RAS–p110α signalling in macrophages is required for effective inflammatory response and resolution of inflammation
Curation statements for this article:-
Curated by eLife

eLife Assessment
This useful study investigates the impact of disrupting the interaction of RAS with the PI3K subunit p110α in macrophage function in vitro and inflammatory responses in vivo. Solid data overall supports a role for RAS-p110α signalling in regulating macrophage activity and so inflammation, however for many of the readouts presented the magnitude of the phenotype is not particularly pronounced. Further analysis would be required to substantiate the claims that RAS-p110α signalling plays a key role in macrophage function. Of note, the molecular mechanisms of how exactly p110α regulates the functions in macrophages have not yet been established.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Macrophages are crucial in the body’s inflammatory response, with tightly regulated functions for optimal immune system performance. Our study reveals that the RAS–p110α signalling pathway, known for its involvement in various biological processes and tumourigenesis, regulates two vital aspects of the inflammatory response in macrophages: the initial monocyte movement and later-stage lysosomal function. Disrupting this pathway, either in a mouse model or through drug intervention, hampers the inflammatory response, leading to delayed resolution and the development of more severe acute inflammatory reactions in live models. This discovery uncovers a previously unknown role of the p110α isoform in immune regulation within macrophages, offering insight into the complex mechanisms governing their function during inflammation and opening new avenues for modulating inflammatory responses.
Article activity feed
-

-
-

eLife Assessment
This useful study investigates the impact of disrupting the interaction of RAS with the PI3K subunit p110α in macrophage function in vitro and inflammatory responses in vivo. Solid data overall supports a role for RAS-p110α signalling in regulating macrophage activity and so inflammation, however for many of the readouts presented the magnitude of the phenotype is not particularly pronounced. Further analysis would be required to substantiate the claims that RAS-p110α signalling plays a key role in macrophage function. Of note, the molecular mechanisms of how exactly p110α regulates the functions in macrophages have not yet been established.
-
Reviewer #2 (Public review):
Summary:
Cell intrinsic signaling pathways controlling the function of macrophages in inflammatory processes, including in response to infection, injury or in the resolution of inflammation are incompletely understood. In this study, Rosell et al. investigate the contribution of RAS-p110α signaling to macrophage activity. p110α is a ubiquitously expressed catalytic subunit of PI3K with previously described roles in multiple biological processes including in epithelial cell growth and survival, and carcinogenesis. While previous studies have already suggested a role for RAS-p110α signaling in macrophage function, the cell intrinsic impact of disrupting the interaction between RAS and p110α in this central myeloid cell subset is not known.
Strengths:
Exploiting a sound previously described genetically …
Reviewer #2 (Public review):
Summary:
Cell intrinsic signaling pathways controlling the function of macrophages in inflammatory processes, including in response to infection, injury or in the resolution of inflammation are incompletely understood. In this study, Rosell et al. investigate the contribution of RAS-p110α signaling to macrophage activity. p110α is a ubiquitously expressed catalytic subunit of PI3K with previously described roles in multiple biological processes including in epithelial cell growth and survival, and carcinogenesis. While previous studies have already suggested a role for RAS-p110α signaling in macrophage function, the cell intrinsic impact of disrupting the interaction between RAS and p110α in this central myeloid cell subset is not known.
Strengths:
Exploiting a sound previously described genetically engineered mouse model that allows tamoxifen-inducible disruption of the RAS-p110α pathway and using different readouts of macrophage activity in vitro and in vivo, the authors provide data consistent with their conclusion that alteration in RAS-p110α signaling impairs various but selective aspects of macrophage function in a cell-intrinsic manner.
Weaknesses:
My main concern is that for various readouts, the difference between wild-type and mutant macrophages in vitro or between wild-type and Pik3caRBD mice in vivo is modest, even if statistically significant. To further substantiate the extent of macrophage function alteration upon disruption of RAS-p110α signaling and its impact on the initiation and resolution of inflammatory responses, the manuscript would benefit from a more extensive assessment of macrophage activity and inflammatory responses in vivo.
In the in vivo model, all cells have disrupted RAS-p100α signaling, not only macrophages. Given that other myeloid cells besides macrophages contribute to the orchestration of inflammatory responses, it remains unclear whether the phenotype described in vivo results from impaired RAS-p100α signaling within macrophages or from defects in other haematopoietic cells such as neutrophils, dendritic cells, etc.
Inclusion of information on the absolute number of macrophages, and total immune cells (e.g. for the spleen analysis) would help determine if the reduced frequency of macrophages represents an actual difference in their total number or rather reflects a relative decrease due to an increase in the number of other/s immune cell/s.
Comments on revisions:
I thank the authors for addressing my comments.
- I believe that additional in vivo experiments, or the inclusion of controls for the specificity of the inhibitor, which the authors argue are beyond the scope of the current study, are essential to address the weaknesses and limitations stated in my current evaluation.
- While the neutrophil depletion suggests neutrophils are not required for the phenotype, there are multiple other myeloid cells, in addition to macrophages, that could be contributing or accounting for the in vivo phenotype observed in the mutant strain (not macrophage specific).
- Inclusion of absolute cell numbers (in addition to the %) is essential. I do not understand why the authors are not including these data. Have they not counted the cells?
- Lastly, inclusion of representatives staining and gating strategies for all immune profiling measurements carried out by flow cytometry is important. This point has not been addressed, not even in writing. -
Author response:
The following is the authors’ response to the current reviews.
Comments on revisions:
I thank the authors for addressing my comments.
- I believe that additional in vivo experiments, or the inclusion of controls for the specificity of the inhibitor, which the authors argue are beyond the scope of the current study, are essential to address the weaknesses and limitations stated in my current evaluation.
We respectfully acknowledge the reviewer's concern but would like to reiterate that demonstrating the specificity of the inhibitor is beyond the scope of this study. Alpelisib (BYL-719) is a clinically approved drug widely recognized as a specific inhibitor of p110α, primarily used in the treatment of breast cancer. Its selectivity for the p110α isoform has been extensively validated in the literature.
In our study, we …
Author response:
The following is the authors’ response to the current reviews.
Comments on revisions:
I thank the authors for addressing my comments.
- I believe that additional in vivo experiments, or the inclusion of controls for the specificity of the inhibitor, which the authors argue are beyond the scope of the current study, are essential to address the weaknesses and limitations stated in my current evaluation.
We respectfully acknowledge the reviewer's concern but would like to reiterate that demonstrating the specificity of the inhibitor is beyond the scope of this study. Alpelisib (BYL-719) is a clinically approved drug widely recognized as a specific inhibitor of p110α, primarily used in the treatment of breast cancer. Its selectivity for the p110α isoform has been extensively validated in the literature.
