numbers of rDNA repeats, such as in sir2 vs sir2 fun30 mutants. Origin activity is expressed as a ratio of the bubble to the single-ARS signal, effectively measuring the number of active rDNA origins per cell at a given time point.
As seen previously (Foss et al. 2019), deletion of SIR2 increased the number of activated rDNA origins, while deletion of FUN30 suppressed this effect. When analyzed in aggregate at 20, 30, 60 and 90 minutes following release into HU, the average number of activated rDNA origin activity in sir2 mutant was increased 6.3-fold compared to those in WT (5.0±2.3 in sir2 vs 0.8±0.4 in wt, p<0.05 by 2 tailed t-test), and the increased number was reduced upon FUN30 deletion (1.3±0.7 in sir2 fun30, p<0.05 by 2 tailed t-test vs sir2, NS for comparison to WT).”
However, for part 4B, they state (p. 11) that deletion of FUN30 in a SIR2 background had no perceptible effect (on ARS305) but I think the data appear otherwise: the FUN30 cells show more Y-arc than WT.
We now provide the assessment of ARS305 activity in HU cells as a ratio of bubble-arc to 1N signal. The reviewer is right that FUN30 has a more robust bubble arc signal compared to WT.
However, after normalization to 1N this difference did not appear significant (3.7 vs 5.1). Overall the analysis of activity or ARS305 origins demonstrates a reciprocity with the activity of rDNA origins in each of the four genotypes. Furthermore, this observation is confirmed in our EdU-based analysis of 111 genomic origins, with statistical analysis showing a very high level of significance (see below).
Ultimately, analysis of unsynchronized cells would give unambiguous results about origin efficiency. In this regard I note that analysis of rDNA origin firing by 2D gels with HU versus asynchronous gives different results in WT versus sir2∆, with no difference in unsynchronized cells (He et al. 2022). It would be interesting to test the strains here unsynchronized, though copy number size would still be a variable to address.
Origin activity in log cultures is typically assessed by comparing replication initiation within an origin, presenting as a bubble arc, to passively replicated DNA (Y-arc). However, such an analysis at tandemly arrayed origins, such as rDNA, is not feasible, as both active and passive replication are the result of activation of the same origins. This explains the lack of difference between WT and sir2 cells previously reported (He et al. 2022), which we have also observed. Differences in activation of rDNA origins in WT vs sir2 cells is clearly reflected in HU experiments, as was the case in the earlier report (He et al. 2022).
To address the issue of differences in copy number between sir2 and sir2 fun30 cells we have now done experiments in a fob1 background where we can equalize the copy number among the two genotypes. These 2D gels are presented in Figure 4C. We address this issue in the revised manuscript as follows:
“The overall impact of FUN30 deletion on rDNA origin activity in a sir2 background is expected to be a composite of two opposing effects: a suppression of rDNA origin activation and increased rDNA origin activation due to reduced rDNA size (Kwan et al. 2023). To evaluate the effect FUN30 on rDNA origin activation independently of rDNA size, we generated an isogenic set of strains in a fob1 background, all of which contain 35 copies of the rDNA repeat. (Deletion of FOB1 is necessary to stabilize rDNA copy number.) Comparing rDNA origin activity in sir2 versus sir2 fun30 genotypes, we observed a robust and reproducible reduction in rDNA origin activity upon FUN30 deletion. This finding confirms that the FUN30 suppresses rDNA origin firing in sir2 background independently of both rDNA size and FOB1 status.”
-EdU analysis is more convincing regarding relative effects on genome versus rDNA, however, again, the effect of reduced rDNA array size in the sir2 fun30 cells may also be the proximal cause of the reduced effect on genome (early origins) replication rather than a direct effect on origin efficiency. No statistic provided to support that fun30 suppresses sir2 for rDNA activity.
This comment raises three distinct, but related, issues:
First, the reviewer is asking whether the reduced rDNA size, of the magnitude we observed in sir2 fun30 cells, could by itself be responsible for increased origin activity elsewhere in the genome, just because there is less rDNA that needs to be replicated. As noted earlier (Kwan et al. 2023), Kwan et al. examined the effect of rDNA size reduction and observed: 1) marked increased in rDNA origin activity and 2) reciprocal reduction in origin activity elsewhere in the genome. This counterintuitive finding suggests that a smaller rDNA size exerts more competition for limited replication resources compared to a larger rDNA size. In light of this, our findings with FUN30 deletion become even more compelling. The suppression of rDNA firing upon FUN30 deletion is so significant that it overrides the expected effects of rDNA size reduction.
Second, the reviewer points out our lack of statistical analysis to support our contention that fun30 suppresses sir2 with regard to rDNA origin activity. We have now addressed this issue as well, by quantifying 2D gel signals, as described above in the text that begins with "Prior to separation on 2D gels, DNA was digested with NheI ...".
Third, we have now provided a statistical analysis to support our conclusion that EdU-based analysis of activity of 111 early origins shows suppression upon deletion of SIR2 that is largely reversed by additional deletion of FUN30.
"Deletion of FUN30 in a sir2 background partially restored EdU incorporation at early origins, concomitant with reduced EdU incorporation at rDNA origins. In particular, the median value of log10 of read depths at 111 early origins, as the data are shown in Figure 4F, dropped from 6.5 for wild type to 6.2 for sir2 but then returned almost to wild type levels (6.4) in sir2 fun30. The p value obtained by Student's t test, comparing the drop in 111 origins from wild type to sir2 with that from wild type to sir2 fun30 was highly significant (<< 10-16) In contrast, FUN30 deletion in the WT background did not reduce EdU incorporation at genomic origins (median 6.6). These findings highlight that FUN30 deletion-induced suppression of rDNA origins in sir2 is accompanied by the activation of genomic origins."
