An antimicrobial drug recommender system using MALDI-TOF MS and dual-branch neural networks
Curation statements for this article:-
Curated by eLife

eLife Assessment
This valuable study presents a machine learning model to recommend effective antimicrobial drugs from patients' samples analysed with mass spectrometry. The evidence supporting the claims of the authors is convincing. This work will be of interest to computational biologists, microbiologists, and clinicians.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Timely and effective use of antimicrobial drugs can improve patient outcomes, as well as help safeguard against resistance development. Matrix-assisted laser desorption/ionization time-of-flight mass spectrometry (MALDI-TOF MS) is currently routinely used in clinical diagnostics for rapid species identification. Mining additional data from said spectra in the form of antimicrobial resistance (AMR) profiles is, therefore, highly promising. Such AMR profiles could serve as a drop-in solution for drastically improving treatment efficiency, effectiveness, and costs. This study endeavors to develop the first machine learning models capable of predicting AMR profiles for the whole repertoire of species and drugs encountered in clinical microbiology. The resulting models can be interpreted as drug recommender systems for infectious diseases. We find that our dual-branch method delivers considerably higher performance compared to previous approaches. In addition, experiments show that the models can be efficiently fine-tuned to data from other clinical laboratories. MALDI-TOF-based AMR recommender systems can, hence, greatly extend the value of MALDI-TOF MS for clinical diagnostics. All code supporting this study is distributed on PyPI and is packaged at https://github.com/gdewael/maldi-nn.
Article activity feed
-

