Decoupling of the onset of anharmonicity between a protein and its surface water around 200 K
Curation statements for this article:-
Curated by eLife

eLife assessment
The study answers the important question of whether the conformational dynamics of proteins are slaved by the motion of solvent water or are intrinsic to the polypeptide. The results from neutron scattering experiments, involving isotopic labelling, carried out on a set of four structurally different proteins are convincing, showing that protein motions are not coupled to the solvent. A strength of this work is the study of a set of proteins using spectroscopy covering a range of resolutions. The work is of broad interest to researchers in the fields of protein biophysics and biochemistry.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The protein dynamical transition at ~200 K, where the biomolecule transforms from a harmonic, non-functional form to an anharmonic, functional state, has been thought to be slaved to the thermal activation of dynamics in its surface hydration water. Here, by selectively probing the dynamics of protein and hydration water using elastic neutron scattering and isotopic labeling, we found that the onset of anharmonicity in the two components around 200 K is decoupled. The one in protein is an intrinsic transition, whose characteristic temperature is independent of the instrumental resolution time, but varies with the biomolecular structure and the amount of hydration, while the one of water is merely a resolution effect.
Article activity feed
-

-
-
-

eLife assessment
The study answers the important question of whether the conformational dynamics of proteins are slaved by the motion of solvent water or are intrinsic to the polypeptide. The results from neutron scattering experiments, involving isotopic labelling, carried out on a set of four structurally different proteins are convincing, showing that protein motions are not coupled to the solvent. A strength of this work is the study of a set of proteins using spectroscopy covering a range of resolutions. The work is of broad interest to researchers in the fields of protein biophysics and biochemistry.
-
Reviewer #1 (Public Review):
Zheng et al. study the 'glass' transitions that occurs in proteins at ca. 200K using neutron diffraction and differential isotopic labeling (hydrogen/deuterium) of the protein and solvent. To overcome limitations in previous studies, this work is conducted in parallel with 4 proteins (myoglobin, cytochrome P450, lysozyme and green fluorescent protein) and experiments were performed at a range of instrument time resolutions (1ns - 10ps). The author's data looks compelling, and suggests that transitions in the protein and solvent behavior are not coupled and contrary to some previous reports, the apparent water transition temperature is a 'resolution effect'; i.e. instrument response is limited. This is likely to be important in the field, as a reassessment of solvent 'slaving' and the role of the hydration …
Reviewer #1 (Public Review):
Zheng et al. study the 'glass' transitions that occurs in proteins at ca. 200K using neutron diffraction and differential isotopic labeling (hydrogen/deuterium) of the protein and solvent. To overcome limitations in previous studies, this work is conducted in parallel with 4 proteins (myoglobin, cytochrome P450, lysozyme and green fluorescent protein) and experiments were performed at a range of instrument time resolutions (1ns - 10ps). The author's data looks compelling, and suggests that transitions in the protein and solvent behavior are not coupled and contrary to some previous reports, the apparent water transition temperature is a 'resolution effect'; i.e. instrument response is limited. This is likely to be important in the field, as a reassessment of solvent 'slaving' and the role of the hydration shell on protein dynamics should be reassessed in light of these findings.
-
Reviewer #2 (Public Review):
Summary:
The manuscript entitled "Decoupling of the Onset of Anharmonicity between a Protein and Its Surface Water around 200 K" by Zheng et al. presents a neutron scattering study trying to elucidate if at the dynamical transition temperature water and protein motions are coupled. The origin of the dynamical transition temperature has been debated for decades, specifically its relation to hydration.
The study is rather well conducted, with a lot of effort to acquire the perdeuterated proteins, and some results are interesting.
-
Author response:
The following is the authors’ response to the previous reviews.
eLife assessment
The study answers the important question of whether the conformational dynamics of proteins are slaved by the motion of solvent water or are intrinsic to the polypeptide. The results from neutron scattering experiments, involving isotopic labelling, carried out on a set of four structurally different proteins are convincing, showing that protein motions are not coupled to the solvent. A strength of this work is the study of a set of proteins using spectroscopy covering a range of resolutions. A minor weakness is the limited description of computational methods and analysis of data. The work is of broad interest to researchers in the fields of protein biophysics and biochemistry.