Use loss of Mcm-ChEC signal as proxy for origin firing. Reasonably convincing that decrease correlates with origin firing on a one-to-one basis (Fig. 5B), though no statistic given.
We provide the statistical analysis in Figure 5-figure supplement 1.
However, there is no demonstration of ability to observe this correlation with fine resolution as needed for the claims here. It seems equally possible that sir2 deletion causes more firing by repositioning MCMs to a better location or that the prior location, which still contains substantial MCM, becomes more permissive. The MCM signal appears to be mobile, so perhaps the role of FUN30 is to prevent to mobility of MCM away from the original site in WT cells; note that significantly less Mcm signal is at the original position in sir2 fun30. No accumulation of MCM occurs near the RFB in WT (and fun30) cells. I understand that origin firing is lower in WT but raises concerns about sensitivity and dynamic range of this assay and that MCM positions may reflect transcription versus replication.
Please see the section above labeled "Addressing Alternative Explanations".
Is Fig 6A Y-axis correctly labeled? I understand this figure to represent MNase-seq reads; is there any Mcm2-ChEC-seq in part A?
We have corrected the labeling. 6A represent MNase-seq reads. Thank you for pointing this out.
I understand part B to represent nucleosome-sized fragments released by Mcm2-ChEC interpreted to be nucleosomes. But could they be large fragments potentially containing adjacent MCM-double hexamers?
Our representation of ChEC-seq data in Figure 1 supplement 1, where we can see the entire spectrum of fragment sizes, demonstrates two distinct populations of fragments: nucleosome size and MCM-size fragments.
Reviewer #2 (Recommendations For The Authors):
Suggestions for the authors to consider:
(1) The authors make a good case for the importance of replication balance between rDNA and euchromatin in ensuring that the genome is replicated in a timely fashion. This seems to be clearly regulated by Sir2. However, Sir2 also affects rDNA copy number and suppresses unequal cross over events, which are stimulates by Fob1. Does Fun30 suppress Fob1-dependent recombination events in sir2D cells?
It is unclear why FUN30 only affects rDNA repeat copy number in sir2 cells. Why doesn't Fun30 reduce copy number in wild-type cells?
Deletion of SIR2 causes rightward repositioning of MCMs to a position where they are more prone to fire, as shown by our HU ChEC datasets in which we show that the repositioned MCMs are more prone to activation than the non-repositioned ones. FUN30 deletion suppresses activation of these, activation-prone repositioned MCMs, as shown by HU ChEC. This suppression of rDNA origin activation in sir2 cells causes rDNA to shrink. In fun30 single mutants, due to the paucity of non-repositioned MCMs, we do not observe significant suppression of rDNA origin firing, and consequently, there is no reduction in rDNA size in fun30 cells.
(2) The authors use Mcm-MNase to map the location of the MCM helicase. Can these results be confirmed using the more standard and direct ChIP assay to examine changes in MCM localization
We carried out suggested MCM ChIP experiments and present these results in Figure 5 supplement 2 and supplement 3. These ChIP data demonstrate that:
(1) MCM signal disappears preferentially at early origins compared to late origins, as seen in our ChEC results.
(2) The disappearance of ChEC signal at rDNA origins in sir2 mutant is accompanied by the signal accumulation at the RFB, consistent with fork stalling at the RFB mirroring the results we obtained by ChEC. While these results indicate that that ChIP has adequate resolution to detect MCM repositioning at 2 kb, scale, its resolution was insufficient for fine scale discrimination of repositioned and non-repositioned MCMs.
In this regard, the specific role of Fun30 in regulation of MCM firing at rDNA is interesting.
Does Fun30 localize to the ARS region of rDNA? How is Fun30 specifically recruited to rDNA?
We carried out ChIP for Fun30 and observed, similarly to previous reports (Durand-Dubief et al. 2012), a wide distribution of Fun30 throughout the genome and at rDNA. We have elected not to include these results in the current manuscript.
(3) The 2D gels in Figure 4 are difficult to interpret. The bubble to arc ratios in fun30D seem different from both wild-type and sir2D. It may be helpful to the reader to quantify the bubble to arc ratios. fun30D also seems to be affecting ARS305 by itself.
We provide quantification of 2 D gels in Figure 4.
(4) Figure 5.
(4.1) For examining origin firing based on the disappearance of the Mcm-MNase reads, is HU arrest necessary? HU may be causing indirect effects due to replication fork stalling. In principle, the authors should be able to perform this analysis without HU, since their cells are released from synchronized arrest in G1 (and at least for the first cell cycle should proceed synchronously on to S phase). In addition, validation of Mcm-ChEC results using ChIP for one of the subunits of the MCM complex would increase confidence in the results.
The HU arrest allows us to examine early events in DNA replication at much finer spatial and temporal resolution than it would be possible without it.
We have now used Mcm2 ChIP to confirm that the signal disappears at the MCM loading site in HU in sir2 cells as discussed above (Figure 5 figure supplement 3). However, the resolution is inadequate to discriminate non-repositioned vs repositioned MCMs.