In our study, we used Alpelisib to assess whether pharmacological inhibition of p110α would produce effects similar to those observed in our genetic model, which is particularly relevant for the potential translational implications of our findings. Given the well-documented specificity of this inhibitor, we believe that additional controls to confirm its selectivity are unnecessary within the context of this study. Instead, our focus has been to investigate the functional role of p110α activity in macrophage-driven inflammation using the models described.
We appreciate the reviewer’s insight and hope this clarification addresses their concern.
- While the neutrophil depletion suggests neutrophils are not required for the phenotype, there are multiple other myeloid cells, in addition to macrophages, that could be contributing or accounting for the in vivo phenotype observed in the mutant strain (not macrophage specific).
We appreciate the reviewer's observation regarding the potential involvement of other myeloid cells. However, it is important to highlight that the inflammatory process follows a well-characterized sequential pattern. Our data clearly demonstrate that in the paw inflammation model:
· Neutrophils are effectively recruited, as evidenced by the inflammatory abscess filled with polymorphonuclear cells.
· However, macrophages fail to be recruited in the RBD model.
Given that this critical step is disrupted, it is reasonable to expect that any subsequent steps in the inflammatory cascade would also be affected. A precise dissection of the role of other myeloid populations would require additional lineage-specific models to selectively target each subset, which, as we have previously stated, would be the focus of an independent study.
While we cannot entirely exclude the contribution of other myeloid cells, our data strongly support the conclusion that macrophages are, at the very least, a key component of the observed phenotype. We explicitly address this point in the Discussion section, where we acknowledge the potential involvement of other myeloid populations.
- Inclusion of absolute cell numbers (in addition to the %) is essential. I do not understand why the authors are not including these data. Have they not counted the cells?
We appreciate the reviewer’s concern regarding the inclusion of absolute cell numbers. However, as stated in the Materials and Methods section, we analyzed 50,000 cells per sample, and the percentages reported in the manuscript are directly derived from this standardized count.
Our decision to present the data as percentages follows standard practices in flow cytometry-based analyses, as it allows for a clearer and more biologically relevant comparison of relative changes between conditions. This approach ensures consistency across samples and facilitates the interpretation of population dynamics during inflammation.
We would also like to clarify that all data are based on actual counts, and rigorous controls were implemented throughout the study to ensure accuracy and reproducibility. We hope this explanation addresses the reviewer’s concern and provides further clarity on our approach.
- Lastly, inclusion of representatives staining and gating strategies for all immune profiling measurements carried out by flow cytometry is important. This point has not been addressed, not even in writing.
We appreciate the reviewer’s concern regarding the inclusion of absolute cell numbers. However, as stated in the Materials and Methods section, we analyzed 50,000 cells per sample, and the percentages reported in the manuscript are directly derived from this standardized count.
Our decision to present the data as percentages follows standard practices in flow cytometry-based analyses, as it allows for a clearer and more biologically relevant comparison of relative changes between conditions. This approach ensures consistency across samples and facilitates the interpretation of population dynamics during inflammation.
We would also like to clarify that all data are based on actual counts, and rigorous controls were implemented throughout the study to ensure accuracy and reproducibility. We hope this explanation addresses the reviewer’s concern and provides further clarity on our approach.
The following is the authors’ response to the original reviews.
Public Reviews:
Reviewer #1 (Public review):
This study by Alejandro Rosell et al. reveals the immunoregulatory role of the RAS-p110α pathway in macrophages, specifically in regulating monocyte extravasation and lysosomal digestion during inflammation. Disrupting this pathway, through genetic tools or pharmacological intervention in mice, impairs the inflammatory response, leading to delayed resolution and more severe acute inflammation. The authors suggest that activating p110α with small molecules could be a potential therapeutic strategy for treating chronic inflammation. These findings provide important insights into the mechanisms by which p110α regulates macrophage function and the overall inflammatory response.
The updates made by the authors in the revised version have addressed the main points raised in the initial review, further improving the strength of their findings.
Reviewer #2 (Public review):
Summary:
Cell intrinsic signaling pathways controlling the function of macrophages in inflammatory processes, including in response to infection, injury or in the resolution of inflammation are incompletely understood. In this study, Rosell et al. investigate the contribution of RAS-p110α signaling to macrophage activity. p110α is a ubiquitously expressed catalytic subunit of PI3K with previously described roles in multiple biological processes including in epithelial cell growth and survival, and carcinogenesis. While previous studies have already suggested a role for RAS-p110α signaling in macrophage function, the cell intrinsic impact of disrupting the interaction between RAS and p110α in this central myeloid cell subset is not known.
Strengths:
Exploiting a sound previously described genetically engineered mouse model that allows tamoxifen-inducible disruption of the RAS-p110α pathway and using different readouts of macrophage activity in vitro and in vivo, the authors provide data consistent with their conclusion that alteration in RAS-p110α signaling impairs various but selective aspects of macrophage function in a cell-intrinsic manner.
Weaknesses:
My main concern is that for various readouts, the difference between wild-type and mutant macrophages in vitro or between wild-type and Pik3caRBD mice in vivo is modest, even if statistically significant. To further substantiate the extent of macrophage function alteration upon disruption of RAS-p110α signaling and its impact on the initiation and resolution of inflammatory responses, the manuscript would benefit from a more extensive assessment of macrophage activity and inflammatory responses in vivo.
Thank you for raising this point. We understand the reviewer’s concern regarding the modest yet statistically significant differences observed between wild-type and mutant macrophages in vitro, as well as between wild-type and Pik3caRBD mice in vivo. Our current study aimed to provide a foundational exploration of the role of RAS-p110α signaling in macrophage function and inflammatory response, focusing on a set of core readouts that demonstrate the physiological relevance of this pathway. While a more extensive in vivo assessment could offer additional insights into macrophage activity and the nuanced effects of RAS-p110α disruption, it would require an array of new experiments that are beyond the current scope.
However, we believe that the current data provide significant insights into the pathway’s role, highlighting important alterations in macrophage function and inflammatory processes due to RAS-p110α disruption. These findings lay the groundwork for future studies that can build upon our results with a more comprehensive analysis of macrophage activity in various inflammatory contexts.
In the in vivo model, all cells have disrupted RAS-p100α signaling, not only macrophages. Given that other myeloid cells besides macrophages contribute to the orchestration of inflammatory responses, it remains unclear whether the phenotype described in vivo results from impaired RAS-p100α signaling within macrophages or from defects in other haematopoietic cells such as neutrophils, dendritic cells, etc.