We thank the editors and reviewers for the positive and …
Author response:
The following is the authors’ response to the previous reviews.
eLife assessment
The study answers the important question of whether the conformational dynamics of proteins are slaved by the motion of solvent water or are intrinsic to the polypeptide. The results from neutron scattering experiments, involving isotopic labelling, carried out on a set of four structurally different proteins are convincing, showing that protein motions are not coupled to the solvent. A strength of this work is the study of a set of proteins using spectroscopy covering a range of resolutions. A minor weakness is the limited description of computational methods and analysis of data. The work is of broad interest to researchers in the fields of protein biophysics and biochemistry.
We thank the editors and reviewers for the positive and encouraging comments.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
Zheng et al. study the 'glass' transitions that occurs in proteins at ca. 200K using neutron diffraction and differential isotopic labeling (hydrogen/deuterium) of the protein and solvent. To overcome limitations in previous studies, this work is conducted in parallel with 4 proteins (myoglobin, cytochrome P450, lysozyme and green fluorescent protein) and experiments were performed at a range of instrument time resolutions (1ns - 10ps). The author's data looks compelling, and suggests that transitions in the protein and solvent behavior are not coupled and contrary to some previous reports, the apparent water transition temperature is a 'resolution effect'; i.e. instrument response limited. This is likely to be important in the field, as a reassessment of solvent 'slaving' and the role of the hydration shell on protein dynamics should be reassessed in light of these findings.
Strengths:
The use of multiple proteins and instruments with a rate of energy resolution/ timescales.
We thank the reviewer for highlighting our key findings.
Weaknesses:
The paper could be organised to better allow the comparison of the complete dataset collected. The extent of hydration clearly influences the protein transition temperature. The authors suggest that "water can be considered here as lubricant or plasticizer which facilitates the motion of the biomolecule." This may be the case, but the extent of hydration may also alter the protein structure.
Following the reviewer’s suggestion, we studied the secondary structure content and tertiary structure of CYP protein at different hydration levels (h = 0.2 and 0.4) through molecular dynamics simulation. As shown in Table S2 and Fig. S6, the extent of hydration does not alter the protein secondary structure content and overall packing. Thus, this result also suggests that water molecules have more influence on protein dynamics than on protein structure. We added the above results in the revised SI.
Reviewer #2 (Public Review):
Summary:
The manuscript entitled "Decoupling of the Onset of Anharmonicity between a Protein and Its Surface Water around 200 K" by Zheng et al. presents a neutron scattering study trying to elucidate if at the dynamical transition temperature water and protein motions are coupled. The origin of the dynamical transition temperature is highly debated since decades and specifically its relation to hydration.
Strengths:
The study is rather well conducted, with a lot of efforts to acquire the perdeuterated proteins, and some results are interesting.
We thank the reviewer for highlighting our key findings.
Weaknesses:
The MD data presented appears to be missing description of the methods used.
If these data support the authors claim that different levels of hydration do not affect the protein structure, careful analysis of the MD simulation data should be presented that show the systems are properly equilibrated under each condition. Additionally, methods are needed to describe the MD parameters and methods used, and for how long the simulations were run.
We have now added the methods of MD simulation into the revised SI.
“The initial structure of protein cytochrome P450 (CYP) for simulations was taken from PDB crystal structure (2ZAX). Two protein monomers were filled in a cubic box. 1013 and 2025 water molecules were inserted into the box randomly to reach a mass ratio of 0.2 and 0.4 gram water/1 gram protein, respectively, which mimics the experimental condition. Then 34 sodium counter ions were added to keep the system neutral in charge. The CHARMM 27 force field in the GROMACS package was used for CYP, whereas the TIP4P/Ew model was chosen for water. The simulations were carried out at a broad range of temperatures from 360 K to 100 K, with a step of 5 K. At each temperature, after the 5000 steps energy-minimization procedure, a 10 ns NVT is conducted. After that, a 30 ns NPT simulation was carried out at 1 atm with the proper periodic boundary condition. As shown in Fig. S7, 30 ns is sufficient to equilibrate the system. The temperature and pressure of the system is controlled by the velocity rescaling method and the method by Parrinello and Rahman, respectively. All bonds of water in all the simulations were constrained with the LINCS algorithm to maintain their equilibration length. In all the simulations, the system was propagated using the leap-frog integration algorithm with a time step of 2 fs. The electrostatic interactions were calculated using the Particle Mesh Ewalds (PME) method. A non-bond pair-list cutoff of 1 nm was used and the pair-list was updated every 20 fs. All MD simulations were performed using GROMACS 4.5.1 software packages.”
Recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
Response to author's changes:
See public review: The MD data presented appears to be missing description of the methods used.
If these data support the authors claim that different levels of hydration do not affect the protein structure, careful analysis of the MD simulation data should be presented that show the systems are properly equilibrated under each condition. Additionally, methods are needed to describe the MD parameters and methods used, and for how long the simulations were run.
We have now added the methods of MD simulation into the revised SI. Please see Reply 5.
Reviewer #2 (Recommendations For The Authors):
The authors answered my questions and substantially improved the manuscript.
We thank the reviewer for the encouraging comments .
-
-
eLife assessment
The study answers the important question of whether the conformational dynamics of proteins are slaved by the motion of solvent water or are intrinsic to the polypeptide. The results from neutron scattering experiments, involving isotopic labelling, carried out on a set of four structurally different proteins are convincing, showing that protein motions are not coupled to the solvent. A strength of this work is the study of a set of proteins using spectroscopy covering a range of resolutions. A minor weakness is the limited description of computational methods and analysis of data. The work is of broad interest to researchers in the fields of protein biophysics and biochemistry.
-
Reviewer #1 (Public Review):
Summary:
Zheng et al. study the 'glass' transitions that occurs in proteins at ca. 200K using neutron diffraction and differential isotopic labeling (hydrogen/deuterium) of the protein and solvent. To overcome limitations in previous studies, this work is conducted in parallel with 4 proteins (myoglobin, cytochrome P450, lysozyme and green fluorescent protein) and experiments were performed at a range of instrument time resolutions (1ns - 10ps). The author's data looks compelling, and suggests that transitions in the protein and solvent behavior are not coupled and contrary to some previous reports, the apparent water transition temperature is a 'resolution effect'; i.e. instrument response limited. This is likely to be important in the field, as a reassessment of solvent 'slaving' and the role of the …
Reviewer #1 (Public Review):
Summary:
Zheng et al. study the 'glass' transitions that occurs in proteins at ca. 200K using neutron diffraction and differential isotopic labeling (hydrogen/deuterium) of the protein and solvent. To overcome limitations in previous studies, this work is conducted in parallel with 4 proteins (myoglobin, cytochrome P450, lysozyme and green fluorescent protein) and experiments were performed at a range of instrument time resolutions (1ns - 10ps). The author's data looks compelling, and suggests that transitions in the protein and solvent behavior are not coupled and contrary to some previous reports, the apparent water transition temperature is a 'resolution effect'; i.e. instrument response limited. This is likely to be important in the field, as a reassessment of solvent 'slaving' and the role of the hydration shell on protein dynamics should be reassessed in light of these findings.
Strengths:
The use of multiple proteins and instruments with a rate of energy resolution/ timescales.
Weaknesses:
The paper could be organised to better allow the comparison of the complete dataset collected.
The extent of hydration clearly influences the protein transition temperature. The authors suggest that "water can be considered here as lubricant or plasticizer which facilitates the motion of the biomolecule." This may be the case, but the extent of hydration may also alter the protein structure. -
Reviewer #2 (Public Review):
Summary:
The manuscript entitled "Decoupling of the Onset of Anharmonicity between a Protein and Its Surface Water around 200 K" by Zheng et al. presents a neutron scattering study trying to elucidate if at the dynamical transition temperature water and protein motions are coupled. The origin of the dynamical transition temperature is highly debated since decades and specifically its relation to hydration.
Strengths:
The study is rather well conducted, with a lot of efforts to acquire the perdeuterated proteins, and some results are interesting.
Weaknesses:
The MD data presented appears to be missing description of the methods used.
If these data support the authors claim that different levels of hydration do not affect the protein structure, careful analysis of the MD simulation data should be presented …Reviewer #2 (Public Review):
Summary:
The manuscript entitled "Decoupling of the Onset of Anharmonicity between a Protein and Its Surface Water around 200 K" by Zheng et al. presents a neutron scattering study trying to elucidate if at the dynamical transition temperature water and protein motions are coupled. The origin of the dynamical transition temperature is highly debated since decades and specifically its relation to hydration.
Strengths:
The study is rather well conducted, with a lot of efforts to acquire the perdeuterated proteins, and some results are interesting.
Weaknesses:
The MD data presented appears to be missing description of the methods used.