(4.2) The non-displaced Mcm-ChEC signal in sir2D seems like it's decreasing more than in wildtype cells. Explain. It would be helpful to quantify these results by integrating the area under each peek (or based on read numbers). It looks like one of the displaced Mcm signals (the one more distal from the non-displaced) is changing at a similar rate to the non-displaced.
Integrating the area under each Mcm-ChEC peak or using read numbers is superfluous for the following reasons: (1) The rectangular appearance of the peaks in Figure 5 clearly reflects signal intensity, making additional numerical integration redundant. (2) The visual differences between wild-type and sir2D cells are distinct and sufficient for drawing conclusions without further quantification. (3) Keeping the analysis straightforward avoids unnecessary complexity and maintains clarity.
(4.3) Can the authors explain why fun30D seems to be suppressing only one of the 2 displaced Mcms from firing?
We speculate that the local environment is more conductive for firing one of two displaced MCMs, but we do not understand why.
(5) Figure 6. Why would the deletion of SIR2, a silencing factor, results in increased nucleosome occupancy at rDNA?
If we understand correctly, the reviewer is referring to a small increase in +2 and +3 signal in sir2 compared to the WT. In WT G1 cells, there is a single MCM between +1 and +3 nucleosome. This space cannot accommodate a +2 nucleosome in G1 cells because MCM is loaded at that position in most cells (in G2 cells however, this space is occupied by a nucleosome (Foss et al., 2019). MCM repositioning in sir2 mutant would displace MCM from this location making it possible for this space to be now occupied by a nucleosome.
The changes in nuc density seem modest. Also, nucleosome density is similarly increased in sir2D and fun30D cells, but sir2 has a dramatic effect on origin firing but fun30D does not. Explain.
We believe that the FUN30 status makes most of the difference for firing of displaced MCMs.
Since there are few displaced MCMs in SIR2 cells, there is not large impact on origin firing. Furthermore, the rDNA already fires late in WT cells, so our ability to detect further delay upon FUN30 deletion could be more difficult.
(6) Discussion. At rDNA Sir2 may simply act by deacetylating nucleosomes and decreasing their mobility. This is unrelated to compaction which is usually only invoked regarding the activities of the full SIR complex (Sir2/3/4) at telomeres and the mating type locus. The arguments regarding polymerase size, compaction etc may not be relevant to the main point since although the budding yeast Sir2 participates in heterochromatin formation at the mating type loci and telomeres, at rDNA it may act locally near its recruitment site at the RFB.
This is a valid point. We have added this sentence in the discussion to highlight the differences between silencing at rDNA and those at the silent mating loci and telomeres that SIR-complex dependent.
“Steric arguments such as these are even less compelling when made for rDNA than for the silent mating type loci and telomeres, because chromatin compaction has been studied mostly in the context of the complete Sir complex (Sir1-4). In contrast, Sir1, 3, and 4 are not present at the rDNA.”
Minor
It would be interesting to see if deletion of any histone acetyltranferases acts in a similar way to Fun30 to reduce rDNA copy number in sir2D cells.
Thank you for this suggestion.
Reviewer #3 (Recommendations For The Authors):
(1) The design of Figure 3 could be improved. A scheme could help understand the assay without flipping back to Figure 1. The numbers below the gel bands need definition.
We have included the scheme describing the restriction and MCM-MNase cut sites and the location of the probe for the Southern blot.
(2) The design of Figure 4 could be improved by adding a scheme to help interpret the 2d gel picture. The figure also lacks quantitation. Are the results reproducible and the differences significant?
We have added the scheme, quantification and statistics in Figure 4.
(3) Please list in each figure legend the exact strains from Table S1 which were used.
We have included the strain numbers in the Figure legend.
Durand-Dubief M, Will WR, Petrini E, Theodorou D, Harris RR, Crawford MR, Paszkiewicz K, Krueger F, Correra RM, Vetter AT et al. 2012. SWI/SNF-like chromatin remodeling factor Fun30 supports point centromere function in S. cerevisiae. PLoS Genet 8: e1002974.
Foss EJ, Gatbonton-Schwager T, Thiesen AH, Taylor E, Soriano R, Lao U, MacAlpine DM, Bedalov A. 2019. Sir2 suppresses transcription-mediated displacement of Mcm2-7 replicative helicases at the ribosomal DNA repeats. PLoS Genet 15: e1008138.
He Y, Petrie MV, Zhang H, Peace JM, Aparicio OM. 2022. Rpd3 regulates single-copy origins independently of the rDNA array by opposing Fkh1-mediated origin stimulation. Proc Natl Acad Sci U S A 119: e2212134119.
Kwan EX, Alvino GM, Lynch KL, Levan PF, Amemiya HM, Wang XS, Johnson SA, Sanchez JC, Miller MA, Croy M et al. 2023. Ribosomal DNA replication time coordinates completion of genome replication and anaphase in yeast. Cell Rep 42: 112161.