Thank you for raising this point. To address this, we have added a paragraph in the Discussion section acknowledging that RAS-p110α signaling disruption affects all hematopoietic cells (lines 461-470 in the discussion). However, we also provide several lines of evidence that support macrophages as the primary cell type involved in the observed phenotype. Specifically, we note that neutrophil depletion in chimera mice did not alter transendothelial extravasation, and that macrophages were the primary cell type showing significant functional defects in the paw edema model. These findings, combined with specific deficiencies in myeloid populations, suggest a predominant role of macrophages in the impaired inflammatory response, though we acknowledge the potential contributions of other myeloid cells.
Inclusion of information on the absolute number of macrophages, and total immune cells (e.g. for the spleen analysis) would help determine if the reduced frequency of macrophages represents an actual difference in their total number or rather reflects a relative decrease due to an increase in the number of other/s immune cell/s.
Thank you for this suggestion. We understand the value of presenting actual measurements; however, we opted to display normalized data to provide a clearer comparison between WT and RBD mice, as this approach highlights the relative differences in immune populations between the two groups. Normalizing data helps to focus on the specific impact of the RAS-p110α disruption by minimizing inter-sample variability that can obscure these differences.
To further address the reviewer’s concern regarding the interpretation of macrophage frequencies, we have included a pie chart that represents the relative proportions of the various immune cell populations studied within our dataset. Author response image 1 provides a visual overview of the immune cell distribution, enabling a clearer understanding of whether the observed decrease in macrophage frequency represents an actual reduction in total macrophage numbers or a shift in their relative abundance due to changes in other immune populations.
We hope this approach satisfactorily addresses reviewer’s concerns by providing both a normalized dataset for clearer interpretation of genotype-specific effects and an overall immune profile that contextualizes macrophage frequency within the broader immune cell landscape.
Author response image 1.
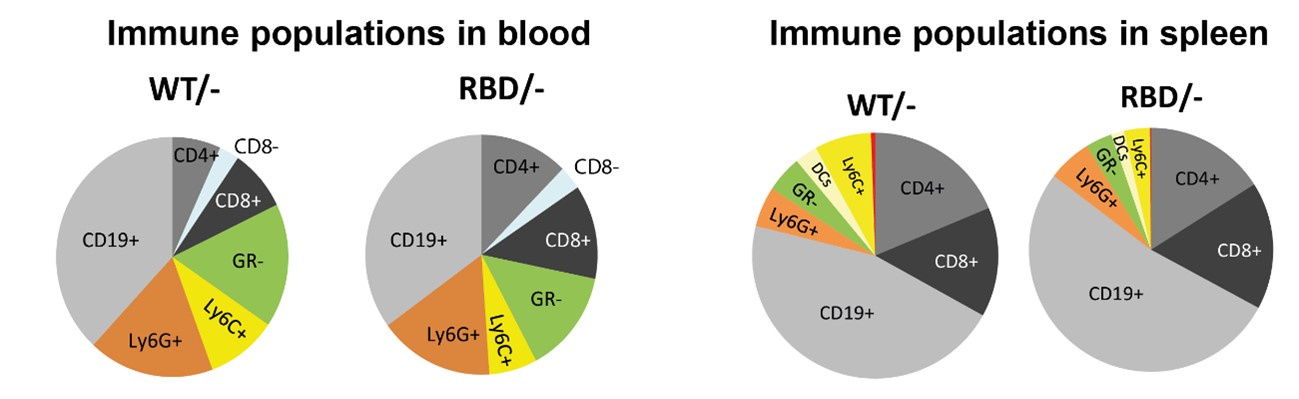
Recommendations for the authors:
Reviewer #2 (Recommendations for the authors):
As proof of concept data that activation of RAS-p110α signaling constitutes indeed a putative approach for treating chronic inflammation is not included in the manuscript, I suggest removing this implication from the abstract.
Thank you for this suggestion. We have now removed this implication from the abstract to maintain clarity and to better reflect the scope of the data presented in the manuscript.
Inclusion of a control in which RBD/- cells are also treated with BYL719, across experiments in which the inhibitor is used, would be important to determine, among other things, the specificity of the inhibitor.
We appreciate the reviewer’s suggestion to include RBD/- cells treated with BYL719 as an additional control. However, we would like to clarify that this approach would raise a different biological question, as treating RBD mice with BYL719 would not only address the specificity of the inhibitor but also examine the combined effects of genetic and pharmacologic disruptions on PI3K pathway signaling. Investigating this dual disruption falls outside the scope of our current study, which is focused specifically on the effects of RAS-p110α disruption.
It is also important to note that our RBD mouse model selectively disrupts RAS-mediated activation of p110α, while PI3K activation can still occur through other pathways, such as receptor tyrosine kinases (RTKs) and G protein-coupled receptors (GPCRs). Thus, inhibiting p110α with BYL719 would produce broader effects beyond the inhibition of RAS-PI3K signaling, impacting PI3K activation regardless of its upstream source.
In addition, incorporating this control would require us to repeat nearly all experiments in the manuscript, as it would necessitate generating and analyzing new samples for each experimental condition. Given the scope and resources involved, we believe this approach is unfeasible at this stage of the revision process.
We hope this explanation is satisfactory and that the current data in the manuscript provide a rigorous assessment of the RAS-p110α signaling pathway within the defined experimental scope.
Figure 3I is missing the statistical analysis (this is mentioned in the legend though).
Thank you for pointing this out. We apologize for the oversight. The statistical analysis for Figure 3I has now been added.
Gating strategies and representative staining should be included more generally across the manuscript.
Thank you for this suggestion. To address this, we have added a new supplementary figure (Figure 2-Supplement Figure 2) that illustrates the gating strategy along with a representative dataset. Additionally, a brief summary of the gating strategy has been included in the main text to further clarify the methodology.
It is recommended that authors show actual measurements rather than only data normalized to the control (or arbitrary units).
Thank you for this suggestion. We understand the value of presenting actual measurements; however, we opted to display normalized data to provide a clearer comparison between WT and RBD mice, as this approach highlights the relative differences in immune populations between the two groups. Normalizing data helps to focus on the specific impact of the RAS-p110α disruption by minimizing inter-sample variability that can obscure these differences.