If these data support the authors claim that different levels of hydration do not affect the protein structure, careful analysis of the MD simulation data should be presented that show the systems are properly equilibrated under each condition. Additionally, methods are needed to describe the MD parameters and methods used, and for how long the simulations were run. -
Author response:
The following is the authors’ response to the original reviews.
eLife assessment:
The study answers the important question of whether the conformational dynamics of proteins are slaved by the motion of solvent water or are intrinsic to the polypeptide. The results from neutron scattering experiments, involving isotopic labelling, carried out on a set of four structurally different proteins are convincing, showing that protein motions are not coupled to the solvent. A strength of this work is the study of a set of proteins using spectroscopy covering a range of resolutions, however, it suffers from some scholarly shortcomings and limited discussion of results. The work is of broad interest to researchers in the fields of protein biophysics and biochemistry.
Reply 1: We thank the editors and reviewers for the positive …
Author response:
The following is the authors’ response to the original reviews.
eLife assessment:
The study answers the important question of whether the conformational dynamics of proteins are slaved by the motion of solvent water or are intrinsic to the polypeptide. The results from neutron scattering experiments, involving isotopic labelling, carried out on a set of four structurally different proteins are convincing, showing that protein motions are not coupled to the solvent. A strength of this work is the study of a set of proteins using spectroscopy covering a range of resolutions, however, it suffers from some scholarly shortcomings and limited discussion of results. The work is of broad interest to researchers in the fields of protein biophysics and biochemistry.
Reply 1: We thank the editors and reviewers for the positive and encouraging comments.
Reviewer #1 (Public Review):
Summary:
Zheng et al. study the 'glass' transitions that occur in proteins at ca. 200K using neutron diffraction and differential isotopic labeling (hydrogen/deuterium) of the protein and solvent. To overcome limitations in previous studies, this work is conducted in parallel with 4 proteins (myoglobin, cytochrome P450, lysozyme, and green fluorescent protein) and experiments were performed at a range of instrument time resolutions (1ns - 10ps). The author's data looks compelling, and suggests that transitions in the protein and solvent behavior are not coupled and contrary to some previous reports, the apparent water transition temperature is a 'resolution effect'; i.e. instrument response is limited. This is likely to be important in the field, as a reassessment of solvent 'slaving' and the role of the hydration shell on protein dynamics should be reassessed in light of these findings.
Strengths:
The use of multiple proteins and instruments with a rate of energy resolution/ timescales.
Reply 2: We thank the reviewer for highlighting our key findings.
Weaknesses:
The paper could be organised to better allow the comparison of the complete dataset collected. The extent of hydration clearly influences the protein transition temperature. The authors suggest that "water can be considered here as lubricant or plasticizer which facilitates the motion of the biomolecule." This may be the case, but the extent of hydration may also alter the protein structure.
Reply 3: Following the reviewer’s suggestion, we studied the secondary structure content and tertiary structure of CYP protein at different hydration levels (h = 0.2 and 0.4) through molecular dynamics simulation. As shown in Table S2 and Figure S6, the extent of hydration does not alter the protein secondary structure content and overall packing. Thus, this result also suggests that water molecules have more influence on protein dynamics than on protein structure.
Reviewer #2 (Public Review):
Summary:
The manuscript entitled "Decoupling of the Onset of Anharmonicity between a Protein and Its Surface Water around 200 K" by Zheng et al. presents a neutron scattering study trying to elucidate if at the dynamical transition temperature water and protein motions are coupled. The origin of the dynamical transition temperature has been highly debated for decades, specifically its relation to hydration.
Strengths:
The study is rather well conducted, with a lot of effort to acquire the perdeuterated proteins, and some results are interesting.
Reply 4: We thank the reviewer for highlighting our key findings.
Weaknesses:
The present work could certainly contribute some arguments, but I have the feeling that not all known facts are properly discussed.
The points the authors should carefully discuss are the following:
(1) Daniel et al. (10.1016/S0006-3495(98)77694-5) have shown that enzymes can be functional below the dynamical transition temperature which is at odds with some of the claims of the authors.
Reply 5: Following the reviewer’s suggestion, we added the following paragraph into the Introduction into the revised main text.