-
-
Author response:
eLife assessment
This study is a detailed investigation of how chromatin structure influences replication origin function in yeast ribosomal DNA, with focus on the role of the histone deacetylase Sir2 and the chromatin remodeler Fun30. Convincing evidence shows that Sir2 does not affect origin licensing but rather affects local transcription and nucleosome positioning which correlates with increased origin firing. However, the evidence remains incomplete as the methods employed do not rigorously establish a key aspect of the mechanism, fully address some alternative models, or sufficiently relate to prior results. Overall, this is a valuable advance for the field that could be improved to establish a more robust paradigm.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
This paper presents a mechanistic …
Author response:
eLife assessment
This study is a detailed investigation of how chromatin structure influences replication origin function in yeast ribosomal DNA, with focus on the role of the histone deacetylase Sir2 and the chromatin remodeler Fun30. Convincing evidence shows that Sir2 does not affect origin licensing but rather affects local transcription and nucleosome positioning which correlates with increased origin firing. However, the evidence remains incomplete as the methods employed do not rigorously establish a key aspect of the mechanism, fully address some alternative models, or sufficiently relate to prior results. Overall, this is a valuable advance for the field that could be improved to establish a more robust paradigm.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
This paper presents a mechanistic study of rDNA origin regulation in yeast by SIR2. Each of the ~180 tandemly repeated rDNA gene copies contains a potential replication origin. Early-efficient initiation of these origins is suppressed by Sir2, reducing competition with origins distributed throughout the genome for rate-limiting initiation factors. Previous studies by these authors showed that SIR2 deletion advances replication timing of rDNA origins by a complex mechanism of transcriptional de-repression of a local PolII promoter causing licensed origin proteins (MCMcomplexes) to re-localize (slide along the DNA) to a different (and altered) chromatin environment. In this study, they identify a chromatin remodeler, FUN30, that suppresses the sir2∆ effect, and remarkably, results in a contraction of the rDNA to about one-quarter it's normal length/number of repeats, implicating replication defects of the rDNA. Through examination of replication timing, MCM occupancy and nucleosome occupancy on the chromatin in sir2, fun30, and double mutants, they propose a model where nucleosome position relative to the licensed origin (MCM complexes) intrinsically determines origin timing/efficiency. While their interpretations of the data are largely reasonable and can be interpreted to support their model, a key weakness is the connection between Mcm ChEC signal disappearance and origin firing. While the cyclical chromatin association-dissociation of MCM proteins with potential origin sequences may be generally interpreted as licensing followed by firing, dissociation may also result from passive replication and as shown here, displacement by transcription and/or chromatin remodeling.
While it is true that both transcription and passive replication can cause the signal of MCM-ChEC to disappear, neither can cause selective disappearance of the displaced complex without affecting the non-displaced complex. Indeed, in the case of transcription, RNA polymerase transcribing C-pro would have to first dislodge the normally positioned MCM complex before even reaching the displaced complex. Furthermore, deletion of FUN30 leads to both more C-pro transcription and less disappearance of the displaced MCM complex. It is important to keep in mind that this cannot somehow reflect continuous replenishment of displaced MCMs with newly loaded MCMs, since the cells are in S phase and licensing is restricted to G1.
Moreover, linking its disappearance from chromatin in the ChEC method with such precise resolution needs to be validated against an independent method to determine the initiation site(s). Differences in rDNA copy number and relative transcription levels also are not directly accounted for, obscuring a clearer interpretation of the results.
Copy number reduction of the magnitude caused by deletion of SIR2 and FUN30 does not suppress the sir2D effect (i.e. early replication of the rDNA), but rather exacerbates it. In particular, deletion of SIR2 and FUN30 causes the rDNA to shrink to approximately 35 copies. Kwan et al., 2023 (PMID: 36842087) have shown that reduction of rDNA copy number to 35 causes a dramatic acceleration of rDNA replication in a SIR2 strain. Thus, the effect of rDNA size on replication timing reinforces our conclusion that deletion of FUN30 suppresses rDNA replication.
However, to address this concern directly, in the revision we will include 2 D gels in fob1 strains with equal number of repeats that allows to conclude that the effect of FUN30 deletion in suppressing rDNA origin firing is independent of either rDNA size or FOB1. The figure of the critical 2 D gels is shown below in the reply to reviewer 2.
Nevertheless, this paper makes a valuable advance with the finding of Fun30 involvement, which substantially reduces rDNA repeat number in sir2∆ background. The model they develop is compelling and I am inclined to agree, but I think the evidence on this specific point is purely correlative and a better method is needed to address the initiation site question. The authors deserve credit for their efforts to elucidate our obscure understanding of the intricacies of chromatin regulation. At a minimum, I suggest their conclusions on these points of concern should be softened and caveats discussed. Statistical analysis is lacking for some claims.
Strengths are the identification of FUN30 as suppressor, examination of specific mutants of FUN30 to distinguish likely functional involvement. Use of multiple methods to analyze replication and protein occupancies on chromatin. Development of a coherent model.
Weaknesses are failure to address copy number as a variable; insufficient validation of ChEC method relationship to exact initiation locus; lack of statistical analysis in some cases.
The two potential initiation sites that one would monitor (non-displaced and displaced) are separated by less than 150 base pairs, and other techniques simply do not have the resolution necessary to distinguish such differences. Furthermore, as we suggest in the manuscript, our results are consistent with a model in which it is only the displaced MCM complex that is activated, whether in sir2 or WT. If no genotype-dependent difference in initiation sites is even expected, it would be hard to interpret even the most precise replication-based assays. However, the reviewer is correct that this is a novel technique and that confirmation with a well-established technique is comforting, therefore we are performing ChIP experiments to corroborate, to the extent possible, the conclusions that we reached with ChEC.
We appreciate the reviewer pointing out that some statistical analyses were lacking, and we will correct this in a revised manuscript.