To further address the reviewer’s concern regarding the interpretation of macrophage frequencies, we have included a pie chart that represents the relative proportions of the various immune cell populations studied within our dataset. Author response image 1 provides a visual overview of the immune cell distribution, enabling a clearer understanding of whether the observed decrease in macrophage frequency represents an actual reduction in total macrophage numbers or a shift in their relative abundance due to changes in other immune populations.
We hope this approach satisfactorily addresses reviewer’s concerns by providing both a normalized dataset for clearer interpretation of genotype-specific effects and an overall immune profile that contextualizes macrophage frequency within the broader immune cell landscape.
-
-

-
-
eLife Assessment
This useful study investigates the impact of disrupting the interaction of RAS with the PI3K subunit p110α in macrophage function in vitro and inflammatory responses in vivo. Solid data overall supports a role for RAS-p110α signalling in regulating macrophage activity and so inflammation, however for many of the readouts presented the magnitude of the phenotype is not particularly pronounced. Further analysis would be required to substantiate the claims that RAS-p110α signalling plays a key role in macrophage function. Of note, the molecular mechanisms of how exactly p110α regulates the functions in macrophages have not yet been established.
-
Reviewer #1 (Public review):
This study by Alejandro Rosell et al. reveals the immunoregulatory role of the RAS-p110α pathway in macrophages, specifically in regulating monocyte extravasation and lysosomal digestion during inflammation. Disrupting this pathway, through genetic tools or pharmacological intervention in mice, impairs the inflammatory response, leading to delayed resolution and more severe acute inflammation. The authors suggest that activating p110α with small molecules could be a potential therapeutic strategy for treating chronic inflammation. These findings provide important insights into the mechanisms by which p110α regulates macrophage function and the overall inflammatory response.
The updates made by the authors in the revised version have addressed the main points raised in the initial review, further improving …
Reviewer #1 (Public review):
This study by Alejandro Rosell et al. reveals the immunoregulatory role of the RAS-p110α pathway in macrophages, specifically in regulating monocyte extravasation and lysosomal digestion during inflammation. Disrupting this pathway, through genetic tools or pharmacological intervention in mice, impairs the inflammatory response, leading to delayed resolution and more severe acute inflammation. The authors suggest that activating p110α with small molecules could be a potential therapeutic strategy for treating chronic inflammation. These findings provide important insights into the mechanisms by which p110α regulates macrophage function and the overall inflammatory response.
The updates made by the authors in the revised version have addressed the main points raised in the initial review, further improving the strength of their findings.
-
Reviewer #2 (Public review):
Summary:
Cell intrinsic signaling pathways controlling the function of macrophages in inflammatory processes, including in response to infection, injury or in the resolution of inflammation are incompletely understood. In this study, Rosell et al. investigate the contribution of RAS-p110α signaling to macrophage activity. p110α is a ubiquitously expressed catalytic subunit of PI3K with previously described roles in multiple biological processes including in epithelial cell growth and survival, and carcinogenesis. While previous studies have already suggested a role for RAS-p110α signaling in macrophage function, the cell intrinsic impact of disrupting the interaction between RAS and p110α in this central myeloid cell subset is not known.
Strengths:
Exploiting a sound previously described genetically …
Reviewer #2 (Public review):
Summary:
Cell intrinsic signaling pathways controlling the function of macrophages in inflammatory processes, including in response to infection, injury or in the resolution of inflammation are incompletely understood. In this study, Rosell et al. investigate the contribution of RAS-p110α signaling to macrophage activity. p110α is a ubiquitously expressed catalytic subunit of PI3K with previously described roles in multiple biological processes including in epithelial cell growth and survival, and carcinogenesis. While previous studies have already suggested a role for RAS-p110α signaling in macrophage function, the cell intrinsic impact of disrupting the interaction between RAS and p110α in this central myeloid cell subset is not known.
Strengths:
Exploiting a sound previously described genetically engineered mouse model that allows tamoxifen-inducible disruption of the RAS-p110α pathway and using different readouts of macrophage activity in vitro and in vivo, the authors provide data consistent with their conclusion that alteration in RAS-p110α signaling impairs various but selective aspects of macrophage function in a cell-intrinsic manner.
Weaknesses:
My main concern is that for various readouts, the difference between wild-type and mutant macrophages in vitro or between wild-type and Pik3caRBD mice in vivo is modest, even if statistically significant. To further substantiate the extent of macrophage function alteration upon disruption of RAS-p110α signaling and its impact on the initiation and resolution of inflammatory responses, the manuscript would benefit from a more extensive assessment of macrophage activity and inflammatory responses in vivo.
In the in vivo model, all cells have disrupted RAS-p100α signaling, not only macrophages. Given that other myeloid cells besides macrophages contribute to the orchestration of inflammatory responses, it remains unclear whether the phenotype described in vivo results from impaired RAS-p100α signaling within macrophages or from defects in other haematopoietic cells such as neutrophils, dendritic cells, etc.
Inclusion of information on the absolute number of macrophages, and total immune cells (e.g. for the spleen analysis) would help determine if the reduced frequency of macrophages represents an actual difference in their total number or rather reflects a relative decrease due to an increase in the number of other/s immune cell/s.
-
Author response:
The following is the authors’ response to the original reviews.
Public Reviews:
Reviewer #1 (Public Review):
In this study, Alejandro Rosell et al. uncovers the immunoregulation functions of RAS-p110α pathway in macrophages, including the extravasation of monocytes from the bloodstream and subsequent lysosomal digestion. Disrupting RAS-p110α pathway by mouse genetic tools or by pharmacological intervention, hampers the inflammatory response, leading to delayed resolution and more severe acute inflammatory reactions. The authors proposed that activating p110α using small molecules could be a promising approach for treating chronic inflammation. This study provides insights into the roles and mechanisms of p110α on macrophage function and the inflammatory response, while some conclusions are still questionable because of …
Author response:
The following is the authors’ response to the original reviews.
Public Reviews:
Reviewer #1 (Public Review):
In this study, Alejandro Rosell et al. uncovers the immunoregulation functions of RAS-p110α pathway in macrophages, including the extravasation of monocytes from the bloodstream and subsequent lysosomal digestion. Disrupting RAS-p110α pathway by mouse genetic tools or by pharmacological intervention, hampers the inflammatory response, leading to delayed resolution and more severe acute inflammatory reactions. The authors proposed that activating p110α using small molecules could be a promising approach for treating chronic inflammation. This study provides insights into the roles and mechanisms of p110α on macrophage function and the inflammatory response, while some conclusions are still questionable because of several issues described below.