“Although exceptions have been reported (Biophys. J. 1998, 75, 2504.), the dynamical transition has been linked to the thermal onset of function in a number of proteins, e.g, myoglobin (Biochemistry, 1975, 14, 5355-5373), ribonuclease (Nature, 1992, 357, 423-424.), elastase ( Biochemistry, 1994, 33, 9285-9293.) and bacteriorhodopsin (PNAS, 1993, 90, 9668-9672.), all of which become inactive below the dynamical transition temperature.”
(2) It is not as easy to say that protonated proteins in D2O reflect protein dynamics while perdeuterated proteins in H2O reflect water dynamics. A recent study by Nidriche et al. (PRX LIFE 2, 013005 (2024)) reveals that H <-> D exchange is much faster than usually assumed and has important consequences for such studies.
Reply 6: For the sample preparation, all the H-proteins were dissolved in D2O to allow full deuterium exchange of all exchangeable hydrogen atoms and then lyophilized for 12 hours to obtain the dry sample. The lyophilized H-protein is then put into a desiccator with D2O, placed in the glove box purged with nitrogen gas, to absorb D2O till the desired hydration level, h (gram water/gram protein). In contrast, the preparation of the deuterated proteins was conducted in the opposite way. The D-proteins were dissolved in H2O to allow full hydrogen exchange of all exchangeable deuterium atoms and then lyophilized for 12 hours to obtain the dry sample. The lyophilized D-protein is then put into a desiccator with H2O to absorb H2O till the desired h. This procedure can avoid H-D exchange during experiments. We added the above methods into the revised SI.
(3) A publication by Jasnin et al. (10.1039/b923878f) on heparin sulfate shows a resolution effect.
Reply 7: Based on the data from Jasnin et al. (10.1039/b923878f), we found that the dynamical transition of heparin sulfate did not exhibit a strong resolution effect. Estimating the dynamical transition of mean square displacement (MSD) for nanosecond motions in all heparan sulfate samples is challenging due to the absence of data on nanosecond motion of HS-dry.
(4) The authors should discuss the impact of the chosen q-range on their findings (see Phys. Chem. Chem. Phys., 2012, 14, 4927-4934, where the authors see a huge effect!).
Reply 8: Following the reviewer's suggestion, we calculated Ton of H-protein in D2O in the q-range from 0.45-0.9 Å⁻¹ and 1.1-1.75 Å⁻¹. The results are summarized in Table S2 and Table S3. As shown in Tables S2-3., the q-range does not alter the Ton of proteins. We added the above results into the revised SI.
(5) The authors underline that the dynamical transition is intrinsic to the protein. However, Cupane et al. (ref 12) have shown that it can also be found in a mixture of amino acids without any protein backbone.
Reply 9: Following the reviewer’s suggestion, we added the following discussion into the revised main text.
“Unfreezing of the protein structural relaxation might facilitate these conformational jumps, turning on its functionality. However, as revealed by Ref (Journal of biological physics, 2010, 36, 291-297.), the denatured form of lysozyme also exhibits a dynamical transition, similar to that seen in its folded native form. Additionally, the dynamical transition also can be found in the mixture of amino acids (Physical Review Letters, 2012, 109, 128102.). Hence, one can argue that the activation of the structural relaxation of the biomolecule above the dynamical transition temperature is a necessary but insufficient condition for the protein to function, as the latter also requires the biomolecule assuming the correctly folded 3-dimensional structure.”
(6) The authors say that they find similar dependences from MSD. They should explain that the MSD is inversely proportional to the summed intensities squared.
Reply 10: Following the reviewer’s suggestion, we added the estimation of mean-squared atomic displacement (MSD) in the revised SI.
“The mean-squared atomic displacement
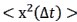 was estimated by performing Gaussian approximation, where
was estimated by performing Gaussian approximation, where 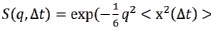 . The values of q used for Gaussian fitting ranges from 0.45 to 0.9 Å (Biophys. J. 2006, 91, 2573.).”
. The values of q used for Gaussian fitting ranges from 0.45 to 0.9 Å (Biophys. J. 2006, 91, 2573.).”(7) A decoupling between water dynamics and membrane dynamics has already been discussed by K. Wood, G. Zaccai et al.