Additional background and discussion for public review:
This paper broadly addresses the mechanism(s) that regulate replication origin firing in different chromatin contexts. The rDNA origin is present in each of ~180 tandem repeats of the rDNA sequence, representing a high potential origin density per length of DNA (9.1kb repeat unit). However, the average origin efficiency of rDNA origins is relatively low (~20% in wild-type cells), which reduces the replication load on the overall genome by reducing competition with origins throughout the genome for limiting replication initiation factors. Deletion of histone deacetylase SIR2, which silences PolII transcription within the rDNA, results in increased early activation or the rDNA origins (and reduced rate of overall genome replication). Previous work by the authors showed that MCM complexes loaded onto the rDNA origins (origin licensing) were laterally displaced (sliding) along the rDNA, away from a well-positioned nucleosome on one side. The authors' major hypothesis throughout this work is that the new MCM location(s) are intrinsically more efficient configurations for origin firing. The authors identify a chromatin remodeling enzyme, FUN30, whose deletion appears to suppress the earlier activation of rDNA origins in sir2∆ cells. Indeed, it appears that the reduction of rDNA origin activity in sir2∆ fun30∆ cells is severe enough to results in a substantial reduction in the rDNA array repeat length (number of repeats); the reduced rDNA length presumably facilitates it's more stable replication and maintenance.
Analysis of replication by 2D gels is marginally convincing, using 2D gels for this purpose is very challenging and tricky to quantify. The more quantitative analysis by EdU incorporation is more convincing of the suppression of the earlier replication caused by SIR2 deletion.
To address the mechanism of suppression, they analyze MCM positioning using ChEC, which in G1 cells shows partial displacement of MCM from normal position A to positions B and C in sir2∆ cells and similar but more complete displacement away from A to positions B and C in sir2fun30 cells. During S-phase in the presence of hydroxyurea, which slows replication progression considerably (and blocks later origin firing) MCM signals redistribute, which is interpreted to represent origin firing and bidirectional movement of MCMs (only one direction is shown), some of which accumulate near the replication fork barrier, consistent with their interpretation. They observe that MCMs displaced (in G1) to sites B or C in sir2∆ cells, disappear more rapidly during S-phase, whereas the similar dynamic is not observed in sir2∆fun30∆. This is the main basis for their conclusion that the B and C sites are more permissive than A. While this may be the simplest interpretation, there are limitations with this assay that undermine a rigorous conclusion (additional points below). The main problem is that we know the MCM complexes are mobile so disappearance may reflect displacement by other means including transcription which is high is the sir2∆ background. Indeed, the double mutant has greater level of transcription per repeat unit which might explain more displaced from A in G1. Thus, displacement might not always represent origin firing. Because the sir2 background profoundly changes transcription, and the double mutant has a much smaller array length associated with higher transcription, how can we rule out greater accessibility at site A, for example in sir2∆, leading to more firing, which is suppressed in sir2 fun30 due to greater MCM displacement away from A?
I think the critical missing data to solidly support their conclusions is a definitive determination of the site(s) of initiation using a more direct method, such as strand specific sequencing of EdU or nascent strand analysis. More direct comparisons of the strains with lower copy number to rule out this facet. As discussed in detail below, copy number reduction is known to suppress at least part of the sir2∆ effect so this looms over the interpretations. I think they are probably correct in their overall model based on the simplest interpretation of the data but I think it remains to be rigorously established. I think they should soften their conclusions in this respect.
Reviewer #2 (Public Review):
Summary:
In this manuscript, the authors follow up on their previous work showing that in the absence of the Sir2 deacetylase the MCM replicative helicase at the rDNA spacer region is repositioned to a region of low nucleosome occupancy. Here they show that the repositioned displaced MCMs have increased firing propensity relative to non-displaced MCMs. In addition, they show that activation of the repositioned MCMs and low nucleosome occupancy in the adjacent region depend on the chromatin remodeling activity of Fun30.
Strengths:
The paper provides new information on the role of a conserved chromatin remodeling protein in the regulation of origin firing and in addition provides evidence that not all loaded MCMs fire and that origin firing is regulated at a step downstream of MCM loading.
Weaknesses:
The relationship between the author's results and prior work on the role of Sir2 (and Fob1) in regulation of rDNA recombination and copy number maintenance is not explored, making it difficult to place the results in a broader context. Sir2 has previously been shown to be recruited by Fob1, which is also required for DSB formation and recombination-mediated changes in rDNA copy number. Are the changes that the authors observe specifically in fun30 sir2 cells related to this pathway? Is Fob1 required for the reduced rDNA copy number in fun30 sir2 double mutant cells?
Strains lacking SIR2 have unstable rDNA size, and FOB1 deletion stabilizes rDNA size in sir2 background. Likewise, FOB1 deletion influences the kinetics rDNA size reduction in sir2 fun30 cells. However, the main effect of Fun30 in sir2 cells we were interested in, suppression of rDNA replication, is preserved in fob1 background, arguing that the observed effect is independent of Fob1 (see figure below). Given that the main focus of the paper is regulation of rDNA origins activity and that these changes were independent of Fob1, we had elected not to include these results in the original manuscript but will gladly include them in the revision.
Besides refuting the possible role of Fob1 in the FUN30-mediated activation of rDNA origin firing in sir2 cells, the use of fob1 background enabled us compare the activation of rDNA origins in the sir2 and sir2 fun30 strains with equally short rDNA size. The 2-D gels demonstrate a dramatic suppression of rDNA origin activity upon deletion of FUN30 in the sir2 fob1 strains with 35 rDNA copies.
Author response image 1.
The deletion of FUN30 diminishes the replication bubble signal in a fob1 sir2 strain with 35 rDNA copies by more than tenfold. The single rARS signal, marked with the arrow, originates from the rightmost rDNA repeat. This specific rightmost rDNA NheI fragment is approximately 25 kb in size, distinctly larger than the 4.7 kb NheI 1N rARS-containing fragments that originate from the internal rDNA repeats.
