Fast evolution of SOS-independent multi-drug resistance in bacteria
Curation statements for this article:-
Curated by eLife

eLife Assessment
This study presents a valuable observation of how deletion of a major repair protein in bacteria can facilitate the rise of mutations that confer resistance against a range of different antibiotics. The data presented are convincing, and the authors addressed the concerns raised by the reviewers in their resubmission, improving the strength of their findings.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The killing mechanism of many antibiotics involves the induction of DNA damage, either directly or indirectly, which activates the SOS response. RecA, the master regulator of the SOS response, has been shown to play a central role in the evolution of resistance to fluoroquinolones, even after short-term exposure. While this paradigm is well established for DNA-damaging antibiotics, it remains unclear whether β-lactams elicit similar resistance dynamics or depend on RecA and SOS-mediated mechanisms. In this study, we observed a rapid and stable evolution of β-lactam resistance (20-fold MIC increase within 8 hr) in Escherichia coli lacking RecA after a single exposure to ampicillin. Contrary to expectation, this resistance emerged through an SOS-independent mechanism involving two distinct evolutionary forces: increased mutational supply and antibiotic-driven selection. Specifically, we found that RecA deletion impaired DNA repair and downregulated base excision repair pathways, while concurrently repressing the transcription of antioxidative defence genes. This dual impairment led to excessive accumulation of reactive oxygen species (ROS), which in turn promoted the emergence of resistance-conferring mutations. While ampicillin treatment did not alter survival, it selectively enriched for rare mutants arising in the RecA-deficient and ROS-elevated background. Collectively, our findings demonstrate that this oxidative environment, together with compromised DNA repair capacity, increases genetic instability and creates a selective landscape favouring the expansion of resistant clones. These results highlight the repair-redox axis as a key determinant of bacterial evolvability under antimicrobial stress.
Article activity feed
-

-
-

eLife Assessment
This study presents a valuable observation of how deletion of a major repair protein in bacteria can facilitate the rise of mutations that confer resistance against a range of different antibiotics. The data presented are convincing, and the authors addressed the concerns raised by the reviewers in their resubmission, improving the strength of their findings.
-
Reviewer #1 (Public review):
Summary:
Jin et al. investigated how the bacterial DNA damage (SOS) response and its regulator protein RecA affects the development of drug resistance under short-term exposure to beta-lactam antibiotics. Canonically, the SOS response is triggered by DNA damage, which results in the induction of error-prone DNA repair mechanisms. These error-prone repair pathways can increase mutagenesis in the cell, leading to the evolution of drug resistance. Thus, inhibiting the SOS regulator RecA has been proposed as means to delay the rise of resistance.
In this paper, the authors deleted the RecA protein from E. coli and exposed this ∆recA strain to selective levels of the beta-lactam antibiotic, ampicillin. After an 8h treatment, they washed the antibiotic away and allowed the surviving cells to recover in regular …
Reviewer #1 (Public review):
Summary:
Jin et al. investigated how the bacterial DNA damage (SOS) response and its regulator protein RecA affects the development of drug resistance under short-term exposure to beta-lactam antibiotics. Canonically, the SOS response is triggered by DNA damage, which results in the induction of error-prone DNA repair mechanisms. These error-prone repair pathways can increase mutagenesis in the cell, leading to the evolution of drug resistance. Thus, inhibiting the SOS regulator RecA has been proposed as means to delay the rise of resistance.
In this paper, the authors deleted the RecA protein from E. coli and exposed this ∆recA strain to selective levels of the beta-lactam antibiotic, ampicillin. After an 8h treatment, they washed the antibiotic away and allowed the surviving cells to recover in regular media. They then measured the minimum inhibitory concentration (MIC) of ampicillin against these treated strains. They note that after just 8 h treatment with ampicillin, the ∆recA had developed higher MICs towards ampicillin, while by contrast, wild-type cells exhibited unchanged MICs. This MIC increase was also observed in subsequent generations of bacteria, suggesting that the phenotype is driven by a genetic change.
The authors then used whole genome sequencing (WGS) to identify mutations that accounted for the resistance phenotype. Within resistant populations, they discovered key mutations in the promoter region of the beta-lactamase gene, ampC; in the penicillin-binding protein PBP3 which is the target of ampicillin; and in the AcrB subunit of the AcrAB-TolC efflux machinery. Importantly, mutations in the efflux machinery can impact the resistance towards other antibiotics, not just beta-lactams. To test this, they repeated the MIC experiments with other classes of antibiotics, including kanamycin, chloramphenicol, and rifampicin. Interestingly, they observed that the ∆recA strains pre-treated with ampicillin showed higher MICs towards all other antibiotics tested. This suggests that the mutations conferring resistance to ampicillin are also increasing resistance to other antibiotics.
The authors then performed an impressive series of genetic, microscopy, and transcriptomic experiments to show that this increase in resistance is not driven by the SOS response, but by independent DNA repair and stress response pathways. Specifically, they show that deletion of the recA reduces the bacterium's ability to process reactive oxygen species (ROS) and repair its DNA. These factors drive the accumulation of mutations that can confer resistance towards different classes of antibiotics. The conclusions are reasonably well-supported by the data, but some aspects of the data and the model need to be clarified and extended.
Strengths:
A major strength of the paper is the detailed bacterial genetics and transcriptomics that the authors performed to elucidate the molecular pathways responsible for this increased resistance. They systemically deleted or inactivated genes involved in the SOS response in E. coli. They then subjected these mutants to the same MIC assays as described previously. Surprisingly, none of the other SOS gene deletions resulted in an increase in drug resistance, suggesting that the SOS response is not involved in this phenotype. This led the authors to focus on the localization of DNA PolI, which also participates in DNA damage repair. Using microscopy, they discovered that in the RecA deletion background, PolI co-localizes with the bacterial chromosome at much lower rates than wild-type. This led the authors to conclude that deletion of RecA hinders PolI and DNA repair. Although the authors do not provide a mechanism, this observation is nonetheless valuable for the field and can stimulate further investigations in the future.
In order to understand how RecA deletion affects cellular physiology, the authors performed RNA-seq on ampicillin-treated strains. Crucially, they discovered that in the RecA deletion strain, genes associated with antioxidative activity (cysJ, cysI, cysH, soda, sufD) and Base Excision Repair repair (mutH, mutY, mutM), which repairs oxidized forms of guanine, were all downregulated. The authors conclude that down-regulation of these genes might result in elevated levels of reactive oxygen species in the cells, which in turn, might drive the rise of resistance. Experimentally, they further demonstrated that treating the ∆recA strain with an antioxidant GSH prevents the rise of MICs. These observations will be useful for more detailed mechanistic follow-ups in the future.
Weaknesses:
Throughout the paper, the authors use language suggesting that ampicillin treatment of the ∆recA strain induces higher levels of mutagenesis inside the cells, leading to the rapid rise of resistance mutations. However, as the authors note, the mutants enriched by ampicillin selection can play a role in efflux and can thus change a bacterium's sensitivity to a wide range of antibiotics, in what is known as cross-resistance. The current data is not clear on whether the elevated "mutagenesis" is driven by ampicillin selection or by a bona fide increase in mutation rate.
Furthermore, on a technical level, the authors employed WGS to identify resistance mutations in the ampicillin-treated wild-type and ∆recA strains. However, the WGS methodology described in the paper is inconsistent. Notably, wild-type WGS samples were picked from non-selective plates, while ΔrecA WGS isolates were picked from selective plates with 50 μg/mL ampicillin. Such an approach biases the frequency and identity of the mutations seen in the WGS and cannot be used to support the idea that ampicillin treatment induces higher levels of mutagenesis.
Finally, it is important to establish what the basal mutation rates of both the WT and ∆recA strains are. Currently, only the ampicillin-treated populations were reported. It is possible that the ∆recA strain has inherently higher mutagenesis than WT, with a larger subpopulation of resistant clones. Thus, ampicillin treatment might not, in fact, induce higher mutagenesis in ∆recA.
Summary of revised manuscript:
In their revisions, the authors have addressed my major concerns with additional experiments and changes to the text. Thank you!
-
Reviewer #3 (Public review):
Summary:
In the present work, Zhang et al investigate the involvement of the bacterial DNA damage repair SOS response in the evolution of beta-lactam drug resistance in Escherichia coli. Using a combination of microbiological, bacterial genetics, laboratory evolution, next-generation, and live-cell imaging approaches, the authors propose short-term (transient) drug resistance evolution can take place in RecA-deficient cells in an SOS response-independent manner. They propose the evolvability of drug resistance is alternatively driven by the oxidative stress imposed by accumulation of reactive oxygen species and compromised DNA repair. Overall, this is a nice study that addresses a growing and fundamental global health challenge (antimicrobial resistance).
Strengths:
The authors introduce new concepts to …
Reviewer #3 (Public review):
Summary:
In the present work, Zhang et al investigate the involvement of the bacterial DNA damage repair SOS response in the evolution of beta-lactam drug resistance in Escherichia coli. Using a combination of microbiological, bacterial genetics, laboratory evolution, next-generation, and live-cell imaging approaches, the authors propose short-term (transient) drug resistance evolution can take place in RecA-deficient cells in an SOS response-independent manner. They propose the evolvability of drug resistance is alternatively driven by the oxidative stress imposed by accumulation of reactive oxygen species and compromised DNA repair. Overall, this is a nice study that addresses a growing and fundamental global health challenge (antimicrobial resistance).
Strengths:
The authors introduce new concepts to antimicrobial resistance evolution mechanisms. They show short-term exposure to beta-lactams can induce durably fixed antimicrobial resistance mutations. They propose this is due to compromised DNA repair and oxidative stress. Antibiotic resistance evolution under transient stress is poorly studied, so the authors' work is a nice mechanistic contribution to this field.
Weaknesses:
The authors revisions have significantly addressed weaknesses previously identified earlier in the review process.
-
Author response:
The following is the authors’ response to the previous reviews
Reviewer #1 (Public review):
Jin et al. investigated how the bacterial DNA damage (SOS) response and its regulator protein RecA affects the development of drug resistance under short-term exposure to beta-lactam antibiotics. Canonically, the SOS response is triggered by DNA damage, which results in the induction of error-prone DNA repair mechanisms. These error-prone repair pathways can increase mutagenesis in the cell, leading to the evolution of drug resistance. Thus, inhibiting the SOS regulator RecA has been proposed as means to delay the rise of resistance.