(1) Fig. 1B showed that disruption of RAS-p110α causes the decrease in the activation of NF-κB, which is a crucial transcription factor that regulates the expression of proinflammatory genes. However, the authors observed that disruption of RAS-p110α interaction results in an exacerbated inflammatory state in vivo, in both localized paw inflammation and systemic inflammatory mediator levels. Also, the authors introduced that "this disruption leads to a change in macrophage polarization, favoring a more proinflammatory M1 state" in introduction according to reference 12. The conclusions drew from the signaling and the models seemed contradictory and puzzling. Besides, it is not clear why the protein level of p65 was decreased at 10' and 30'. Was it attributed to the degradation of p65 or experimental variation?
We thank the reviewer for this insightful comment and apologize for not previously explaining the implications of the observed decrease in NF-κB activation. We found a decrease in NF-κB activation in response to LPS + IFN-γ stimulation in macrophages lacking RAS-PI3K interaction. As the reviewer pointed out, NF-κB is a key transcription factor that regulates the expression of various proinflammatory genes. To better characterize whether the decrease in p-p65 would lead to a reduction in the expression of specific cytokines, we performed a cytokine array using unstimulated and LPS + IFN-γ stimulated macrophages. The results indicated a small number of cytokines with altered expression, validating that RAS-p110α activation of p-p65 regulates the expression of some inflammatory cytokines. These results have been added to the manuscript and to Figure 1 (panels C and D). In brief, the data suggest an impairment in recruitment factors and inflammatory regulators following the disruption of RAS-p110α signaling in macrophages, which aligns with the observed in vivo phenotype.
Our findings indicate that the disruption of RAS-p110α signaling has a complex and multifaceted role in BMDMs. Specifically, monocytes lacking RAS-PI3K are unable to reach the inflamed area due to an impaired ability to extravasate, caused by altered actin cytoskeleton dynamics. Consequently, inflammation is sustained over time, continuously releasing inflammatory mediators. Moreover, we have shown that macrophages deficient in RAS-p110α interaction fail to mount a full inflammatory response due to decreased activation of p-p65, leading to reduced production of a set of inflammatory regulators. Additionally, these macrophages are unable to effectively process phagocytosed material and activate the resolutive phase of inflammation. As a result of these defects, an exacerbated and sustained inflammatory response occurs.
Our in vivo data, showing an increase in systemic inflammatory mediators, might be a consequence of the accumulation of monocytes produced by bone marrow progenitors in response to sensed inflammatory stimuli, but unable to extravasate.
Regarding the sentence in the introduction: "this disruption leads to a change in macrophage polarization, favoring a more proinflammatory M1 state" (reference 12), this was observed in an oncogenic context, which might differ from the role of RAS-p110α in a non-oncogenic situation, as analyzed in this work. We introduced these results as an example to establish the role of RAS-p110α in macrophages, demonstrating its participation in macrophage-dependent responses. Together with our study, these findings clearly indicate that p110α signaling is critical when analyzing full immune responses. Previously, little was known about the role of this PI3K isoform in immune responses. Our data, along with those presented by Murillo et al. (ref. 12), demonstrate that p110α plays a significant role in macrophage function in both oncogenic and inflammatory contexts. Additionally, our results suggest that this role is complex and multifaceted, warranting further investigation to fully understand the complexity of p110α signaling in macrophages.
Regarding decreased levels of p65 at 10’ and 30’ in RBD cells we are still uncertain about the possible molecular mechanism leading to the observed decrease. No changes in p65 mRNA levels were observed after 30 minutes of LPS+IFNγ treatment as shown in Author response image 1.
Author response image 1.
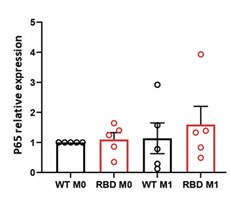
Preliminary data not shown here suggest that treating macrophages with BYL exhibits a similar effect, indicating a potential pathway for investigation. Considering that the decrease in protein levels is not due to lower mRNA expression, we may infer that post-translational mechanisms are leading to early protein degradation in RAS-p110α deficient macrophages. This could explain the observed decrease in protein activation. However, the specific molecular mechanism responsible for this degradation remains unclear, and further research is necessary to elucidate it.
(2) In Fig 3, the authors used bone-marrow derived macrophages (BMDMs) instead of isolated monocytes to evaluate the ability of monocyte transendothelial migration, which is not sufficiently convincing. In Fig. 3B, the authors evaluated the migration in Pik3caWT/- BMDMs, and Pik3caWT/WT BMDMs treated with BYL-719'. Given that the dose effect of gene expression, the best control is Pik3caWT/- BMDMs treated with BYL-719.
We thank reviewer for this comment. While we agree that using BMDMs might not be the most conventional approach for studying monocyte migration, there were several reasons why we still considered them a valid method. While isolated monocytes are the initial cell type involved in transendothelial migration, bone marrow-derived macrophages (BMDMs) provide a relevant and practical model for studying this process. BMDMs are differentiated from the same bone marrow precursors as monocytes and retain the ability to respond to chemotactic signals, adhere to endothelial cells, and migrate through the endothelium. This makes them a suitable tool for examining the cellular and molecular mechanisms underlying monocyte migration and subsequent macrophage infiltration into tissues. Additionally, BMDMs offer experimental consistency and are easier to manipulate in vitro, enabling more controlled and reproducible studies.
In response to the comment regarding Fig. 3B, we appreciate the suggestion to use Pik3ca WT/- BMDMs treated with BYL-719 as a control. However, our rationale for using Pik3ca WT/WT BMDMs treated with BYL-719 was based on a conceptual approach rather than a purely experimental control. The BYL-719 treatment in Pik3ca WT/WT cells was intended to simulate the inhibition of p110α in a fully functional, wild-type context. This allows us to directly assess the impact of p110α inhibition under normal physiological conditions, which is more representative of what would occur in an organism where the full dose of Pik3ca is present. Using Pik3ca WT/- BMDMs treated with BYL-719 as a control may not accurately reflect the in vivo scenario, where any therapeutic intervention would likely occur in the context of a fully functional, wild-type background. Our approach aims to provide a clearer understanding of how p110α inhibition affects cell functionality in a wild-type setting, which is relevant for potential therapeutic applications. Therefore, we considered the use of Pik3ca WT/WT BMDMs with BYL-719 treatment to be a more appropriate control for testing the effects of p110α inhibition in normal conditions.
(3) In Fig. 4E-4G, the authors observed that elevated levels of serine 3 phosphorylated Cofilin in Pik3caRBD/- BMDMs both in unstimulated and in proinflammatory conditions, and phosphorylation of Cofilin at Ser3 increase actin stabilization, it is not clear why disruption of RAS-p110α binding caused a decrease in the F-actin pool in unstimulated BMDMs?