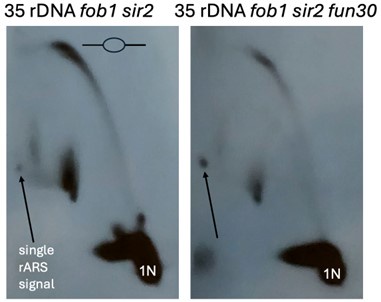
Reviewer #3 (Public Review):
Summary:
Heterochromatin is characterized by low transcription activity and late replication timing, both dependent on the NAD-dependent protein deacetylase Sir2, the founding member of the sirtuins. This manuscript addresses the mechanism by which Sir2 delays replication timing at the rDNA in budding yeast. Previous work from the same laboratory (Foss et al. PLoS Genetics 15, e1008138) showed that Sir2 represses transcription-dependent displacement of the Mcm helicase in the rDNA. In this manuscript, the authors show convincingly that the repositioned Mcms fire earlier and that this early firing partly depends on the ATPase activity of the nucleosome remodeler Fun30. Using read-depth analysis of sorted G1/S cells, fun30 was the only chromatin remodeler mutant that somewhat delayed replication timing in sir2 mutants, while nhp10, chd1, isw1, htl1, swr1, isw2, and irc5 had not effect. The conclusion was corroborated with orthogonal assays including two-dimensional gel electrophoresis and analysis of EdU incorporation at early origins. Using an insightful analysis with an Mcm-MNase fusion (Mcm-ChEC), the authors show that the repositioned Mcms in sir2 mutants fire earlier than the Mcm at the normal position in wild type. This early firing at the repositioned Mcms is partially suppressed by Fun30. In addition, the authors show Fun30 affects nucleosome occupancy at the sites of the repositioned Mcm, providing a plausible mechanism for the effect of Fun30 on Mcm firing at that position. However, the results from the MNAse-seq and ChEC-seq assays are not fully congruent for the fun30 single mutant. Overall, the results support the conclusions providing a much better mechanistic understanding how Sir2 affects replication timing at rDNA.
The reason that the results for the fun30 single mutant appear incongruent, with a larger signal of the +2 nucleosome in the MNase-seq plot but a negligible signal in the ChEC-seq plot is the paucity of displaced Mcm in the fun30 single mutant. Given the relative absence of displaced MCMs, the MCM-MNase fusion protein can't "light up" the +2 nucleosome. We will comment on this in the revision to clarify this.
Strengths
(1) The data clearly show that the repositioned Mcm helicase fires earlier than the Mcm in the wild type position.
(2) The study identifies a specific role for Fun30 in replication timing and an effect on nucleosome occupancy around the newly positioned Mcm helicase in sir2 cells.
Weaknesses
(1) It is unclear which strains were used in each experiment.
(2) The relevance of the fun30 phospho-site mutant (S20AS28A) is unclear.
(3) For some experiments (Figs. 3, 4, 6) it is unclear whether the data are reproducible and the differences significant. Information about the number of independent experiments and quantitation is lacking. This affects the interpretation, as fun30 seems to affect the +3 nucleosome much more than let on in the description.
We appreciate the reviewer pointing out places in which our manuscript omitted key pieces of information (items 1 and 3), and we will fix these oversights in our revision.
With regard to point 2, we had written:
“Fun30 is also known to play a role in the DNA damage response; specifically, phosphorylation of Fun30 on S20 and S28 by CDK1 targets Fun30 to sites of DNA damage, where it promotes DNA resection (Chen et al. 2016; Bantele et al. 2017). To determine whether the replication phenotype that we observed might be a consequence of Fun30's role in the DNA damage response, we tested non-phosphorylatable mutants for the ability to suppress early replication of the rDNA in sir2; these mutations had no effect on the replication phenotype (Figure 2B), arguing against a primary role for Fun30
in DNA damage repair that somehow manifests itself in replication.”
We will expand on this to clarify our point in the revision.
-
eLife assessment
This study is a detailed investigation of how chromatin structure influences replication origin function in yeast ribosomal DNA, with focus on the role of the histone deacetylase Sir2 and the chromatin remodeler Fun30. Convincing evidence shows that Sir2 does not affect origin licensing but rather affects local transcription and nucleosome positioning which correlates with increased origin firing. However, the evidence remains incomplete as the methods employed do not rigorously establish a key aspect of the mechanism, fully address some alternative models, or sufficiently relate to prior results. Overall, this is a valuable advance for the field that could be improved to establish a more robust paradigm.