In this paper, the authors deleted the RecA protein from E. coli and exposed this ∆recA strain to selective levels of the beta-lactam antibiotic, ampicillin. After an 8h treatment, they washed the …
Author response:
The following is the authors’ response to the previous reviews
Reviewer #1 (Public review):
Jin et al. investigated how the bacterial DNA damage (SOS) response and its regulator protein RecA affects the development of drug resistance under short-term exposure to beta-lactam antibiotics. Canonically, the SOS response is triggered by DNA damage, which results in the induction of error-prone DNA repair mechanisms. These error-prone repair pathways can increase mutagenesis in the cell, leading to the evolution of drug resistance. Thus, inhibiting the SOS regulator RecA has been proposed as means to delay the rise of resistance.
In this paper, the authors deleted the RecA protein from E. coli and exposed this ∆recA strain to selective levels of the beta-lactam antibiotic, ampicillin. After an 8h treatment, they washed the antibiotic away and allowed the surviving cells to recover in regular media. They then measured the minimum inhibitory concentration (MIC) of ampicillin against these treated strains. They note that after just 8 h treatment with ampicillin, the ∆recA had developed higher MICs towards ampicillin, while by contrast, wild-type cells exhibited unchanged MICs. This MIC increase was also observed subsequent generations of bacteria, suggesting that the phenotype is driven by a genetic change.
The authors then used whole genome sequencing (WGS) to identify mutations that accounted for the resistance phenotype. Within resistant populations, they discovered key mutations in the promoter region of the beta-lactamase gene, ampC; in the penicillin-binding protein PBP3 which is the target of ampicillin; and in the AcrB subunit of the AcrAB-TolC efflux machinery. Importantly, mutations in the efflux machinery can impact the resistances towards other antibiotics, not just beta-lactams. To test this, they repeated the MIC experiments with other classes of antibiotics, including kanamycin, chloramphenicol, and rifampicin. Interestingly, they observed that the ∆recA strains pre-treated with ampicillin showed higher MICs towards all other antibiotic tested. This suggests that the mutations conferring resistance to ampicillin are also increasing resistance to other antibiotics.
The authors then performed an impressive series of genetic, microscopy, and transcriptomic experiments to show that this increase in resistance is not driven by the SOS response, but by independent DNA repair and stress response pathways. Specifically, they show that deletion of the recA reduces the bacterium's ability to process reactive oxygen species (ROS) and repair its DNA. These factors drive accumulation of mutations that can confer resistance towards different classes of antibiotics. The conclusions are reasonably well-supported by the data, but some aspects of the data and the model need to be clarified and extended.
Strengths:
A major strength of the paper is the detailed bacterial genetics and transcriptomics that the authors performed to elucidate the molecular pathways responsible for this increased resistance. They systemically deleted or inactivated genes involved in the SOS response in E. coli. They then subjected these mutants the same MIC assays as described previously. Surprisingly, none of the other SOS gene deletions resulted an increase in drug resistance, suggesting that the SOS response is not involved in this phenotype. This led the authors to focus on the localization of DNA PolI, which also participates in DNA damage repair. Using microscopy, they discovered that in the RecA deletion background, PolI co-localizes with the bacterial chromosome at much lower rates than wild-type. This led the authors to conclude that deletion of RecA hinders PolI and DNA repair. Although the authors do not provide a mechanism, this observation is nonetheless valuable for the field and can stimulate further investigations in the future.
In order to understand how RecA deletion affects cellular physiology, the authors performed RNA-seq on ampicillin-treated strains. Crucially, they discovered that in the RecA deletion strain, genes associated with antioxidative activity (cysJ, cysI, cysH, soda, sufD) and Base Excision Repair repair (mutH, mutY, mutM), which repairs oxidized forms of guanine, were all downregulated. The authors conclude that down-regulation of these genes might result in elevated levels of reactive oxygen species in the cells, which in turn, might drive the rise of resistance. Experimentally, they further demonstrated that treating the ∆recA strain with an antioxidant GSH prevents the rise of MICs. These observations will be useful for more detailed mechanistic follow-ups in the future.
Weaknesses:
Throughout the paper, the authors use language suggesting that ampicillin treatment of the ∆recA strain induces higher levels of mutagenesis inside the cells, leading to the rapid rise of resistance mutations. However, as the authors note, the mutants enriched by ampicillin selection can play a role in efflux and can thus change a bacterium's sensitivity to a wide range of antibiotics, in what is known as cross-resistance. The current data is not clear on whether the elevated "mutagenesis" is driven ampicillin selection or by a bona fide increase in mutation rate.
Furthermore, on a technical level, the authors employed WGS to identify resistance mutations in the treated ampicillin-treated wild-type and ∆recA strains. However, the WGS methodology described in the paper is inconsistent. Notably, wild-type WGS samples were picked from non-selective plates, while ΔrecA WGS isolates were picked from selective plates with 50 μg/mL ampicillin. Such an approach biases the frequency and identity of the mutations seen in the WGS and cannot be used to support the idea that ampicillin treatment induces higher levels of mutagenesis.
Finally, it is important to establish what the basal mutation rates of both the WT and ∆recA strains are. Currently, only the ampicillin-treated populations were reported. It is possible that the ∆recA strain has inherently higher mutagenesis than WT, with a larger subpopulation of resistant clones. Thus, ampicillin treatment might not in fact induce higher mutagenesis in ∆recA.
Comments on revisions:
Thank you for responding to the concerns raised previously. The manuscript overall has improved.
We sincerely thank the reviewer for raising this important point. In our initial submission, we acknowledge that our mutation analysis was based on a limited number of replicates (n=6), which may not have been sufficient to robustly distinguish between mutation induction and selection. In response to this concern, we have substantially expanded our experimental dataset. Specifically, we redesigned the mutation rate validation experiment by increasing the number of biological replicates in each condition to 96 independent parallel cultures. This enabled us to systematically assess mutation frequency distributions under four conditions (WT, WT+ampicillin, ΔrecA, ΔrecA+ampicillin), using both maximum likelihood estimation (MLE) and distribution-based fluctuation analysis (new Figure 1F, 1G, and Figure S5).
These expanded datasets revealed that:
(1) While the estimated mutation rate was significantly elevated in ΔrecA+ampicillin compared to ΔrecA alone (Fig. 1G),
(2) The distribution of mutation frequencies in ΔrecA+ampicillin was highly skewed with evident jackpot cultures (Fig. 1F), and
(3) The observed pattern significantly deviated from Poisson expectations, which is inconsistent with uniform mutagenesis and instead supports clonal selection from an early-arising mutational pool (Fig. S5).
Importantly, these new results do not contradict our original conclusions but rather extend and refine them. The previous evidence for ROS-mediated mutagenesis remains valid and is supported by our GSH experiments, transcriptomic analysis of oxidative stress genes, and DNA repair pathway repression. However, the additional data now indicate that ROS-induced variants are not uniformly induced after antibiotic exposure but are instead generated stochastically under the stress-prone ΔrecA background and then selectively enriched upon ampicillin treatment.
Taken together, we now propose a two-step model of resistance evolution in ΔrecA cells (new Figure 5):
Step i: RecA deficiency creates a hypermutable state through impaired repair and elevated ROS, increasing the probability of resistance-conferring mutations.
Step ii: β-lactam exposure acts as a selective bottleneck, enriching early-arising mutants that confer resistance.
We have revised both the Results and Discussion sections to clearly articulate this complementary relationship between mutational supply and selection, and we believe this integrated model better explains the observed phenotypes and mechanistic outcomes.
Reviewer #2 (Public review):
This study aims to demonstrate that E. coli can acquire rapid antibiotic resistance mutations in the absence of a DNA damage response. The authors employed a modified Adaptive Laboratory Evolution (ALE) workflow to investigate this, initiating the process by diluting an overnight culture 50-fold into an ampicillin selection medium. They present evidence that a recA- strain develops ampicillin resistance mutations more rapidly than the wild-type, as indicated by the Minimum Inhibitory Concentration (MIC) and mutation frequency. Whole-genome sequencing of recA- colonies resistant to ampicillin showed predominant inactivation of genes involved in the multi-drug efflux pump system, contrasting with wild-type mutations that seem to activate the chromosomal ampC cryptic promoter. Further analysis of mutants, including a lexA3 mutant incapable of inducing the SOS response, led the authors to conclude that the rapid evolution of antibiotic resistance occurs via an SOS-independent mechanism in the absence of recA. RNA sequencing suggests that antioxidative response genes drive the rapid evolution of antibiotic resistance in the recA- strain. They assert that rapid evolution is facilitated by compromised DNA repair, transcriptional repression of antioxidative stress genes, and excessive ROS accumulation.
Strengths:
The experiments are well-executed and the data appear reliable. It is evident that the inactivation of recA promotes faster evolutionary responses, although the exact mechanisms driving this acceleration remain elusive and deserve further investigation.
Weaknesses:
Some conclusions are overstated. For instance, the conclusion regarding the LexA3 allele, indicating that rapid evolution occurs in an SOS-independent manner (line 217), contradicts the introductory statement that attributes evolution to compromised DNA repair.
We thank the reviewer for this insightful observation, which highlights a central conceptual advance of our study. Our data indeed indicate that resistance evolution in ΔrecA occurs independently of canonical SOS induction (as shown by the lack of resistance in lexA3, dpiBA, and translesion polymerase mutants), yet is clearly associated with impaired DNA repair capacity (e.g., downregulation of polA, mutH, mutY).
This apparent “contradiction” reflects the dual role of RecA: it functions both as the master activator of the SOS response and as a key factor in SOS-independent repair processes. Thus, the rapid resistance evolution in ΔrecA is not due to loss of SOS, but rather due to the broader suppression of DNA repair pathways that RecA coordinates, which elevates mutational load under stress (This point is discussed in further detail in our response to Reviewer 1).
The claim made in the discussion of Figure 3 that the hindrance of DNA repair in recA- is crucial for rapid evolution is at best suggestive, not demonstrative. Additionally, the interpretation of the PolI data implies its role, yet it remains speculative.
We appreciate this comment and would like to respectfully clarify that our conclusion regarding the role of DNA repair impairment is supported by several independent lines of mechanistic evidence.