We thank the reviewer for this insightful comment. During the review process, we have carefully quantified all the Western blots conducted. While we did observe an increase in phospho-Cofilin (Ser3) levels in RBD BMDMs, this increase did not reach statistical significance. As a result, we cannot confidently attribute the observed increase in F-actin to this proposed mechanism. We apologize for any confusion this may have caused. Consequently, we have removed these data from Figure 4G and the associated discussion.
Unfortunately, we have not yet identified the underlying mechanism responsible for this phenotype. Future experiments will focus on exploring potential alterations in other actin-nucleating, regulating, and stabilizing proteins that could account for the observed changes in F-actin levels.
Reviewer #2 (Public Review):
Summary:
Cell intrinsic signaling pathways controlling the function of macrophages in inflammatory processes, including in response to infection, injury or in the resolution of inflammation are incompletely understood. In this study, Rosell et al. investigate the contribution of RAS-p110α signaling to macrophage activity. p110α is a ubiquitously expressed catalytic subunit of PI3K with previously described roles in multiple biological processes including in epithelial cell growth and survival, and carcinogenesis. While previous studies have already suggested a role for RAS-p110α signaling in macrophages function, the cell intrinsic impact of disrupting the interaction between RAS and p110α in this central myeloid cell subset is not known.
Strengths:
Exploiting a sound previously described genetically mouse model that allows tamoxifen-inducible disruption of the RAS-p110α pathway and using different readouts of macrophage activity in vitro and in vivo, the authors provide data consistent with their conclusion that alteration in RAS-p110α signaling impairs the function of macrophages in a cell intrinsic manner. The study is well designed, clearly written with overall high-quality figures.
Weaknesses:
My main concern is that for many of the readouts, the difference between wild-type and mutant macrophages in vitro or between wild-type and Pik3caRBD mice in vivo is rather modest, even if statistically significant (e.g. Figure 1A, 1C, 2A, 2F, 3B, 4B, 4C). In other cases, such as for the analysis of the H&E images (Figure 1D-E, S1E), the images are not quantified, and it is hard to appreciate what the phenotype in samples from Pik3caRBD mice is or whether this is consistently observed across different animals. Also, the authors claim there is a 'notable decrease' in Akt activation but 'no discernible chance' in ERK activation based on the western blot data presented in Figure 1A. I do not think the data shown supports this conclusion.
We appreciate the reviewer's careful examination of our data and their observation regarding the modest differences between wild-type and mutant macrophages in vitro, as well as between wild-type and Pik3caRBD mice in vivo. While the differences observed in Figures 1A, 1C, 2A, 2F, 3B, 4B, and 4C are statistically significant but modest, our data demonstrate that they are biologically relevant and should be interpreted within the specific nature of our model. Our study focuses on the disruption of the RASp110α interaction, but it should be noted that alternative pathways for p110α activation, independent of RAS, remain functional in this model. Additionally, the model retains the expression of other p110 isoforms, such as p110β, p110γ, and p110δ, which are known to have significant roles in immune responses. Given the overlapping functions of these p110 isoforms, and the fact that our model involves a subtle modification that specifically affects the RAS-p110α interaction without completely abrogating p110α activity, it is understandable that only modest effects are observed in some readouts. The redundancy and compensation by other p110 isoforms likely mitigate the impact of disrupting RAS-mediated p110α activation.
However, despite these modest in vitro differences, it is crucial to highlight that the in vivo effects on inflammation are both clear and consistent. The persistence of inflammation in our model suggests that the RAS-p110α interaction plays a specific, non-redundant role in resolving inflammation, which cannot be fully compensated by other signaling pathways or p110 isoforms. These findings underscore the importance of RAS-p110α signaling in immune homeostasis and suggest that even subtle disruptions in this pathway can lead to significant physiological consequences over time, particularly in the context of inflammation. The modest differences observed may represent early or subtle alterations that could lead to more pronounced phenotypes under specific stress or stimulation conditions. This could be tested across all the figures mentioned. For instance, in Fig. 1A, the Western blot for AKT has been quantified, demonstrating a significant decrease in AKT levels; in Fig. 1C, although the difference in paw inflammation was only a few millimeters in thickness, considering the size of a mouse paw, those millimeters were very noticeable by eye. Furthermore, pathological examination of the tissue consistently showed an increase in inflammation in RBD mice. Furthermore, the consistency of the observed differences across different readouts and experimental setups reinforces the reliability and robustness of our findings. Even modest changes that are consistently observed across different assays and conditions are indicative of genuine biological effects. The statistical significance of the differences indicates that they are unlikely to be due to random variation. This statistical rigor supports the conclusion that the observed effects, albeit modest, are real and warrant further exploration.
Regarding the analysis of H&E images, we have now quantified the changes with the assistance of the pathologist, Mª Carmen García Macías, who has been added to the author list. We removed the colored arrows from the images and instead quantified fibrin and chromatin remnants as markers of inflammation staging. Loose chromatin, which increases as a consequence of cell death, is higher in the early phases of inflammation and decreases as macrophages phagocytose cell debris to initiate tissue healing. Chromatin content was scored on a scale from 1 to 3, where 1 represents the lowest amount and 3 the highest. The scoring was based on the area within the acute inflammatory abscess where chromatin could be found: 3 for less than 30%, 2 for 30-60%, and 1 for over 60%. Graphs corresponding to this quantification have now been added to Figure 1 and an explanation of the scale has been added to Material and Methods.
To further substantiate the extent of macrophage function alteration upon disruption of RAS-p110α signaling, the manuscript would benefit from testing macrophage activity in vitro and in vivo across other key macrophage activities such as bacteria phagocytosis, cytokine/chemokine production in response to titrating amounts of different PAMPs, inflammasome function, etc. This would be generally important overall but also useful to determine whether the defects in monocyte motility or macrophage lysosomal function are selectively controlled downstream of RAS-p110α signaling.
We thank reviewer #2 for this comment. In order to better address the role of RAS-PI3K in macrophage function, we have performed some additional experiments, some of which have been added to the revised version of the manuscript.
(1) We have performed cytokine microarrays of RAS-p110α deficient macrophages unstimulated and stimulated with LPS+IFN-g. Results have been added to the manuscript and to Supplementary Figure S1E and S1F. In brief, the data obtained suggest an impairment in recruitment factors, as well as in inflammatory regulators after disruption of RAS-p110α signaling in macrophages, which align with the in vivo observed phenotype.