-
Reviewer #1 (Public Review):
Summary:
This paper presents a mechanistic study of rDNA origin regulation in yeast by SIR2. Each of the ~180 tandemly repeated rDNA gene copies contains a potential replication origin. Early-efficient initiation of these origins is suppressed by Sir2, reducing competition with origins distributed throughout the genome for rate-limiting initiation factors. Previous studies by these authors showed that SIR2 deletion advances replication timing of rDNA origins by a complex mechanism of transcriptional de-repression of a local PolII promoter causing licensed origin proteins (MCMcomplexes) to re-localize (slide along the DNA) to a different (and altered) chromatin environment. In this study, they identify a chromatin remodeler, FUN30, that suppresses the sir2∆ effect, and remarkably, results in a contraction of …
Reviewer #1 (Public Review):
Summary:
This paper presents a mechanistic study of rDNA origin regulation in yeast by SIR2. Each of the ~180 tandemly repeated rDNA gene copies contains a potential replication origin. Early-efficient initiation of these origins is suppressed by Sir2, reducing competition with origins distributed throughout the genome for rate-limiting initiation factors. Previous studies by these authors showed that SIR2 deletion advances replication timing of rDNA origins by a complex mechanism of transcriptional de-repression of a local PolII promoter causing licensed origin proteins (MCMcomplexes) to re-localize (slide along the DNA) to a different (and altered) chromatin environment. In this study, they identify a chromatin remodeler, FUN30, that suppresses the sir2∆ effect, and remarkably, results in a contraction of the rDNA to about one-quarter it's normal length/number of repeats, implicating replication defects of the rDNA. Through examination of replication timing, MCM occupancy and nucleosome occupancy on the chromatin in sir2, fun30, and double mutants, they propose a model where nucleosome position relative to the licensed origin (MCM complexes) intrinsically determines origin timing/efficiency. While their interpretations of the data are largely reasonable and can be interpreted to support their model, a key weakness is the connection between Mcm ChEC signal disappearance and origin firing. While the cyclical chromatin association-dissociation of MCM proteins with potential origin sequences may be generally interpreted as licensing followed by firing, dissociation may also result from passive replication and as shown here, displacement by transcription and/or chromatin remodeling. Moreover, linking its disappearance from chromatin in the ChEC method with such precise resolution needs to be validated against an independent method to determine the initiation site(s). Differences in rDNA copy number and relative transcription levels also are not directly accounted for, obscuring a clearer interpretation of the results. Nevertheless, this paper makes a valuable advance with the finding of Fun30 involvement, which substantially reduces rDNA repeat number in sir2∆ background. The model they develop is compelling and I am inclined to agree, but I think the evidence on this specific point is purely correlative and a better method is needed to address the initiation site question. The authors deserve credit for their efforts to elucidate our obscure understanding of the intricacies of chromatin regulation. At a minimum, I suggest their conclusions on these points of concern should be softened and caveats discussed. Statistical analysis is lacking for some claims.
Strengths are the identification of FUN30 as suppressor, examination of specific mutants of FUN30 to distinguish likely functional involvement. Use of multiple methods to analyze replication and protein occupancies on chromatin. Development of a coherent model.
Weaknesses are failure to address copy number as a variable; insufficient validation of ChEC method relationship to exact initiation locus; lack of statistical analysis in some cases.
Additional background and discussion for public review:
This paper broadly addresses the mechanism(s) that regulate replication origin firing in different chromatin contexts. The rDNA origin is present in each of ~180 tandem repeats of the rDNA sequence, representing a high potential origin density per length of DNA (9.1kb repeat unit). However, the average origin efficiency of rDNA origins is relatively low (~20% in wild-type cells), which reduces the replication load on the overall genome by reducing competition with origins throughout the genome for limiting replication initiation factors. Deletion of histone deacetylase SIR2, which silences PolII transcription within the rDNA, results in increased early activation or the rDNA origins (and reduced rate of overall genome replication). Previous work by the authors showed that MCM complexes loaded onto the rDNA origins (origin licensing) were laterally displaced (sliding) along the rDNA, away from a well-positioned nucleosome on one side. The authors' major hypothesis throughout this work is that the new MCM location(s) are intrinsically more efficient configurations for origin firing. The authors identify a chromatin remodeling enzyme, FUN30, whose deletion appears to suppress the earlier activation of rDNA origins in sir2∆ cells. Indeed, it appears that the reduction of rDNA origin activity in sir2∆ fun30∆ cells is severe enough to results in a substantial reduction in the rDNA array repeat length (number of repeats); the reduced rDNA length presumably facilitates it's more stable replication and maintenance.
Analysis of replication by 2D gels is marginally convincing, using 2D gels for this purpose is very challenging and tricky to quantify. The more quantitative analysis by EdU incorporation is more convincing of the suppression of the earlier replication caused by SIR2 deletion.
To address the mechanism of suppression, they analyze MCM positioning using ChEC, which in G1 cells shows partial displacement of MCM from normal position A to positions B and C in sir2∆ cells and similar but more complete displacement away from A to positions B and C in sir2fun30 cells. During S-phase in the presence of hydroxyurea, which slows replication progression considerably (and blocks later origin firing) MCM signals redistribute, which is interpreted to represent origin firing and bidirectional movement of MCMs (only one direction is shown), some of which accumulate near the replication fork barrier, consistent with their interpretation. They observe that MCMs displaced (in G1) to sites B or C in sir2∆ cells, disappear more rapidly during S-phase, whereas the similar dynamic is not observed in sir2∆fun30∆. This is the main basis for their conclusion that the B and C sites are more permissive than A. While this may be the simplest interpretation, there are limitations with this assay that undermine a rigorous conclusion (additional points below). The main problem is that we know the MCM complexes are mobile so disappearance may reflect displacement by other means including transcription which is high is the sir2∆ background. Indeed, the double mutant has greater level of transcription per repeat unit which might explain more displaced from A in G1. Thus, displacement might not always represent origin firing. Because the sir2 background profoundly changes transcription, and the double mutant has a much smaller array length associated with higher transcription, how can we rule out greater accessibility at site A, for example in sir2∆, leading to more firing, which is suppressed in sir2 fun30 due to greater MCM displacement away from A?