First, our RNA-seq analysis revealed transcriptional suppression of multiple DNA repair genes in ΔrecA cells following ampicillin treatment, including polA (DNA Pol I) and the base excision repair genes mutH, mutY, and mutM (Fig. 4K). This indicates that multiple repair pathways, including those responsible for correcting oxidative DNA lesions, are downregulated under these conditions.
Second, we observed a significant reduction in DNA Pol I protein expression as well as reduced colocalization with chromosomal DNA in ΔrecA cells, suggesting impaired engagement of repair machinery (Fig. 3C-E). These phenotypes are not limited to transcriptional signatures but extend to functional protein localization.
Third, and most importantly, resistance evolution was fully suppressed in ΔrecA cells upon co-treatment with glutathione (GSH), which reduces ROS levels. As GSH did not affect ampicillin killing (Fig. 4J), these findings suggest that mutagenesis and thus the emergence of resistance requires both ROS accumulation and the absence of efficient repair.
Therefore, we believe these data go beyond correlation and demonstrate a mechanistic role for DNA repair impairment in driving stress-associated resistance evolution in ΔrecA. We have revised the Discussion to emphasize the strength of this evidence while avoiding overstatement.
In Figure 2A table, mutations in amp promoters are leading to amino acid changes.
We thank the reviewer for spotting this inconsistency. Indeed, the ampC promoter mutations we identified reside in non-coding regulatory regions and do not result in amino acid substitutions. We have corrected the annotation in Fig. 2A and clarified in the main text that these mutations likely affect gene expression through transcriptional regulation, rather than protein sequence alteration.
The authors' assertion that ampicillin significantly influences persistence pathways in the wild-type strain, affecting quorum sensing, flagellar assembly, biofilm formation, and bacterial chemotaxis, lacks empirical validation.
We thank the reviewer for pointing this out. In the original version, we acknowledged transcriptional enrichment of genes related to quorum sensing, flagellar assembly, and chemotaxis in the wild-type strain upon ampicillin treatment. However, as we did not directly assess persistence phenotypes (e.g., biofilm formation or persister levels), we agree that such functional inferences were not fully supported. We have revised the relevant statements to focus solely on transcriptomic changes and have removed language suggesting direct effects on persistence pathways.
Figure 1G suggests that recA cells treated with ampicillin exhibit a strong mutator phenotype; however, it remains unclear if this can be linked to the mutations identified in Figure 2's sequencing analysis.
We appreciate the reviewer’s comment. This point is discussed in further detail in our response to Reviewer 1.
Reviewer #3 (Public review):
In the present work, Zhang et al investigate involvement of the bacterial DNA damage repair SOS response in the evolution of beta-lactam drug resistance evolution in Escherichia coli. Using a combination of microbiological, bacterial genetics, laboratory evolution, next-generation, and live-cell imaging approaches, the authors propose short-term (transient) drug resistance evolution can take place in RecA-deficient cells in an SOS response-independent manner. They propose the evolvability of drug resistance is alternatively driven by the oxidative stress imposed by accumulation of reactive oxygen species and compromised DNA repair. Overall, this is a nice study that addresses a growing and fundamental global health challenge (antimicrobial resistance).
Strengths:
The authors introduce new concepts to antimicrobial resistance evolution mechanisms. They show short-term exposure to beta-lactams can induce durably fixed antimicrobial resistance mutations. They propose this is due to comprised DNA repair and oxidative stress. Antibiotic resistance evolution under transient stress is poorly studied, so the authors' work is a nice mechanistic contribution to this field.
Weaknesses:
The authors do not show any direct evidence of altered mutation rate or accumulated DNA damage in their model.
We appreciate the reviewer’s comment. This point is discussed in further detail in our response to Reviewer 1.
Recommendations for the authors:
Reviewer #1 (Recommendations for the authors):
I would like to suggest two minor changes to the text.
(1) Re. WGS data.
The authors write in their response "We appreciate your concern regarding potential inconsistencies in the WGS methodology. However, we would like to clarify that the primary aim of the WGS experiment was to identify the types of mutations present in the wild type and ΔrecA strains after treatment of ampicillin, rather than to quantify or compare mutation frequencies. This purpose was explicitly stated in the manuscript.
I think the source of my confusion stemmed from this part in the text:
"In bacteria, resistance to most antibiotics requires the accumulation of drug resistance associated DNA mutations developed over time to provide high levels of resistance (29). To verify whether drug resistance associated DNA mutations have led to the rapid development of antibiotic resistance in recA mutant strain, we..."
I would change the phrase "verify whether drug resistance associated DNA mutations have led to the rapid development of antibiotic resistance in recA mutant strain" to "identify the types of mutations present in the wild type and ΔrecA strains after treatment of ampicillin." This would explicitly state what the sequencing was for (ie. ID-ing mutations). The current phrase can give the impression that WGS was used to validate rapid or high mutagenesis.
Thanks for this suggestion. We have revised this description to “In bacteria, resistance to most antibiotics requires the accumulation of drug resistance associated DNA mutations that can arise stochastically and, under stress conditions, become enriched through selection over time to confer high levels of resistance (33). Having observed a non-random and right-skewed distribution of mutation frequencies in ΔrecA isolates following ampicillin exposure, we next sought to determine whether specific resistance-conferring mutations were enriched in ΔrecA isolates following antibiotic exposure.”
(2) Re. whether the mutations are "induced" or "pre-existing."
The authors write:
"We appreciate your detailed feedback on the language used to describe our data. We understand the concern regarding the use of the term "induced" in relation to beta-lactam exposure. To clarify, we employed not only beta-lactam antibiotics but also other antibiotics, such as ciprofloxacin and chloramphenicol, in our experiments (data not shown). However, we observed that beta-lactam antibiotics specifically induced the emergence of resistance or altered the MIC in our bacterial populations. If resistance had pre-existed before antibiotic exposure, we would expect other antibiotics to exhibit a similar selective effect, particularly given the potential for cross-resistance to multiple antibiotics."
I think it is important to discuss the negative data for the other antibiotics (along with the other points made in your Reviewer response) in the main text.
This point is discussed in further detail in our response to Reviewer 1 (Public Review).
-
-
-
eLife Assessment
This useful study examines how deletion of a major DNA repair gene in bacteria may facilitate the rise of mutations that confer resistance against a range of different antibiotics. Although the phenotypic evidence is intriguing, the interpretation of the phenotypic data presented and the proposed mechanism by which these mutations are generated are incomplete, relying on untested assumptions and methodology that merits optimization. For instance, the authors cannot fully rule out the possibility that the resistance mutations are the result of selection. Nevertheless, this work could be of interest to microbiologists studying antibiotic resistance, genome integrity, and evolution, but the significance remains uncertain.
-
Reviewer #1 (Public review):
Summary:
Jin et al. investigated how the bacterial DNA damage (SOS) response and its regulator protein RecA affects the development of drug resistance under short-term exposure to beta-lactam antibiotics. Canonically, the SOS response is triggered by DNA damage, which results in the induction of error-prone DNA repair mechanisms. These error-prone repair pathways can increase mutagenesis in the cell, leading to the evolution of drug resistance. Thus, inhibiting the SOS regulator RecA has been proposed as means to delay the rise of resistance.
In this paper, the authors deleted the RecA protein from E. coli and exposed this ∆recA strain to selective levels of the beta-lactam antibiotic, ampicillin. After an 8h treatment, they washed the antibiotic away and allowed the surviving cells to recover in regular …
Reviewer #1 (Public review):
Summary:
Jin et al. investigated how the bacterial DNA damage (SOS) response and its regulator protein RecA affects the development of drug resistance under short-term exposure to beta-lactam antibiotics. Canonically, the SOS response is triggered by DNA damage, which results in the induction of error-prone DNA repair mechanisms. These error-prone repair pathways can increase mutagenesis in the cell, leading to the evolution of drug resistance. Thus, inhibiting the SOS regulator RecA has been proposed as means to delay the rise of resistance.
In this paper, the authors deleted the RecA protein from E. coli and exposed this ∆recA strain to selective levels of the beta-lactam antibiotic, ampicillin. After an 8h treatment, they washed the antibiotic away and allowed the surviving cells to recover in regular media. They then measured the minimum inhibitory concentration (MIC) of ampicillin against these treated strains. They note that after just 8 h treatment with ampicillin, the ∆recA had developed higher MICs towards ampicillin, while by contrast, wild-type cells exhibited unchanged MICs. This MIC increase was also observed subsequent generations of bacteria, suggesting that the phenotype is driven by a genetic change.
The authors then used whole genome sequencing (WGS) to identify mutations that accounted for the resistance phenotype. Within resistant populations, they discovered key mutations in the promoter region of the beta-lactamase gene, ampC; in the penicillin-binding protein PBP3 which is the target of ampicillin; and in the AcrB subunit of the AcrAB-TolC efflux machinery. Importantly, mutations in the efflux machinery can impact the resistances towards other antibiotics, not just beta-lactams. To test this, they repeated the MIC experiments with other classes of antibiotics, including kanamycin, chloramphenicol, and rifampicin. Interestingly, they observed that the ∆recA strains pre-treated with ampicillin showed higher MICs towards all other antibiotic tested. This suggests that the mutations conferring resistance to ampicillin are also increasing resistance to other antibiotics.
The authors then performed an impressive series of genetic, microscopy, and transcriptomic experiments to show that this increase in resistance is not driven by the SOS response, but by independent DNA repair and stress response pathways. Specifically, they show that deletion of the recA reduces the bacterium's ability to process reactive oxygen species (ROS) and repair its DNA. These factors drive accumulation of mutations that can confer resistance towards different classes of antibiotics. The conclusions are reasonably well-supported by the data, but some aspects of the data and the model need to be clarified and extended.
Strengths:
A major strength of the paper is the detailed bacterial genetics and transcriptomics that the authors performed to elucidate the molecular pathways responsible for this increased resistance. They systemically deleted or inactivated genes involved in the SOS response in E. coli. They then subjected these mutants the same MIC assays as described previously. Surprisingly, none of the other SOS gene deletions resulted an increase in drug resistance, suggesting that the SOS response is not involved in this phenotype. This led the authors to focus on the localization of DNA PolI, which also participates in DNA damage repair. Using microscopy, they discovered that in the RecA deletion background, PolI co-localizes with the bacterial chromosome at much lower rates than wild-type. This led the authors to conclude that deletion of RecA hinders PolI and DNA repair. Although the authors do not provide a mechanism, this observation is nonetheless valuable for the field and can stimulate further investigations in the future.