(2) We also conducted phagocytosis assays to analyze the ability of RAS-p110α deficient macrophages to phagocytose 1 µm Sepharose beads, Borrelia burgdorferi, and apoptotic cells. The data reveal varied behavior of RAS-p110α deficient bone marrow-derived macrophages (BMDMs) depending on the target:
• Engulfment of Non-biological Particles: RAS-p110α deficient macrophages showed a decreased ability to engulf 1 µm Sepharose beads. This suggests that RAS-p110α signaling is important for the effective phagocytosis of non-biological particles. These findings have now been added to the text and figures have been added to supplementary Fig. S4A
• Response to Bacterial Pathogens: When exposed to Borrelia burgdorferi, RAS-p110α deficient macrophages did not exhibit a change in bacterial uptake. This indicates that RAS-p110α may not play a critical role in the initial phagocytosis of this bacterial pathogen. The observed increase in the phagocytic index, although not statistically significant, might imply a compensatory mechanism or a more complex interaction that warrants further investigation. These findings have now been added to the text and figures have been added to supplementary Fig. S4B. These experiments were performed in collaboration with Dr. Anguita, from CICBioBune (Bilbao, Spain) and, as a consequence, he has been added as an author in the paper.
• Phagocytosis of Apoptotic Cells: There were no differences in the phagocytosis rate of apoptotic cells between RAS-p110α deficient and control macrophages at early time points. However, the accumulation of engulfed material at later time points suggests a possible delay in the processing and degradation of apoptotic cells in the absence of RAS-p110α signaling.
These findings highlight the complexity of RAS-p110α's involvement in phagocytic processes and suggest that its role may vary with different types of phagocytic targets.
Furthermore, given the key role of other myeloid cells besides macrophages in inflammation and immunity it remains unclear whether the phenotype observed in vivo can be attributed to impaired macrophage function. Is the function of neutrophils, dendritic cells or other key innate immune cells not affected?
Thank you for this insightful comment. We understand the key role of other myeloid cells in inflammation and immunity. However, our study specifically focuses on the role of macrophages. Our data show that disruption of RAS-PI3K leads to a clear defect in macrophage extravasation, and our in vitro data demonstrate issues in macrophage cytoskeleton and phagocytosis, aligning with the in vivo phenotype.
Experiments investigating the role of RAS-PI3K in neutrophils, dendritic cells, or other innate immune cells are beyond the scope of this study. Understanding these interactions would indeed require separate, comprehensive studies and the generation of new mouse models to disrupt RAS-PI3K exclusively in specific cell types.
Furthermore, during paw inflammation experiments, polymorphonuclear cells were present from the initial phases of the inflammatory response. What caught our attention was the prolonged presence of these cells. In conversation with our in-house pathologist, she mentioned the lack of macrophages to remove dead polymorphonuclear cells in our RAS-PI3K mutant mice. Specific staining for macrophages confirmed the absence of macrophages in the inflamed node of mutant mice.
We acknowledge that further research is necessary to elucidate the effects on other myeloid cells. However, our current findings provide clear evidence of a decrease in inflammatory monocytes and defective macrophage responses to inflammation, both in vivo and in vitro. We believe these results significantly contribute to understanding the role of RAS-PI3K in macrophage function during inflammation.
Compelling proof of concept data that targeting RAS-p110α signalling constitutes indeed a putative approach for modulation of chronic inflammation is lacking. Addressing this further would increase the conceptual advance of the manuscript and provide extra support to the authors' suggestion that p110α inhibition or activation constitute promising approaches to manage inflammation.
We thank Reviewer #2 for this insightful comment. In our manuscript, we have demonstrated through multiple experiments that the inhibition of p110α, either by disrupting RAS-p110α signaling or through the use of Alpelisib (BYL-719), has a modulatory effect on inflammatory responses. However, we acknowledge that we have not activated the pathway due to the unavailability of a suitable p110α activator until the concluding phase of our study.
We recognize the importance of this point and are eager about investigating both the inhibition and activation of p110α as potential approaches to managing inflammation in well-established inflammatory disease models. We believe that such comprehensive studies would significantly enhance the conceptual advance and translational relevance of our findings.
However, it is essential to note that the primary aim of our current work was to demonstrate the role of RAS-p110α in the inflammatory responses of macrophages. We have successfully shown that RASp110α influences macrophage behavior and inflammatory signaling. Expanding the scope to include disease models and pathway activation studies would be an extensive project that goes beyond the current objectives of this manuscript. While our present study establishes the foundational role of RASp110α in macrophage-mediated inflammatory responses, we agree that further investigation into both p110α inhibition and activation in disease models is crucial. We are keen to pursue this line of research in future studies, which we believe will provide robust evidence supporting the therapeutic potential of targeting RAS-p110α signaling in chronic inflammation.
Finally, the analysis by FACS should also include information about the total number of cells, not just the percentage, which is affected by the relative change in other populations. On this point, Figure S2B shows a substantial, albeit not significant (with less number of mice analysed), increase in the percentage of CD3+ cells. Is there an increase in the absolute number of T cells or does this apparent relative increase reflect a reduction in myeloid cells?
We thank the reviewer for this comment, which we have addressed in the revised version of the manuscript. Regarding the total number of cells analyzed, we have added to the Materials and Methods section that in all our studies, a total of 50,000 cells were analyzed (line 749). The percentages of cells are related to these 50,000 events. Additionally, we have increased the number of mice analyzed by including new mice for CD3+ cell analysis. Despite this, the results remain not significant.
Recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
(1) It is recommended to provide a graphical abstract to summarize the multiple functions of RAS-p110α pathway in monocyte/macrophages that the authors proposed
We thank reviewer for this useful recommendation. A graphical abstract has now been added to the study.
(2) Western blots in this paper need quantification and a measure of reproducibility
We have now added a graph with the quantification of the western blots performed in this work as a measure of reproducibility.
(3) Representative flow data and gating strategy should be included
We have now added the description of the gating strategy followed to material and methods section.
-
-
-
-
eLife assessment
This useful study investigates the impact of disrupting the interaction of RAS with the PI3K subunit p110α in macrophage function in vitro and inflammatory responses in vivo. Solid data overall supports a role for RAS-p110α signalling in regulating macrophage activity and so inflammation, however for many of the readouts presented the magnitude of the phenotype is not particularly pronounced. Further analysis would be required to substantiate the claims that RAS-p110α signalling plays a key role in macrophage function. Of note, the molecular mechanisms of how exactly p110α regulating the functions in macrophage have not yet been established.