I think the critical missing data to solidly support their conclusions is a definitive determination of the site(s) of initiation using a more direct method, such as strand specific sequencing of EdU or nascent strand analysis. More direct comparisons of the strains with lower copy number to rule out this facet. As discussed in detail below, copy number reduction is known to suppress at least part of the sir2∆ effect so this looms over the interpretations. I think they are probably correct in their overall model based on the simplest interpretation of the data but I think it remains to be rigorously established. I think they should soften their conclusions in this respect.
-
Reviewer #2 (Public Review):
Summary:
In this manuscript, the authors follow up on their previous work showing that in the absence of the Sir2 deacetylase the MCM replicative helicase at the rDNA spacer region is repositioned to a region of low nucleosome occupancy. Here they show that the repositioned displaced MCMs have increased firing propensity relative to non-displaced MCMs. In addition, they show that activation of the repositioned MCMs and low nucleosome occupancy in the adjacent region depend on the chromatin remodeling activity of Fun30.
Strengths:
The paper provides new information on the role of a conserved chromatin remodeling protein in the regulation of origin firing and in addition provides evidence that not all loaded MCMs fire and that origin firing is regulated at a step downstream of MCM loading.
Weaknesses:
The …
Reviewer #2 (Public Review):
Summary:
In this manuscript, the authors follow up on their previous work showing that in the absence of the Sir2 deacetylase the MCM replicative helicase at the rDNA spacer region is repositioned to a region of low nucleosome occupancy. Here they show that the repositioned displaced MCMs have increased firing propensity relative to non-displaced MCMs. In addition, they show that activation of the repositioned MCMs and low nucleosome occupancy in the adjacent region depend on the chromatin remodeling activity of Fun30.
Strengths:
The paper provides new information on the role of a conserved chromatin remodeling protein in the regulation of origin firing and in addition provides evidence that not all loaded MCMs fire and that origin firing is regulated at a step downstream of MCM loading.
Weaknesses:
The relationship between the author's results and prior work on the role of Sir2 (and Fob1) in regulation of rDNA recombination and copy number maintenance is not explored, making it difficult to place the results in a broader context. Sir2 has previously been shown to be recruited by Fob1, which is also required for DSB formation and recombination-mediated changes in rDNA copy number. Are the changes that the authors observe specifically in fun30 sir2 cells related to this pathway? Is Fob1 required for the reduced rDNA copy number in fun30 sir2 double mutant cells?
-
Reviewer #3 (Public Review):
Summary:
Heterochromatin is characterized by low transcription activity and late replication timing, both dependent on the NAD-dependent protein deacetylase Sir2, the founding member of the sirtuins. This manuscript addresses the mechanism by which Sir2 delays replication timing at the rDNA in budding yeast. Previous work from the same laboratory (Foss et al. PLoS Genetics 15, e1008138) showed that Sir2 represses transcription-dependent displacement of the Mcm helicase in the rDNA. In this manuscript, the authors show convincingly that the repositioned Mcms fire earlier and that this early firing partly depends on the ATPase activity of the nucleosome remodeler Fun30. Using read-depth analysis of sorted G1/S cells, fun30 was the only chromatin remodeler mutant that somewhat delayed replication timing in sir2 …
Reviewer #3 (Public Review):
Summary:
Heterochromatin is characterized by low transcription activity and late replication timing, both dependent on the NAD-dependent protein deacetylase Sir2, the founding member of the sirtuins. This manuscript addresses the mechanism by which Sir2 delays replication timing at the rDNA in budding yeast. Previous work from the same laboratory (Foss et al. PLoS Genetics 15, e1008138) showed that Sir2 represses transcription-dependent displacement of the Mcm helicase in the rDNA. In this manuscript, the authors show convincingly that the repositioned Mcms fire earlier and that this early firing partly depends on the ATPase activity of the nucleosome remodeler Fun30. Using read-depth analysis of sorted G1/S cells, fun30 was the only chromatin remodeler mutant that somewhat delayed replication timing in sir2 mutants, while nhp10, chd1, isw1, htl1, swr1, isw2, and irc5 had not effect. The conclusion was corroborated with orthogonal assays including two-dimensional gel electrophoresis and analysis of EdU incorporation at early origins. Using an insightful analysis with an Mcm-MNase fusion (Mcm-ChEC), the authors show that the repositioned Mcms in sir2 mutants fire earlier than the Mcm at the normal position in wild type. This early firing at the repositioned Mcms is partially suppressed by Fun30. In addition, the authors show Fun30 affects nucleosome occupancy at the sites of the repositioned Mcm, providing a plausible mechanism for the effect of Fun30 on Mcm firing at that position. However, the results from the MNAse-seq and ChEC-seq assays are not fully congruent for the fun30 single mutant. Overall, the results support the conclusions providing a much better mechanistic understanding how Sir2 affects replication timing at rDNA,
Strengths
(1) The data clearly show that the repositioned Mcm helicase fires earlier than the Mcm in the wild type position.
(2) The study identifies a specific role for Fun30 in replication timing and an effect on nucleosome occupancy around the newly positioned Mcm helicase in sir2 cells.Weaknesses
(1) It is unclear which strains were used in each experiment.
(2) The relevance of the fun30 phospho-site mutant (S20AS28A) is unclear.
(3) For some experiments (Figs. 3, 4, 6) it is unclear whether the data are reproducible and the differences significant. Information about the number of independent experiments and quantitation is lacking. This affects the interpretation, as fun30 seems to affect the +3 nucleosome much more than let on in the description. -