In order to understand how RecA deletion affects cellular physiology, the authors performed RNA-seq on ampicillin-treated strains. Crucially, they discovered that in the RecA deletion strain, genes associated with antioxidative activity (cysJ, cysI, cysH, soda, sufD) and Base Excision Repair repair (mutH, mutY, mutM), which repairs oxidized forms of guanine, were all downregulated. The authors conclude that down-regulation of these genes might result in elevated levels of reactive oxygen species in the cells, which in turn, might drive the rise of resistance. Experimentally, they further demonstrated that treating the ∆recA strain with an antioxidant GSH prevents the rise of MICs. These observations will be useful for more detailed mechanistic follow-ups in the future.
Weaknesses:
Throughout the paper, the authors use language suggesting that ampicillin treatment of the ∆recA strain induces higher levels of mutagenesis inside the cells, leading to the rapid rise of resistance mutations. However, as the authors note, the mutants enriched by ampicillin selection can play a role in efflux and can thus change a bacterium's sensitivity to a wide range of antibiotics, in what is known as cross-resistance. The current data is not clear on whether the elevated "mutagenesis" is driven ampicillin selection or by a bona fide increase in mutation rate.
Furthermore, on a technical level, the authors employed WGS to identify resistance mutations in the treated ampicillin-treated wild-type and ∆recA strains. However, the WGS methodology described in the paper is inconsistent. Notably, wild-type WGS samples were picked from non-selective plates, while ΔrecA WGS isolates were picked from selective plates with 50 μg/mL ampicillin. Such an approach biases the frequency and identity of the mutations seen in the WGS and cannot be used to support the idea that ampicillin treatment induces higher levels of mutagenesis.
Finally, it is important to establish what the basal mutation rates of both the WT and ∆recA strains are. Currently, only the ampicillin-treated populations were reported. It is possible that the ∆recA strain has inherently higher mutagenesis than WT, with a larger subpopulation of resistant clones. Thus, ampicillin treatment might not in fact induce higher mutagenesis in ∆recA.
Comments on revisions:
Thank you for responding to the concerns raised previously. The manuscript overall has improved.
-
Reviewer #2 (Public review):
Summary:
This study aims to demonstrate that E. coli can acquire rapid antibiotic resistance mutations in the absence of a DNA damage response. The authors employed a modified Adaptive Laboratory Evolution (ALE) workflow to investigate this, initiating the process by diluting an overnight culture 50-fold into an ampicillin selection medium. They present evidence that a recA- strain develops ampicillin resistance mutations more rapidly than the wild-type, as indicated by the Minimum Inhibitory Concentration (MIC) and mutation frequency. Whole-genome sequencing of recA- colonies resistant to ampicillin showed predominant inactivation of genes involved in the multi-drug efflux pump system, contrasting with wild-type mutations that seem to activate the chromosomal ampC cryptic promoter. Further analysis of …
Reviewer #2 (Public review):
Summary:
This study aims to demonstrate that E. coli can acquire rapid antibiotic resistance mutations in the absence of a DNA damage response. The authors employed a modified Adaptive Laboratory Evolution (ALE) workflow to investigate this, initiating the process by diluting an overnight culture 50-fold into an ampicillin selection medium. They present evidence that a recA- strain develops ampicillin resistance mutations more rapidly than the wild-type, as indicated by the Minimum Inhibitory Concentration (MIC) and mutation frequency. Whole-genome sequencing of recA- colonies resistant to ampicillin showed predominant inactivation of genes involved in the multi-drug efflux pump system, contrasting with wild-type mutations that seem to activate the chromosomal ampC cryptic promoter. Further analysis of mutants, including a lexA3 mutant incapable of inducing the SOS response, led the authors to conclude that the rapid evolution of antibiotic resistance occurs via an SOS-independent mechanism in the absence of recA. RNA sequencing suggests that antioxidative response genes drive the rapid evolution of antibiotic resistance in the recA- strain. They assert that rapid evolution is facilitated by compromised DNA repair, transcriptional repression of antioxidative stress genes, and excessive ROS accumulation.
Strengths:
The experiments are well-executed and the data appear reliable. It is evident that the inactivation of recA promotes faster evolutionary responses, although the exact mechanisms driving this acceleration remain elusive and deserve further investigation.
Weaknesses:
Some conclusions are overstated. For instance, the conclusion regarding the LexA3 allele, indicating that rapid evolution occurs in an SOS-independent manner (line 217), contradicts the introductory statement that attributes evolution to compromised DNA repair. The claim made in the discussion of Figure 3 that the hindrance of DNA repair in recA- is crucial for rapid evolution is at best suggestive, not demonstrative. Additionally, the interpretation of the PolI data implies its role, yet it remains speculative. In Figure 2A table, mutations in amp promoters are leading to amino acid changes! The authors' assertion that ampicillin significantly influences persistence pathways in the wild-type strain, affecting quorum sensing, flagellar assembly, biofilm formation, and bacterial chemotaxis, lacks empirical validation. Figure 1G suggests that recA cells treated with ampicillin exhibit a strong mutator phenotype; however, it remains unclear if this can be linked to the mutations identified in Figure 2's sequencing analysis.
-
Reviewer #3 (Public review):
Summary:
In the present work, Zhang et al investigate involvement of the bacterial DNA damage repair SOS response in the evolution of beta-lactam drug resistance evolution in Escherichia coli. Using a combination of microbiological, bacterial genetics, laboratory evolution, next-generation, and live-cell imaging approaches, the authors propose short-term (transient) drug resistance evolution can take place in RecA-deficient cells in an SOS response-independent manner. They propose the evolvability of drug resistance is alternatively driven by the oxidative stress imposed by accumulation of reactive oxygen species and compromised DNA repair. Overall, this is a nice study that addresses a growing and fundamental global health challenge (antimicrobial resistance).
Strengths:
The authors introduce new concepts …
Reviewer #3 (Public review):
Summary:
In the present work, Zhang et al investigate involvement of the bacterial DNA damage repair SOS response in the evolution of beta-lactam drug resistance evolution in Escherichia coli. Using a combination of microbiological, bacterial genetics, laboratory evolution, next-generation, and live-cell imaging approaches, the authors propose short-term (transient) drug resistance evolution can take place in RecA-deficient cells in an SOS response-independent manner. They propose the evolvability of drug resistance is alternatively driven by the oxidative stress imposed by accumulation of reactive oxygen species and compromised DNA repair. Overall, this is a nice study that addresses a growing and fundamental global health challenge (antimicrobial resistance).
Strengths:
The authors introduce new concepts to antimicrobial resistance evolution mechanisms. They show short-term exposure to beta-lactams can induce durably fixed antimicrobial resistance mutations. They propose this is due to comprised DNA repair and oxidative stress. Antibiotic resistance evolution under transient stress is poorly studied, so the authors' work is a nice mechanistic contribution to this field.
Weaknesses:
The authors do not show any direct evidence of altered mutation rate or accumulated DNA damage in their model.
-
Author response:
The following is the authors’ response to the original reviews.
Public Reviews:
Review #1:
Summary:
Jin et al. investigated how the bacterial DNA damage (SOS) response and its regulator protein RecA affect the development of drug resistance under short-term exposure to beta-lactam antibiotics. Canonically, the SOS response is triggered by DNA damage, which results in the induction of error-prone DNA repair mechanisms. These error-prone repair pathways can increase mutagenesis in the cell, leading to the evolution of drug resistance. Thus, inhibiting the SOS regulator RecA has been proposed as a means to delay the rise of resistance.
In this paper, the authors deleted the RecA protein from E. coli and exposed this ∆recA strain to selective levels of the beta-lactam antibiotic, ampicillin. After an 8-hour treatment, they …
Author response:
The following is the authors’ response to the original reviews.
Public Reviews:
Review #1:
Summary:
Jin et al. investigated how the bacterial DNA damage (SOS) response and its regulator protein RecA affect the development of drug resistance under short-term exposure to beta-lactam antibiotics. Canonically, the SOS response is triggered by DNA damage, which results in the induction of error-prone DNA repair mechanisms. These error-prone repair pathways can increase mutagenesis in the cell, leading to the evolution of drug resistance. Thus, inhibiting the SOS regulator RecA has been proposed as a means to delay the rise of resistance.
In this paper, the authors deleted the RecA protein from E. coli and exposed this ∆recA strain to selective levels of the beta-lactam antibiotic, ampicillin. After an 8-hour treatment, they washed the antibiotic away and allowed the surviving cells to recover in regular media. They then measured the minimum inhibitory concentration (MIC) of ampicillin against these treated strains. They note that after just 8-hour treatment with ampicillin, the ∆recA had developed higher MICs towards ampicillin, while by contrast, wild-type cells exhibited unchanged MICs. This MIC increase was also observed in subsequent generations of bacteria, suggesting that the phenotype is driven by a genetic change.
The authors then used whole genome sequencing (WGS) to identify mutations that accounted for the resistance phenotype. Within resistant populations, they discovered key mutations in the promoter region of the beta-lactamase gene, ampC; in the penicillin-binding protein PBP3 which is the target of ampicillin; and in the AcrB subunit of the AcrAB-TolC efflux machinery. Importantly, mutations in the efflux machinery can impact the resistance towards other antibiotics, not just beta-lactams. To test this, they repeated the MIC experiments with other classes of antibiotics, including kanamycin, chloramphenicol, and rifampicin. Interestingly, they observed that the ∆recA strains pre-treated with ampicillin showed higher MICs towards all other antibiotics tested. This suggests that the mutations conferring resistance to ampicillin are also increasing resistance to other antibiotics.
The authors then performed an impressive series of genetic, microscopy, and transcriptomic experiments to show that this increase in resistance is not driven by the SOS response, but by independent DNA repair and stress response pathways. Specifically, they show that deletion of the recA reduces the bacterium's ability to process reactive oxygen species (ROS) and repair its DNA. These factors drive the accumulation of mutations that can confer resistance to different classes of antibiotics. The conclusions are reasonably well-supported by the data, but some aspects of the data and the model need to be clarified and extended.
We sincerely appreciate your overall summary of the manuscript and their positive evaluation of our work.