-
Reviewer #1 (Public Review):
In this study, Alejandro Rosell et al. uncovers the immunoregulation functions of RAS-p110α pathway in macrophages, including the extravasation of monocytes from the bloodstream and subsequent lysosomal digestion. Disrupting RAS-p110α pathway by mouse genetic tools or by pharmacological intervention, hampers the inflammatory response, leading to delayed resolution and more severe acute inflammatory reactions. The authors proposed that activating p110α using small molecules could be a promising approach for treating chronic inflammation. This study provides insights into the roles and mechanisms of p110α on macrophage function and the inflammatory response, while some conclusions are still questionable because of several issues described below.
(1) Fig. 1B showed that disruption of RAS-p110α causes the …
Reviewer #1 (Public Review):
In this study, Alejandro Rosell et al. uncovers the immunoregulation functions of RAS-p110α pathway in macrophages, including the extravasation of monocytes from the bloodstream and subsequent lysosomal digestion. Disrupting RAS-p110α pathway by mouse genetic tools or by pharmacological intervention, hampers the inflammatory response, leading to delayed resolution and more severe acute inflammatory reactions. The authors proposed that activating p110α using small molecules could be a promising approach for treating chronic inflammation. This study provides insights into the roles and mechanisms of p110α on macrophage function and the inflammatory response, while some conclusions are still questionable because of several issues described below.
(1) Fig. 1B showed that disruption of RAS-p110α causes the decrease in the activation of NF-κB, which is a crucial transcription factor that regulates the expression of proinflammatory genes. However, the authors observed that disruption of RAS-p110α interaction results in an exacerbated inflammatory state in vivo, in both localized paw inflammation and systemic inflammatory mediator levels. Also, the authors introduced that "this disruption leads to a change in macrophage polarization, favouring a more proinflammatory M1 state" in introduction according to reference 12. The conclusions drew from the signaling and the models seemed contradictory and puzzling. Besides, it is not clear why the protein level of p65 was decreased at 10' and 30'. Was it attributed to the degradation of p65 or experimental variation?
(2) In Fig 3, the authors used bone-marrow derived macrophages (BMDMs) instead of isolated monocytes to evaluate the ability of monocyte transendothelial migration, which is not sufficiently convincing. In Fig. 3B, the authors evaluated the migration in Pik3caWT/- BMDMs, and Pik3caWT/WT BMDMs treated with BYL-719'. Given that the dose effect of gene expression, the best control is Pik3caWT/- BMDMs treated with BYL-719.
(3) In Fig. 4E-4G, the authors observed that elevated levels of serine 3 phosphorylated Cofilin in Pik3caRBD/- BMDMs both in unstimulated and in proinflammatory conditions, and phosphorylation of Cofilin at Ser3 increase actin stabilization, it is not clear why disruption of RAS-p110α binding caused a decrease in the F-actin pool in unstimulated BMDMs?
-
Reviewer #2 (Public Review):
Summary:
Cell intrinsic signaling pathways controlling the function of macrophages in inflammatory processes, including in response to infection, injury or in the resolution of inflammation are incompletely understood. In this study, Rosell et al. investigate the contribution of RAS-p110α signaling to macrophage activity. p110α is a ubiquitously expressed catalytic subunit of PI3K with previously described roles in multiple biological processes including in epithelial cell growth and survival, and carcinogenesis. While previous studies have already suggested a role for RAS-p110α signaling in macrophages function, the cell intrinsic impact of disrupting the interaction between RAS and p110α in this central myeloid cell subset is not known.
Strengths:
Exploiting a sound previously described genetically mouse …
Reviewer #2 (Public Review):
Summary:
Cell intrinsic signaling pathways controlling the function of macrophages in inflammatory processes, including in response to infection, injury or in the resolution of inflammation are incompletely understood. In this study, Rosell et al. investigate the contribution of RAS-p110α signaling to macrophage activity. p110α is a ubiquitously expressed catalytic subunit of PI3K with previously described roles in multiple biological processes including in epithelial cell growth and survival, and carcinogenesis. While previous studies have already suggested a role for RAS-p110α signaling in macrophages function, the cell intrinsic impact of disrupting the interaction between RAS and p110α in this central myeloid cell subset is not known.
Strengths:
Exploiting a sound previously described genetically mouse model that allows tamoxifen-inducible disruption of the RAS-p110α pathway and using different readouts of macrophage activity in vitro and in vivo, the authors provide data consistent with their conclusion that alteration in RAS-p110α signaling impairs the function of macrophages in a cell intrinsic manner. The study is well designed, clearly written with overall high-quality figures.
Weaknesses:
My main concern is that for many of the readouts, the difference between wild-type and mutant macrophages in vitro or between wild-type and Pik3caRBD mice in vivo is rather modest, even if statistically significant (e.g. Figure 1A, 1C, 2A, 2F, 3B, 4B, 4C). In other cases, such as for the analysis of the H&E images (Figure 1D-E, S1E), the images are not quantified, and it is hard to appreciate what the phenotype in samples from Pik3caRBD mice is or whether this is consistently observed across different animals. Also, the authors claim there is a 'notable decrease' in Akt activation but 'no discernible chance' in ERK activation based on the western blot data presented in Figure 1A. I do not think the data shown supports this conclusion.
To further substantiate the extent of macrophage function alteration upon disruption of RAS-p110α signaling, the manuscript would benefit from testing macrophage activity in vitro and in vivo across other key macrophage activities such as bacteria phagocytosis, cytokine/chemokine production in response to titrating amounts of different PAMPs, inflammasome function, etc. This would be generally important overall but also useful to determine whether the defects in monocyte motility or macrophage lysosomal function are selectively controlled downstream of RAS-p110α signaling.
Furthermore, given the key role of other myeloid cells besides macrophages in inflammation and immunity it remains unclear whether the phenotype observed in vivo can be attributed to impaired macrophage function. Is the function of neutrophils, dendritic cells or other key innate immune cells not affected?
Compelling proof of concept data that targeting RAS-p110α signalling constitutes indeed a putative approach for modulation of chronic inflammation is lacking. Addressing this further would increase the conceptual advance of the manuscript and provide extra support to the authors' suggestion that p110α inhibition or activation constitute promising approaches to manage inflammation.
Finally, the analysis by FACS should also include information about the total number of cells, not just the percentage, which is affected by the relative change in other populations. On this point, Figure S2B shows a substantial, albeit not significant (with less number of mice analysed), increase in the percentage of CD3+ cells. Is there an increase in the absolute number of T cells or does this apparent relative increase reflect a reduction in myeloid cells?
-
-