Reply 11: Following the reviewer’s suggestion, we added the discussion in revised main text. “The results from the neutron scattering experiments suggest that the dynamical transition in proteins is an intrinsic property of the biomolecule and strongly depends on the amount of water surrounding it. Such an intrinsic transition can result either from a critical phase transition, e.g., water to ice (PNAS 2007, 104, 18049-18054.; JPCB, 1999, 103, 8036-8050), or from freezing of the structural relaxation of the system beyond the equilibrium time (~100-1000 s) of the experiment, in analogy to the glass transition in polymers from rubbery state to the glass form (Philosophical Magazine, 2004, 84, 1341-1353.; Science, 1995, 267, 1939-1945.; Colloid and Polymer Science, 1995, 273, 413-420.).”
(8) The fact that transition temperature in lipid membranes is higher when the membrane is dry is also well known (A.V. Popova, D.K. Hincha, BMC Biophys. 4, 11 (2011)).
Reply 12: We agree with the reviewer that transition temperature in lipid membranes is higher when the membrane is dry is well known. We cited this work as reference.
(9) The authors should mention the slope (K/min) they used for DSC and discuss the impact of it on the results.
Reply 13: Following the reviewer’s suggestion, we added DSC measurements in revised SI. “DSC measurements were performed by using the METTLER instruments DSC3+. The sample was sealed in a pan of aluminum. An empty pan was used as a reference. All the experiments were carried out in the temperature range from 150 to 300 K with a heating rate of 1 K/min. The heating rate of DSC is the same as neutron experiments.”
(10) In the introduction, the authors should present the different explanations forwarded for the dynamical transition.
Reply 14: Following the reviewer’s suggestion, we added different explanations forwarded for the dynamical transition in the Introduction in revised main text.
“The dynamical transition of protein represents a significant change in the internal mobility of proteins, which has garnered various explanations. One theory suggests it's due to the behavior of water in the hydration shell, transitioning from rigid to fluid at certain temperatures, thus influencing protein flexibility. Another theory considers the transition as an inherent property of the protein, where thermal energy allows the protein to access a wider range of conformations. ”
Reviewer #1 (Recommendations For The Authors):
A major strength of the work is the parallel experiments performed on each of the 4 proteins. To allow better comparison of these it would be helpful to present these combined data in relevant figures to make a side-by-side comparison easier. A summary table of Ton (and potentially TDSC) values would also be helpful.
Reply 15: Following the reviewer’s suggestion, we summarized the Ton of proteins in Table S5 and Table S6.
The effect of hydration on protein structure should be considered. Alterations in protein secondary and tertiary structure would be expected to alter dynamics and thus could be seen as a change in Ton.
Reply 16: The detailed analysis and discussion are presented in Reply 3.
No uncertainty (error) in Ton values is presented. Could these be estimated from e.g. a comparison of protein Ton values measured under identical sample conditions with different spectrometers?
Reply 17: It would be hard to compare Ton of proteins measured with different spectrometers because different spectrometers have different energy resolutions. For example, the energy resolutions of HFBS, DNA and OSIRIS are 1 μeV, 13 μeV, 25.4 μeV and 100 μeV, respectively.
More detail is needed to correctly describe/define the proteins used for the study - e.g. P450 is a family of enzymes, so which one was used?
Reply 18: We used P450 from Pseudomonas putida for the study. The PDB ID is 2ZAX. We added this information in the revised SI.
P450 and myoglobin also have heme cofactors. Were these deuterated as part of the protein preparation?
Reply 19: The heme cofactors were deuterated as part of the protein preparation. For D-protein, all the cell culture for E.coli is deuterated.
-
-
eLife assessment:
The study answers the important question of whether the conformational dynamics of proteins are slaved by the motion of solvent water or are intrinsic to the polypeptide. The results from neutron scattering experiments, involving isotopic labelling, carried out on a set of four structurally different proteins are convincing, showing that protein motions are not coupled to the solvent. A strength of this work is the study of a set of proteins using spectroscopy covering a range of resolutions, however, it suffers from some scholarly shortcomings and limited discussion of results. The work is of broad interest to researchers in the fields of protein biophysics and biochemistry.