Strengths:
A major strength of the paper is the detailed bacterial genetics and transcriptomics that the authors performed to elucidate the molecular pathways responsible for this increased resistance. They systemically deleted or inactivated genes involved in the SOS response in E. coli. They then subjected these mutants to the same MIC assays as described previously. Surprisingly, none of the other SOS gene deletions resulted in an increase in drug resistance, suggesting that the SOS response is not involved in this phenotype. This led the authors to focus on the localization of DNA PolI, which also participates in DNA damage repair. Using microscopy, they discovered that in the RecA deletion background, PolI co-localizes with the bacterial chromosome at much lower rates than wild-type. This led the authors to conclude that deletion of RecA hinders PolI and DNA repair. Although the authors do not provide a mechanism, this observation is nonetheless valuable for the field and can stimulate further investigations in the future.
In order to understand how RecA deletion affects cellular physiology, the authors performed RNA-seq on ampicillin-treated strains. Crucially, they discovered that in the RecA deletion strain, genes associated with antioxidative activity (cysJ, cysI, cysH, soda, sufD) and Base Excision Repair repair (mutH, mutY, mutM), which repairs oxidized forms of guanine, were all downregulated. The authors conclude that down-regulation of these genes might result in elevated levels of reactive oxygen species in the cells, which in turn, might drive the rise of resistance. Experimentally, they further demonstrated that treating the ∆recA strain with an antioxidant GSH prevents the rise of MICs. These observations will be useful for more detailed mechanistic follow-ups in the future.
We are grateful to you for your positive assessment of the strengths of our manuscript and your recognition of its potential future applications.
Weaknesses:
Throughout the paper, the authors use language suggesting that ampicillin treatment of the ∆recA strain induces higher levels of mutagenesis inside the cells, leading to the rapid rise of resistance mutations. However, as the authors note, the mutants enriched by ampicillin selection can play a role in efflux and can thus change a bacterium's sensitivity to a wide range of antibiotics, in what is known as cross-resistance. The current data is not clear on whether the elevated "mutagenesis" is driven ampicillin selection or by a bona fide increase in mutation rate.
We greatly appreciate you for raising this issue, as it is an important premise that must be clearly stated throughout the entire manuscript. To verify that the observed increase in mutation rate is a bona fide increase and not due to experimental error, we used a non-selective antibiotic, rifampicin, to evaluate the mutation frequency after drug induction, as it is a gold-standard method documented in other studies [Heterogeneity in efflux pump expression predisposes antibiotic-resistant cells to mutation, Science, 362, 6415, 686-690, 2018.]. In the absence of ampicillin treatment, the natural mutation rates detected using rifampicin were consistent between the wild-type and the ΔrecA strain. However, after ampicillin treatment, the mutation rate detected using rifampicin was significantly elevated only in the ΔrecA strain (Fig. 1G). We also employed other antibiotics, such as ciprofloxacin and chloramphenicol, in our experiments to treat the cells (data not shown). However, we observed that beta-lactam antibiotics specifically induced the emergence of resistance or altered the MIC in our bacterial populations. If resistance had pre-existed before antibiotic exposure or a bona fide increase in mutation rate, we would expect other antibiotics to exhibit a similar selective effect, particularly given the potential for cross-resistance to multiple antibiotics.
Furthermore, on a technical level, the authors employed WGS to identify resistance mutations in the treated ampicillin-treated wild-type and ∆recA strains. However, the WGS methodology described in the paper is inconsistent. Notably, wild-type WGS samples were picked from non-selective plates, while ΔrecA WGS isolates were picked from selective plates with 50 μg/mL ampicillin. Such an approach biases the frequency and identity of the mutations seen in the WGS and cannot be used to support the idea that ampicillin treatment induces higher levels of mutagenesis.
We appreciate your concern regarding potential inconsistencies in the WGS methodology. However, we would like to clarify that the primary aim of the WGS experiment was to identify the types of mutations present in the wild-type and ΔrecA strains after treatment of ampicillin, rather than to quantify or compare mutation frequencies. This purpose was explicitly stated in the manuscript.
Furthermore, the choice of selective and non-selective conditions was made to ensure the successful isolation of mutants in both strains. Specifically, if selective conditions (50 μg/mL ampicillin) were applied to the wild-type strain, it would have been nearly impossible to recover colonies for WGS analysis, as wild-type cells are highly susceptible to ampicillin at this concentration (Top, Author response image 1). Conversely, under non-selective conditions, ΔrecA mutants carrying resistance mutations may not have been effectively isolated, which would have limited our ability to identify resistance mutations in these strains (Bottom, Author response image 1 Thus, the use of different selection pressures was essential for achieving the objective of mutation identification in this study.
Author response image 1.
After 8 hours of antibiotic treatment, the wild type or the ΔrecA cells were plated on agar plates either without ampicillin or with 50 μg/mL ampicillin and incubated for 24-48 hours. Top: Under selective conditions, no wild type colonies were recovered, indicating high susceptibility to the antibiotic, preventing further analysis. Bottom: In non-selective conditions, both ΔrecA resistant mutants and non-resistant cells grew, making it difficult to distinguish and isolate the mutants carrying resistance mutations.
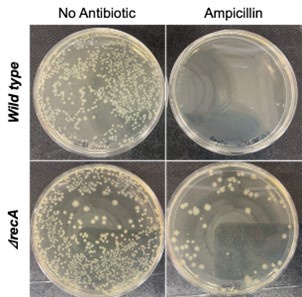
Finally, it is important to establish what the basal mutation rates of both the WT and ∆recA strains are. Currently, only the ampicillin-treated populations were reported. It is possible that the ∆recA strain has inherently higher mutagenesis than WT, with a larger subpopulation of resistant clones. Thus, ampicillin treatment might not in fact induce higher mutagenesis in ∆recA.
Thanks for this suggestion. The basal mutation frequency of the wild-type and the ∆recA strain have been measured using rifampicin (Fig. 1G), and there is no significant difference between them.
Reviewer #2:
Summary:
This study aims to demonstrate that E. coli can acquire rapid antibiotic resistance mutations in the absence of a DNA damage response. To investigate this, the authors employed a sophisticated experimental framework based on a modified Adaptive Laboratory Evolution (ALE) workflow. This workflow involves numerous steps culminating in the measurement of antibiotic resistance. The study presents evidence that a recA strain develops ampicillin resistance mutations more quickly than the wild-type, as shown by measuring the Minimum Inhibitory Concentration (MIC) and mutation frequency. Whole-genome sequencing of 15 recA-colonies resistant to ampicillin revealed predominantly inactivation of genes involved in the multi-drug efflux pump system, whereas, in the wild-type, mutations appear to enhance the activity of the chromosomal ampC cryptic promoter. By analyzing mutants involved in the SOS response, including a lexA3 mutant incapable of inducing the SOS response, the authors conclude that the rapid evolution of antibiotic resistance occurs in an SOS-independent manner when recA is absent.
Furthermore, RNA sequencing (RNA-seq) of the four experimental conditions suggests that genes related to antioxidative responses drive the swift evolution of antibiotic resistance in the recA-strain.
We greatly appreciate your overall summary of the manuscript and their positive evaluation of our work.
Weaknesses:
However, a potential limitation of this study is the experimental design used to determine the 'rapid' evolution of antibiotic resistance. It may introduce a significant bottleneck in selecting ampicillin-resistant mutants early on. A recA mutant could be more susceptible to ampicillin than the wild-type, and only resistant mutants might survive after 8 hours, potentially leading to their enrichment in subsequent steps. To address this concern, it would be critical to perform a survival analysis at various time points (0h, 2h, 4h, 6h, and 8h) during ampicillin treatment for both recA and wild-type strains, ensuring there is no difference in viability.
We appreciate your suggestion. We measured the survival fraction at 0, 2, 4, 6, and 8 hours after ampicillin treatment. The results show no significant difference in antibiotic sensitivity between the wild-type and ΔrecA strain (Fig. S2). We therefore added a description int the main text, “Meanwhile, after 8 hours of treatment with 50 μg/mL ampicillin, the survival rates of both wild type and ΔrecA strain were consistent (Fig. S2)”.
The observation that promoter mutations are absent in ΔrecA strains could be explained by previous research indicating that amplification of the AmpC genes is a mechanism for E. coli resistance to ampicillin, which does not occur in a recA-deficient background (PMID# 19474201).
We are very grateful to you for providing this reference. We did examine the amplification of the ampC gene in both wild-type and _recA-_deficient strains, but we found no significant changes in its copy number after ampicillin treatment (Author response image 2). Therefore, the results and discussion regarding gene copy number were not included in this manuscript.
Author response image 2.
Copy number variations of genes in the chromosome before and after exposure to ampicillin at 50 µg/mL for 8 hours in the wild type and ΔrecA strain.
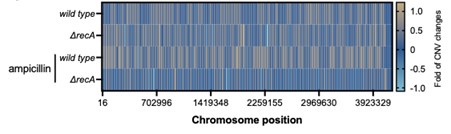
The section describing Figure 3 is poorly articulated, and the conclusions drawn are apparent. The inability of a recA strain to induce the SOS response is well-documented (lines 210 and 278). The data suggest that merely blocking SOS induction is insufficient to cause 'rapid' evolution in their experimental conditions. To investigate whether SOS response can be induced independently of lexA cleavage by recA, alternative experiments, such as those using a sulA-GFP fusion, might be more informative.
Thanks for your suggestion. We agree that detecting the expression level of SulA can provide valuable information to reveal the impact of the SOS system on rapid drug resistance. In addition to fluorescence visualization and quantification of SulA expression, regulating the transcription level of the sulA gene can achieve the same objective. Therefore, in our transcriptome sequencing analysis, we focused on evaluating the transcription level of sulA (Fig. 4E).
In Figure 4E, the lack of increased SulA gene expression in the wild-type strain treated with ampicillin is unexpected, given that SulA is an SOS-regulated gene. The fact that polA (Pol I) is going down should be taken into account in the interpretation of Figures 2D and 2E.
Thank you for your observation regarding the lack of increased SulA gene expression in the wild-type strain treated with ampicillin in Figure 4E. We agree that SulA is typically an SOS-regulated gene, and its expression is expected to increase in response to DNA damage induced by antibiotics like ampicillin. However, in our experimental conditions, the observed lack of increased SulA expression could be due to different factors. One possibility is that the concentration of ampicillin used, or the duration of treatment, was not applicable to induce a strong SOS response in the wild type strain under the specific conditions tested. Additionally, differences in experimental setups such as timing, sampling, or cellular stress responses could account for the lack of a pronounced upregulation of SulA.
You may state that the fact that polA (Pol I) is going down should be taken into account in the interpretation of Figures 3D and 3E, and we agree with you.
The connection between compromised DNA repair, the accumulation of Reactive Oxygen Species (ROS) based on RNA-seq data, and accelerated evolution is merely speculative at this point and not experimentally established.
We greatly appreciate your comments. First, the correlation between DNA mutations and the accumulation of reactive oxygen species (ROS) has been experimentally confirmed. As shown in Fig. 4I, after the addition of the antioxidant GSH, DNA resistance mutations were not detected in the ΔrecA strain treated with ampicillin for 8 hours, compared to those without the addition of GSH, proving that the rapid accumulation of ROS induces the enhancement of DNA resistance mutations. Second, the enhancement of DNA resistance mutations in relation to bacterial resistance has been widely validated and is generally accepted. Finally, we appreciate the your suggestion to strengthen the evidence supporting ROS enhancement. To address this, we have added an experiment to measure ROS levels. Through flow cytometry, we found that ROS levels significantly increased in both the wild-type and ΔrecA strain after 8 hours of ampicillin treatment. However, ROS levels in the ΔrecA strain showed a significant further increase compared to the wild-type strain (Fig. 4G). Additionally, with the addition of 50 mM glutathione, no significant change in ROS levels was observed in either the wild-type or ΔrecA strain before and after ampicillin treatment (Fig. 4H). This result further confirms our finding in Fig. 4I, where adding GSH inhibited the development of antibiotic resistance.
Reviewer #3:
Summary:
In the present work, Zhang et al investigate the involvement of the bacterial DNA damage repair SOS response in the evolution of beta-lactam drug resistance evolution in Escherichia coli. Using a combination of microbiological, bacterial genetics, laboratory evolution, next-generation, and live-cell imaging approaches, the authors propose short-term drug resistance evolution that can take place in RecA-deficient cells in an SOS response-independent manner. They propose the evolvability of drug resistance is alternatively driven by the oxidative stress imposed by the accumulation of reactive oxygen species and inhibition of DNA repair. Overall, this is a nice study that addresses a growing and fundamental global health challenge (antimicrobial resistance). However, although the authors perform several multi-disciplinary experiments, there are several caveats to the authors' proposal that ultimately do not fully support their interpretation that the observed antimicrobial resistance evolution phenotype is due to compromised DNA repair.
We greatly appreciate your overall summary of the manuscript and positive evaluation of our work.
Strengths:
The authors introduce new concepts to antimicrobial resistance evolution mechanisms. They show short-term exposure to beta-lactams can induce durably fixed antimicrobial resistance mutations. They propose this is due to comprised DNA repair and oxidative stress. This is primarily supported by their observations that resistance evolution phenotypes only exist for recA deletion mutants and not other genes in the SOS response.
Thanks for your positive comments.
Weaknesses:
The authors do not show any direct evidence (1) that these phenotypes exist in strains harboring deletions in other DNA repair genes outside of the SOS response, (2) that DNA damage is increased, (3) that reactive oxygen species accumulate, (4) that accelerated resistance evolution can be reversed by anything other than recA complementation. The authors do not directly test alternative hypotheses. The conclusions drawn are therefore premature.
We sincerely thank you for your insightful comments. First, in this study, our primary focus is on the role of recA deficiency in bacterial antibiotic resistance evolution. Therefore, we conducted an in-depth investigation on E. coli strains lacking RecA and found that its absence promotes resistance evolution through mechanisms involving increased ROS accumulation and downregulation of DNA repair pathways. While we acknowledge the importance of other DNA repair genes outside of the SOS response, exploring them is beyond the scope of this paper. However, in a separate unpublished study, we have identified the involvement of another DNA recombination protein, whose role in resistance evolution is not yet fully elucidated, in promoting resistance development. This finding is part of another independent investigation.
Regarding DNA damage and repair, our paper emphasizes that resistance-related mutations in DNA are central to the development of antibiotic resistance. These mutations are a manifestation of DNA damage. To demonstrate this, we measured mutation frequency and performed whole-genome sequencing, both of which confirmed an increase in DNA mutations.
We appreciate the reviewer's suggestion to provide additional evidence for ROS accumulation, and we have now supplemented our manuscript with relevant experiments. Through flow cytometry, we found that ROS levels significantly increased in both the wild type and ΔrecA strains after 8 hours of ampicillin treatment. However, ROS levels in the ΔrecA strain showed a significant further increase compared to the wild-type strain (Fig. 4G). Additionally, with the addition of 50 mM glutathione, no significant change in ROS levels was observed in either the wild-type or ΔrecA strain before and after ampicillin treatment (Fig. 4H). This result further confirms our finding in Fig. 4I, where adding GSH inhibited the development of antibiotic resistance.
Finally, in response to your question about reversing accelerated resistance evolution, we would like to highlight that, in addition to recA complementation, we successfully suppressed rapid resistance evolution by supplementing with an antioxidant, GSH (Fig. 4I). This further supports our hypothesis that increased ROS levels play a key role in driving accelerated resistance evolution in the absence of RecA.
Recommendations for the authors:
Reviewer #1:
The author's model asserts that deletion of recA impairs DNA repair in E. coli, leading to an accumulation of ROS in the cell, and ultimately driving the rapid rise of resistance mutations. However, the experimental evidence does not adequately address whether the resistance mutations are true, de novo mutations that arose due to beta-lactam treatment, or mutations that confer cross-resistance enriched by ampicillin selection.
a. Major: In Figure 1F & G, the authors show that the ∆recA strain, following ampicillin treatment, has higher resistance and mutation frequency towards rifampicin than WT. However, it is not clear whether the elevated resistance and mutagenesis are driven by mutations enriched by the ampicillin treatment (e.g. mutations in acrB, as seen in Figure 2) or by "new" mutations in the rpoB gene. As the authors note, the mutants enriched by ampicillin selection can play a role in efflux and can thus change a bacterium's sensitivity to a wide range of antibiotics, including rifampicin, in what is known as cross-resistance. Therefore, the mutation frequency calculation, which relies on quantifying rifampicin-resistant clones, might be confounded by bacteria with mutations that confer cross-resistance. A better approach to calculate mutation frequency would be to employ an assay that does not require antibiotic selection, such as a lac-reversion assay. This would mitigate the confounding effects of cross-resistance of drug-resistant mutations.
We appreciate your thoughtful comments regarding the potential for cross-resistance to confound the mutation frequency calculation based on rifampicin-resistant clones. Indeed, as noted, ampicillin selection can enrich for mutants with enhanced efflux activity, which may confer cross-resistance to a range of antibiotics, including rifampicin.
However, we believe that the current approach of calculating mutation frequency using rifampicin-resistant mutants is still valid in our specific context. Rifampicin targets the RNA polymerase β subunit, and resistance typically arises from specific mutations in the rpoB gene. These mutations are well-characterized and distinct from those typically associated with efflux-related cross-resistance. Thus, the likelihood of cross-resistance affecting our mutation frequency calculation is minimized in this scenario.
Additionally, while the lac-reversion assay could be an alternative, it focuses on specific metabolic pathway mutations (such as those affecting lacZ) and would not necessarily capture the same types of mutations relevant to rifampicin resistance or antibiotic-induced mutagenesis. Given our experimental objective of understanding how ampicillin induces mutations that confer antibiotic resistance, the current approach of using rifampicin selection provides a direct and relevant measurement of mutation frequency under antibiotic stress.
b. Major: It is important to establish what the basal mutation frequencies/rates of both the WT and ∆recA strains are. Currently, only the ampicillin-treated populations were reported. It is possible that the ∆recA strain has an inherently higher mutagenesis than WT. Thus, ampicillin treatment might not in fact induce higher mutagenesis in ∆recA.
Thanks for your suggestion. The basal mutation frequency of the wild-type and the ∆recA strain have been measured using rifampicin (Fig. 1G), and there is no significant difference between them.
c. Major: In the text, the authors write, "To verify whether drug resistance associated DNA mutations have led to the rapid development of antibiotic resistance in recA mutant strain, we randomly selected 15 colonies on non-selected LB agar plates from the wild type surviving isolates, and antibiotic screening plates containing 50 μg/mL ampicillin from the ΔrecA resistant isolates, respectively." Why were the WT clones picked from non-selective plates and the recA mutant from selective ones for WGS? It appears that such a procedure would bias the recA mutant clones to show more mutations (caused by selection on the ampicillin plate). The authors need to address this discrepancy.
We appreciate your concern regarding potential inconsistencies in the WGS methodology. However, we would like to clarify that the primary aim of the WGS experiment was to identify the types of mutations present in the wild-type and ΔrecA strains after treatment of ampicillin, rather than to quantify or compare mutation frequencies. This purpose was explicitly stated in the manuscript.
Furthermore, the choice of selective and non-selective conditions was made to ensure the successful isolation of mutants in both strains. Specifically, if selective conditions (50 μg/mL ampicillin) were applied to the wild type strain, it would have been nearly impossible to recover colonies for WGS analysis, as wild-type cells are highly susceptible to ampicillin at this concentration (Top, Author response image 1). Conversely, under non-selective conditions, ΔrecA mutants carrying resistance mutations may not have been effectively isolated, which would have limited our ability to identify resistance mutations in these strains (Bottom, Author response image 1). Thus, the use of different selection pressures was essential for achieving the objective of mutation identification in this study.
d. Major: In some instances, the authors do not use accurate language to describe their data. In Figure 2A, the authors randomly selected 15 ∆recA clones from a selective plate with 50 µg/mL of ampicillin. These clones were then subjected to WGS, which subsequently identified resistant mutations. Based on the described methods, these mutations are a result of selection: in other words, resistant mutations were preexisting in the bacterial population, and the addition of ampicillin selection killed off the sensitive cells, enabling the proliferation of the resistant clones. However, the in Figure 2 legend and associated text, the authors suggest that these mutations were "induced" by beta-lactam exposure, which is misleading. The data does not support that.