-
Reviewer #1 (Public Review):
Summary:
Zheng et al. study the 'glass' transitions that occur in proteins at ca. 200K using neutron diffraction and differential isotopic labeling (hydrogen/deuterium) of the protein and solvent. To overcome limitations in previous studies, this work is conducted in parallel with 4 proteins (myoglobin, cytochrome P450, lysozyme, and green fluorescent protein) and experiments were performed at a range of instrument time resolutions (1ns - 10ps). The author's data looks compelling, and suggests that transitions in the protein and solvent behavior are not coupled and contrary to some previous reports, the apparent water transition temperature is a 'resolution effect'; i.e. instrument response is limited. This is likely to be important in the field, as a reassessment of solvent 'slaving' and the role of the …
Reviewer #1 (Public Review):
Summary:
Zheng et al. study the 'glass' transitions that occur in proteins at ca. 200K using neutron diffraction and differential isotopic labeling (hydrogen/deuterium) of the protein and solvent. To overcome limitations in previous studies, this work is conducted in parallel with 4 proteins (myoglobin, cytochrome P450, lysozyme, and green fluorescent protein) and experiments were performed at a range of instrument time resolutions (1ns - 10ps). The author's data looks compelling, and suggests that transitions in the protein and solvent behavior are not coupled and contrary to some previous reports, the apparent water transition temperature is a 'resolution effect'; i.e. instrument response is limited. This is likely to be important in the field, as a reassessment of solvent 'slaving' and the role of the hydration shell on protein dynamics should be reassessed in light of these findings.
Strengths:
The use of multiple proteins and instruments with a rate of energy resolution/ timescales.
Weaknesses:
The paper could be organised to better allow the comparison of the complete dataset collected.
The extent of hydration clearly influences the protein transition temperature. The authors suggest that "water can be considered here as lubricant or plasticizer which facilitates the motion of the biomolecule." This may be the case, but the extent of hydration may also alter the protein structure. -
Reviewer #2 (Public Review):
Summary:
The manuscript entitled "Decoupling of the Onset of Anharmonicity between a Protein and Its Surface Water around 200 K" by Zheng et al. presents a neutron scattering study trying to elucidate if at the dynamical transition temperature water and protein motions are coupled. The origin of the dynamical transition temperature has been highly debated for decades, specifically its relation to hydration.
Strengths:
The study is rather well conducted, with a lot of effort to acquire the perdeuterated proteins, and some results are interesting.
Weaknesses:
The present work could certainly contribute some arguments, but I have the feeling that not all known facts are properly discussed.
The points the authors should carefully discuss are the following:
(1) Daniel et al. (10.1016/S0006-3495(98)77694-5) have …
Reviewer #2 (Public Review):
Summary:
The manuscript entitled "Decoupling of the Onset of Anharmonicity between a Protein and Its Surface Water around 200 K" by Zheng et al. presents a neutron scattering study trying to elucidate if at the dynamical transition temperature water and protein motions are coupled. The origin of the dynamical transition temperature has been highly debated for decades, specifically its relation to hydration.
Strengths:
The study is rather well conducted, with a lot of effort to acquire the perdeuterated proteins, and some results are interesting.
Weaknesses:
The present work could certainly contribute some arguments, but I have the feeling that not all known facts are properly discussed.
The points the authors should carefully discuss are the following:
(1) Daniel et al. (10.1016/S0006-3495(98)77694-5) have shown that enzymes can be functional below the dynamical transition temperature which is at odds with some of the claims of the authors.
(2) It is not as easy to say that protonated proteins in D2O reflect protein dynamics while perdeuterated proteins in H2O reflect water dynamics. A recent study by Nidriche et al. (PRX LIFE 2, 013005 (2024)) reveals that H <-> D exchange is much faster than usually assumed and has important consequences for such studies.
(3) A publication by Jasnin et al. (10.1039/b923878f) on heparin sulfate shows a resolution effect.
(4) The authors should discuss the impact of the chosen q-range on their findings (see Phys. Chem. Chem. Phys., 2012, 14, 4927-4934, where the authors see a huge effect !).
(5) The authors underline that the dynamical transition is intrinsic to the protein. However, Cupane et al. (ref 12) have shown that it can also be found in a mixture of amino acids without any protein backbone.
(6) The authors say that they find similar dependences from MSD. They should explain that the MSD is inversely proportional to the summed intensities squared.
(7) A decoupling between water dynamics and membrane dynamics has already been discussed by K. Wood, G. Zaccai et al.
(8) The fact that transition temperature in lipid membranes is higher when the membrane is dry is also well known (A.V. Popova, D.K. Hincha, BMC Biophys. 4, 11 (2011)).
(9) The authors should mention the slope (K/min) they used for DSC and discuss the impact of it on the results.
(10) In the introduction, the authors should present the different explanations forwarded for the dynamical transition.
-