We appreciate your detailed feedback on the language used to describe our data. We understand the concern regarding the use of the term "induced" in relation to beta-lactam exposure. To clarify, we employed not only beta-lactam antibiotics but also other antibiotics, such as ciprofloxacin and chloramphenicol, in our experiments (data not shown). However, we observed that beta-lactam antibiotics specifically induced the emergence of resistance or altered the MIC in our bacterial populations. If resistance had pre-existed before antibiotic exposure, we would expect other antibiotics to exhibit a similar selective effect, particularly given the potential for cross-resistance to multiple antibiotics.
Furthermore, we used two different ∆recA strains, and the results were consistent between the strains (Fig. S3). Given that spontaneous mutations can occur with significant variability in populations, if resistance mutations pre-existed before antibiotic exposure, the selective outcomes should have varied between the two strains.
Most importantly, we found that the addition of anti-oxidative compound GSH prevented the evolution of antibiotic from the treatment of ampicillin in the ΔrecA strain. If we assume that resistant bacteria preexist in the ∆recA strain, then the addition of GSH should not affect the evolution of resistance. Therefore, we believe that the resistance mutations we detected were not simply the result of selection from preexisting mutations but were indeed induced by beta-lactam exposure.
e. Major: For Figure 4J, using WGS the authors show that the addition of GSH to WT and ∆recA cells inhibited the rise of resistance mutations; no resistance mutations were reported. However, in the "Whole genome sequencing" section under "Materials and Methods", they state that "Resistant clones were isolated by selection using LB agar plates with the supplementation of ampicillin at 50 μg/mL". These clones were then genome-extracted and sequenced. Given the methodology, it is surprising that the WGS did not reveal any resistance mutations in the GSH-treated cells. How were these cells able to grow on 50 μg/mL ampicillin plates for isolation in the first place? The authors need to address this.
We sincerely apologize for the confusion caused by the incorrect expression in the "Materials and Methods" section. Indeed, when bacteria were treated with the combination of antibiotics and GSH, resistance was significantly suppressed, and no resistant clones could be isolated from selective plates (i.e., LB agar supplemented with 50 μg/mL ampicillin).
To address this, we instead plated the bacteria treated with antibiotics and GSH onto non-selective plates (without ampicillin) and randomly selected 15 colonies for WGS. None of them showed resistance mutations. We will revise the text in the "Materials and Methods" section to accurately reflect this procedure and provide clarity.
f. Minor: for Figure 1G, it is misleading to have both "mutation frequency" and "mutant rate" in the y-axis; the two are defined and calculated differently. Based on the Materials and Materials, "mutation frequency" would be the appropriate term. Also, for the ∆recA strain, it is a bit unusual to see mutation frequencies that are tightly clustered. Usually, mutation frequencies follow the Luria-Delbruck distribution. Can the authors explain why the ∆recA data looks so different compared to, say, the WT mutation frequencies?
Thank you for your insightful feedback. We agree that having both "mutation frequency" and "mutant rate" on the y-axis is misleading, as these terms are defined and calculated differently. To avoid confusion, we will revise Figure 1G to use only "mutation frequency" as the correct term, in line with the methods described in the Materials and Methods section.
Regarding the ∆recA strain's mutation frequencies, we acknowledge that the data appear more tightly clustered compared to the expected Luria-Delbruck distribution seen in the wild type strain. In fact, the y-axis of the Figure 1G is logarithmic, this causes the data to appear more clustered.
We further added the basal mutation frequency in the wild type and ∆recA strains before the exposure to ampicillin. The basal mutation frequency of the wild-type and the ∆recA strain have been measured using rifampicin (Fig. 1G), and there is no significant difference between them.
g. Minor: It needs to be made clear in the Main Text what the selective antibiotic agar plate used was, rifampicin or ampicillin. I am assuming it was rifampicin, as ampicillin plates would yield resistance frequencies close to 100%, given the prior treatment of the culture with ampicillin.
Thanks for your comments. Depending on the objective, we used different selective plates. For example, when testing the mutation frequency of antibiotic resistance, we used a selective plate containing rifampicin in order to utilize a non-inducing antibiotic, which is the standard method for calculating resistance mutation frequency. In the WGS experiment, to obtain mutations specific to ampicillin resistance, we selected a selective plate containing ampicillin.
Reviewer #2:
The Y-axis label (log10 mutant rate) in Figure 1G is misleading or incorrect.
Thanks for your comments and we apologize for this misleading information. The Figure 1G has been revised accordingly.
In line 393 of the discussion, the authors claim that excessive ROS accumulation drives the evolution of ampicillin resistance, which has not been conclusively demonstrated. Additional experiments are needed to support this statement.
We greatly appreciate your comments. First, the correlation between DNA mutations and the accumulation of reactive oxygen species (ROS) has been experimentally confirmed. As shown in Fig. 4I, after the addition of the antioxidant GSH, DNA resistance mutations were not detected in the ΔrecA strain treated with ampicillin for 8 hours, compared to those without the addition of GSH, proving that the rapid accumulation of ROS induces the enhancement of DNA resistance mutations. Second, the enhancement of DNA resistance mutations in relation to bacterial resistance has been widely validated and is generally accepted. Finally, we appreciate the your suggestion to strengthen the evidence supporting ROS enhancement. To address this, we have added an experiment to measure ROS levels. Through flow cytometry, we found that ROS levels significantly increased in both the wild-type and ΔrecA strain after 8 hours of ampicillin treatment. However, ROS levels in the ΔrecA strain showed a significant further increase compared to the wild-type strain (Fig. 4G). Additionally, with the addition of 50 mM glutathione, no significant change in ROS levels was observed in either the wild-type or ΔrecA strain before and after ampicillin treatment (Fig. 4H). This result further confirms our finding in Fig. 4I, where adding GSH inhibited the development of antibiotic resistance.
The abstract is overly complex and difficult to read, e.g. "Contrary to previous findings, it is shown that this accelerated resistance development process is dependent on the hindrance of DNA repair, which is completely orthogonal to the SOS response").
Thank you for the valuable feedback regarding the complexity of the abstract. We agree that certain sections could be simplified for clarity. In response, we have revised the abstract to make it more concise and easier to understand. For example, the sentence “Contrary to previous findings, it is shown that this accelerated resistance development process is dependent on the hindrance of DNA repair, which is completely orthogonal to the SOS response” has been rewritten as: "Unlike earlier studies, we found that the rapid development of resistance relies on the hindrance of DNA repair, a mechanism that operates independently of the SOS response."
Reviewer #3:
As indicated above, direct evidence is needed to show (1) that these phenotypes exist in strains harboring deletions in other DNA repair genes outside of the SOS response, (2) that DNA damage is increased, (3) that reactive oxygen species accumulate, (4) that accelerated resistance evolution can be reversed by anything other than recA complementation. There are also other resistance evolution mechanisms untested here, including transcription-coupled repair (TCR) mechanisms involving Mfd. These need to be shown in order to draw the conclusions proposed.
We sincerely thank you for your insightful comments. First, in this study, our primary focus is on the role of recA deficiency in bacterial antibiotic resistance evolution. Therefore, we conducted an in-depth investigation on E. coli strains lacking RecA and found that its absence promotes resistance evolution through mechanisms involving increased ROS accumulation and downregulation of DNA repair pathways. While we acknowledge the importance of other DNA repair genes outside of the SOS response and other resistance evolution mechanisms including the TCR mechanism, exploring them is beyond the scope of this paper. However, in a separate unpublished study, we have identified the involvement of another DNA recombination protein, whose role in resistance evolution is not yet fully elucidated, in promoting resistance development. This finding is part of another independent investigation.
Regarding DNA damage and repair, our paper emphasizes that resistance-related mutations in DNA are central to the development of antibiotic resistance. These mutations are a manifestation of DNA damage. To demonstrate this, we measured mutation frequency and performed whole-genome sequencing, both of which confirmed an increase in DNA mutations.
We appreciate the reviewer's suggestion to provide additional evidence for ROS accumulation, and we have now supplemented our manuscript with relevant experiments. Through flow cytometry, we found that ROS levels significantly increased in both the wild type and ΔrecA strains after 8 hours of ampicillin treatment. However, ROS levels in the ΔrecA strain showed a significant further increase compared to the wild-type strain (Fig. 4G). Additionally, with the addition of 50 mM glutathione, no significant change in ROS levels was observed in either the wild-type or ΔrecA strain before and after ampicillin treatment (Fig. 4H). This result further confirms our finding in Fig. 4I, where adding GSH inhibited the development of antibiotic resistance.
Finally, in response to your question about reversing accelerated resistance evolution, we would like to highlight that, in addition to recA complementation, we successfully suppressed rapid resistance evolution by supplementing with an antioxidant, GSH (Fig. 4I). This further supports our hypothesis that increased ROS levels play a key role in driving accelerated resistance evolution in the absence of RecA.
-
-
eLife assessment
This useful study examines how deletion of a major DNA repair gene in bacteria may facilitate the rise of mutations that confer resistance against a range of different antibiotics. Although the phenotypic evidence is intriguing, the interpretation of the phenotypic data presented and the proposed mechanism by which these mutations are generated are incomplete, relying on untested assumptions and suboptimal methodology. If substantially improved, this work could be of interest to microbiologists studying antibiotic resistance, genome integrity, and evolution, but as yet is of unclear significance.
-
Reviewer #1 (Public Review):
Summary:
Jin et al. investigated how the bacterial DNA damage (SOS) response and its regulator protein RecA affect the development of drug resistance under short-term exposure to beta-lactam antibiotics. Canonically, the SOS response is triggered by DNA damage, which results in the induction of error-prone DNA repair mechanisms. These error-prone repair pathways can increase mutagenesis in the cell, leading to the evolution of drug resistance. Thus, inhibiting the SOS regulator RecA has been proposed as a means to delay the rise of resistance.In this paper, the authors deleted the RecA protein from E. coli and exposed this ∆recA strain to selective levels of the beta-lactam antibiotic, ampicillin. After an 8-hour treatment, they washed the antibiotic away and allowed the surviving cells to recover in …
Reviewer #1 (Public Review):
Summary:
Jin et al. investigated how the bacterial DNA damage (SOS) response and its regulator protein RecA affect the development of drug resistance under short-term exposure to beta-lactam antibiotics. Canonically, the SOS response is triggered by DNA damage, which results in the induction of error-prone DNA repair mechanisms. These error-prone repair pathways can increase mutagenesis in the cell, leading to the evolution of drug resistance. Thus, inhibiting the SOS regulator RecA has been proposed as a means to delay the rise of resistance.In this paper, the authors deleted the RecA protein from E. coli and exposed this ∆recA strain to selective levels of the beta-lactam antibiotic, ampicillin. After an 8-hour treatment, they washed the antibiotic away and allowed the surviving cells to recover in regular media. They then measured the minimum inhibitory concentration (MIC) of ampicillin against these treated strains. They note that after just 8-hour treatment with ampicillin, the ∆recA had developed higher MICs towards ampicillin, while by contrast, wild-type cells exhibited unchanged MICs. This MIC increase was also observed in subsequent generations of bacteria, suggesting that the phenotype is driven by a genetic change.
The authors then used whole genome sequencing (WGS) to identify mutations that accounted for the resistance phenotype. Within resistant populations, they discovered key mutations in the promoter region of the beta-lactamase gene, ampC; in the penicillin-binding protein PBP3 which is the target of ampicillin; and in the AcrB subunit of the AcrAB-TolC efflux machinery. Importantly, mutations in the efflux machinery can impact the resistance towards other antibiotics, not just beta-lactams. To test this, they repeated the MIC experiments with other classes of antibiotics, including kanamycin, chloramphenicol, and rifampicin. Interestingly, they observed that the ∆recA strains pre-treated with ampicillin showed higher MICs towards all other antibiotics tested. This suggests that the mutations conferring resistance to ampicillin are also increasing resistance to other antibiotics.
The authors then performed an impressive series of genetic, microscopy, and transcriptomic experiments to show that this increase in resistance is not driven by the SOS response, but by independent DNA repair and stress response pathways. Specifically, they show that deletion of the recA reduces the bacterium's ability to process reactive oxygen species (ROS) and repair its DNA. These factors drive the accumulation of mutations that can confer resistance to different classes of antibiotics. The conclusions are reasonably well-supported by the data, but some aspects of the data and the model need to be clarified and extended.
Strengths:
A major strength of the paper is the detailed bacterial genetics and transcriptomics that the authors performed to elucidate the molecular pathways responsible for this increased resistance. They systemically deleted or inactivated genes involved in the SOS response in E. coli. They then subjected these mutants to the same MIC assays as described previously. Surprisingly, none of the other SOS gene deletions resulted in an increase in drug resistance, suggesting that the SOS response is not involved in this phenotype. This led the authors to focus on the localization of DNA PolI, which also participates in DNA damage repair. Using microscopy, they discovered that in the RecA deletion background, PolI co-localizes with the bacterial chromosome at much lower rates than wild-type. This led the authors to conclude that deletion of RecA hinders PolI and DNA repair. Although the authors do not provide a mechanism, this observation is nonetheless valuable for the field and can stimulate further investigations in the future.In order to understand how RecA deletion affects cellular physiology, the authors performed RNA-seq on ampicillin-treated strains. Crucially, they discovered that in the RecA deletion strain, genes associated with antioxidative activity (cysJ, cysI, cysH, soda, sufD) and Base Excision Repair repair (mutH, mutY, mutM), which repairs oxidized forms of guanine, were all downregulated. The authors conclude that down-regulation of these genes might result in elevated levels of reactive oxygen species in the cells, which in turn, might drive the rise of resistance. Experimentally, they further demonstrated that treating the ∆recA strain with an antioxidant GSH prevents the rise of MICs. These observations will be useful for more detailed mechanistic follow-ups in the future.
Weaknesses:
Throughout the paper, the authors use language suggesting that ampicillin treatment of the ∆recA strain induces higher levels of mutagenesis inside the cells, leading to the rapid rise of resistance mutations. However, as the authors note, the mutants enriched by ampicillin selection can play a role in efflux and can thus change a bacterium's sensitivity to a wide range of antibiotics, in what is known as cross-resistance. The current data is not clear on whether the elevated "mutagenesis" is driven ampicillin selection or by a bona fide increase in mutation rate.Furthermore, on a technical level, the authors employed WGS to identify resistance mutations in the treated ampicillin-treated wild-type and ∆recA strains. However, the WGS methodology described in the paper is inconsistent. Notably, wild-type WGS samples were picked from non-selective plates, while ΔrecA WGS isolates were picked from selective plates with 50 μg/mL ampicillin. Such an approach biases the frequency and identity of the mutations seen in the WGS and cannot be used to support the idea that ampicillin treatment induces higher levels of mutagenesis.
Finally, it is important to establish what the basal mutation rates of both the WT and ∆recA strains are. Currently, only the ampicillin-treated populations were reported. It is possible that the ∆recA strain has inherently higher mutagenesis than WT, with a larger subpopulation of resistant clones. Thus, ampicillin treatment might not in fact induce higher mutagenesis in ∆recA.
-
Reviewer #2 (Public Review):
Summary:
This study aims to demonstrate that E. coli can acquire rapid antibiotic resistance mutations in the absence of a DNA damage response. To investigate this, the authors employed a sophisticated experimental framework based on a modified Adaptive Laboratory Evolution (ALE) workflow. This workflow involves numerous steps culminating in the measurement of antibiotic resistance. The study presents evidence that a recA strain develops ampicillin resistance mutations more quickly than the wild-type, as shown by measuring the Minimum Inhibitory Concentration (MIC) and mutation frequency. Whole-genome sequencing of 15 recA- colonies resistant to ampicillin revealed predominantly inactivation of genes involved in the multi-drug efflux pump system, whereas, in the wild-type, mutations appear to enhance the …Reviewer #2 (Public Review):
Summary:
This study aims to demonstrate that E. coli can acquire rapid antibiotic resistance mutations in the absence of a DNA damage response. To investigate this, the authors employed a sophisticated experimental framework based on a modified Adaptive Laboratory Evolution (ALE) workflow. This workflow involves numerous steps culminating in the measurement of antibiotic resistance. The study presents evidence that a recA strain develops ampicillin resistance mutations more quickly than the wild-type, as shown by measuring the Minimum Inhibitory Concentration (MIC) and mutation frequency. Whole-genome sequencing of 15 recA- colonies resistant to ampicillin revealed predominantly inactivation of genes involved in the multi-drug efflux pump system, whereas, in the wild-type, mutations appear to enhance the activity of the chromosomal ampC cryptic promoter. By analyzing mutants involved in the SOS response, including a lexA3 mutant incapable of inducing the SOS response, the authors conclude that the rapid evolution of antibiotic resistance occurs in an SOS-independent manner when recA is absent.Furthermore, RNA sequencing (RNA-seq) of the four experimental conditions suggests that genes related to antioxidative responses drive the swift evolution of antibiotic resistance in the recA- strain.
Weaknesses:
However, a potential limitation of this study is the experimental design used to determine the 'rapid' evolution of antibiotic resistance. It may introduce a significant bottleneck in selecting ampicillin-resistant mutants early on. A recA mutant could be more susceptible to ampicillin than the wild-type, and only resistant mutants might survive after 8 hours, potentially leading to their enrichment in subsequent steps. To address this concern, it would be critical to perform a survival analysis at various time points (0h, 2h, 4h, 6h, and 8h) during ampicillin treatment for both recA and wild-type strains, ensuring there is no difference in viability.The observation that promoter mutations are absent in recA strains could be explained by previous research indicating that amplification of the AmpC genes is a mechanism for E. coli resistance to ampicillin, which does not occur in a recA-deficient background (PMID# 19474201).
The section describing Figure 3 is poorly articulated, and the conclusions drawn are apparent. The inability of a recA strain to induce the SOS response is well-documented (lines 210 and 278). The data suggest that merely blocking SOS induction is insufficient to cause 'rapid' evolution in their experimental conditions. To investigate whether SOS response can be induced independently of lexA cleavage by recA, alternative experiments, such as those using a sulA-GFP fusion, might be more informative.
In Figure 4E, the lack of increased SulA gene expression in the wild-type strain treated with ampicillin is unexpected, given that SulA is an SOS-regulated gene. The fact that polA (Pol I) is going down should be taken into account in the interpretation of Figures 2D and 2E.
The connection between compromised DNA repair, the accumulation of Reactive Oxygen Species (ROS) based on RNA-seq data, and accelerated evolution is merely speculative at this point and not experimentally established.
-
Reviewer #3 (Public Review):
Summary:
In the present work, Zhang et al investigate the involvement of the bacterial DNA damage repair SOS response in the evolution of beta-lactam drug resistance evolution in Escherichia coli. Using a combination of microbiological, bacterial genetics, laboratory evolution, next-generation, and live-cell imaging approaches, the authors propose short-term drug resistance evolution that can take place in RecA-deficient cells in an SOS response-independent manner. They propose the evolvability of drug resistance is alternatively driven by the oxidative stress imposed by the accumulation of reactive oxygen species and inhibition of DNA repair. Overall, this is a nice study that addresses a growing and fundamental global health challenge (antimicrobial resistance). However, although the authors perform …Reviewer #3 (Public Review):
Summary:
In the present work, Zhang et al investigate the involvement of the bacterial DNA damage repair SOS response in the evolution of beta-lactam drug resistance evolution in Escherichia coli. Using a combination of microbiological, bacterial genetics, laboratory evolution, next-generation, and live-cell imaging approaches, the authors propose short-term drug resistance evolution that can take place in RecA-deficient cells in an SOS response-independent manner. They propose the evolvability of drug resistance is alternatively driven by the oxidative stress imposed by the accumulation of reactive oxygen species and inhibition of DNA repair. Overall, this is a nice study that addresses a growing and fundamental global health challenge (antimicrobial resistance). However, although the authors perform several multi-disciplinary experiments, there are several caveats to the authors' proposal that ultimately do not fully support their interpretation that the observed antimicrobial resistance evolution phenotype is due to compromised DNA repair.Strengths:
The authors introduce new concepts to antimicrobial resistance evolution mechanisms. They show short-term exposure to beta-lactams can induce durably fixed antimicrobial resistance mutations. They propose this is due to comprised DNA repair and oxidative stress. This is primarily supported by their observations that resistance evolution phenotypes only exist for recA deletion mutants and not other genes in the SOS responseWeaknesses:
The authors do not show any direct evidence (1) that these phenotypes exist in strains harboring deletions in other DNA repair genes outside of the SOS response, (2) that DNA damage is increased, (3) that reactive oxygen species accumulate, (4) that accelerated resistance evolution can be reversed by anything other than recA complementation. The authors do not directly test alternative hypotheses. The conclusions drawn are therefore premature. -