-
-
-

eLife Assessment
This valuable study presents a machine learning model to recommend effective antimicrobial drugs from patients' samples analysed with mass spectrometry. The evidence supporting the claims of the authors is convincing. This work will be of interest to computational biologists, microbiologists, and clinicians.
-
Joint Public Reviews:
De Waele et al. framed the mass-spectrum-based prediction of antimicrobial resistance (AMR) prediction as a drug recommendation task. Neural networks were trained on the recently available DRIAMS database of MALDI-TOF (matrix-assisted laser desorption/ionization time-of-flight) mass spectrometry data and their associated antibiotic susceptibility profiles (Weis et al. 2022). Weis et al. (2022) also introduced the benchmark models which take as the input a single species and are trained to predict resistance to a single drug. Instead here, a pair of drugs and spectrum are fed to two neural network models to predict a resistance probability. In this manner, knowledge from different drugs and species can be shared through the model parameters. Questions asked: What is the best way to encode the drugs? Does the dual …
Joint Public Reviews:
De Waele et al. framed the mass-spectrum-based prediction of antimicrobial resistance (AMR) prediction as a drug recommendation task. Neural networks were trained on the recently available DRIAMS database of MALDI-TOF (matrix-assisted laser desorption/ionization time-of-flight) mass spectrometry data and their associated antibiotic susceptibility profiles (Weis et al. 2022). Weis et al. (2022) also introduced the benchmark models which take as the input a single species and are trained to predict resistance to a single drug. Instead here, a pair of drugs and spectrum are fed to two neural network models to predict a resistance probability. In this manner, knowledge from different drugs and species can be shared through the model parameters. Questions asked: What is the best way to encode the drugs? Does the dual neural network outperform the single spectrum-drug network?
The authors showed consistent performance of their strategy to predict antibiotic susceptibility for different spectrum and antibiotic representations (i.e., embedders). Remarkably, the authors showed how small datasets collected at one location can improve the performance of a model trained with limited data collected at a second location. The authors also showed that species-specific models (trained in multiple antibiotic resistance profiles) outperformed both the single recommender model and the individual species-antibiotic combination models.
Strengths:
• A single antimicrobial resistance recommender system could potentially facilitate the adoption of MALDI-TOF based antibiotic susceptibility profiling into clinical practices by reducing the number of models to be considered, and the efforts that may be required to periodically update them.
• The authors tested multiple combinations of embedders for the mass spectra and antibiotics while using different metrics to evaluate the performance of the resulting models. Models trained using different spectrum embedder-antibiotic embedder combinations had remarkably good performance for all tested metrics. The average ROC AUC scores for global and species-specific evaluations were above 0.8.
• Authors developed species-specific recommenders as an intermediate layer between the single recommender system and single species-antibiotic models. This intermediate approach achieved maximum performance (with one type of the species-specific recommender achieving a 0.9 ROC AUC), outlining the potential of this type of recommenders for frequent pathogens.
• Authors showed that data collected in one location can be leveraged to improve the performance of models generated using a smaller number of samples collected at a different location. This result may encourage researchers to optimize data integration to reduce the burden of data generation for institutions interested in testing this method.Weaknesses:
• Authors do not offer information about the model features associated with resistance. While reviewers understand that it is difficult to map mass spectra to specific pathways or metabolites, mechanistic insights are much more important in the context of AMR than in the context of bacterial identification. For example, this information may offer additional antimicrobial targets. Thus, authors should at least identify mass spectra peaks highly associated with resistance profiles. Are those peaks consistent across species? This would be a key step towards a proteomic survey of mechanisms of AMR. See previous work on this topic (Hrabak et al. 2013, Torres-Sangiao et al. 2022).
References:
Hrabak et al. (2013). Clin Microbiol Rev 26. doi: 10.1128/CMR.00058-12.
Torres-Sangiao et al. (2022). Front Med 9. doi: 10.3389/fmed.2022.850374.
Weis et al. (2022). Nat Med 28. doi: 10.1038/s41591-021-01619-9. -
Author response:
The following is the authors’ response to the previous reviews.
Reviewer #1:
Section 4.3 ("expert baseline model"): the authors need to explain how the probabilities defined as baselines were exactly used to predict individual patient susceptible profiles.
We have added a more detailed and mathematically formal explanation of the “simulated expert’s best guess” in Section 4.3.
This section now reads:
“More formally, considering all training spectra as Strain, all training labels corresponding to one drug j and species t are gathered:
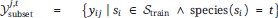
The "simulated expert's best guess" predicted probability for any spectrum si and drug dj, then, corresponds to, the fraction of positive labels in their corresponding training label set
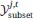 :”
:”Authors should explain in more detail how a ROC curve is generated from a single spectrum …
Author response:
The following is the authors’ response to the previous reviews.
Reviewer #1:
Section 4.3 ("expert baseline model"): the authors need to explain how the probabilities defined as baselines were exactly used to predict individual patient susceptible profiles.
We have added a more detailed and mathematically formal explanation of the “simulated expert’s best guess” in Section 4.3.
This section now reads:
“More formally, considering all training spectra as Strain, all training labels corresponding to one drug j and species t are gathered:
The "simulated expert's best guess" predicted probability for any spectrum si and drug dj, then, corresponds to, the fraction of positive labels in their corresponding training label set
:”Authors should explain in more detail how a ROC curve is generated from a single spectrum (i.e., per patient) and then average across spectra. I have an idea of how it's done but I am not completely sure.
We have added a more detailed explanation in Section 3.2. It reads:
To compute the (per-patient average) ROC-AUC, for any spectrum/patient, all observed drug resistance labels and their corresponding predictions are gathered. Then, the patient-specific ROC-AUC is computed on that subset of labels and predictions. Finally, all ROC-AUCs per patient are averaged to a "spectrum-macro" ROC-AUC.
In addition, our description under Supplementary Figure 8 (showing the ROC curve) provides additional clarification:
Note that this ROC curve is not a traditional ROC curve constructed from one single label set and one corresponding prediction set. Rather, it is constructed from spectrum-macro metrics as follows: for any possible threshold value, binarize all predictions. Then, for every spectrum/patient independently, compute the sensitivity and specificity for the subset of labels corresponding to that spectrum/patient. Finally, those sensititivies and specificities are averaged across patients to obtain one point on above ROC curve.
Section 3.2 & reply # 1: can the authors compute and apply the Youden cutoff that gives max precision-sensitivity for each ROC curve? In that way the authors could report those values.
We have computed this cut-off on the curve shown in Supplementary Figure 8. The Figure now shows the sensitivity and specificity at the Youden cutoff in addition to the ROC. We have chosen only to report these values for this model as we did not want to inflate our manuscript with additional metrics (especially since the ROC-AUC already captures sensitivities and specificities). We do, however, see the value of adding this once, so that biologists have an indication of what kind of values to expect for these metrics.
Related to reply #5: assuming that different classifiers are trained in the same data, with the same number of replicates, could authors use the DeLong test compare ROC curves? If not, please explain why.
We thank the reviewer for bringing our attention to the DeLong’s test. It does indeed seem true that this test is appropriate for comparing two ROC-AUCs using the same ground truth values.
We have chosen not to use this test for one conceptual and one practical reason:
(1) Our point still stands that in machine learning one chooses the test set, and hence one can artificially increase statistical power by simply allocating a larger fraction of the data to test.
(2) DeLong’s test is defined for single AUCs (i.e. to compare two lists of predictions against one list of ground truths), but here we report the spectrum/patient-macro ROC-AUC. It is not clear how to adjust the test to macro-evaluated AUCs. One option may be to apply the test per patient ROC curve, and perform multiple testing correction, but then we are not comparing models, but models per patient. In addition, the number of labels/predictions per patient is prohibitively small for statistical power.
Reviewer #2 (Recommendations For The Authors):
After revision, all issues were been resolved.
-
-
eLife assessment
This valuable study presents a machine learning model to recommend effective antimicrobial drugs from patients' samples analysed with mass spectrometry. The evidence supporting the claims of the authors is convincing, although including a measure of statistical significance to compare different proposed models would further strengthen the support. This work will be of interest to computational biologists, microbiologists, and clinicians.
-
Reviewer #1 (Public Review):
Summary:
De Waele et al. reported a dual-branch neural network model for predicting antibiotic resistance profiles using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry data. Neural networks were trained on the recently available DRIAMS database of MALDI-TOF mass spectrometry data and their associated antibiotic susceptibility profiles. The authors used dual branch neural network to simultaneously represent information about mass spectra and antibiotics for a wide range of species and antibiotic combinations. The authors showed consistent performance of their strategy to predict antibiotic susceptibility for different spectrum and antibiotic representations (i.e., embedders). Remarkably, the authors showed how small datasets collected at one location can improve the …
Reviewer #1 (Public Review):
Summary:
De Waele et al. reported a dual-branch neural network model for predicting antibiotic resistance profiles using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry data. Neural networks were trained on the recently available DRIAMS database of MALDI-TOF mass spectrometry data and their associated antibiotic susceptibility profiles. The authors used dual branch neural network to simultaneously represent information about mass spectra and antibiotics for a wide range of species and antibiotic combinations. The authors showed consistent performance of their strategy to predict antibiotic susceptibility for different spectrum and antibiotic representations (i.e., embedders). Remarkably, the authors showed how small datasets collected at one location can improve the performance of a model trained with limited data collected at a second location. The authors also showed that species-specific models (trained in multiple antibiotic resistance profiles) outperformed both the single recommender model and the individual species-antibiotic combination models. Despite the promising results, the authors should explain in more detail some of the analyses reported in the manuscript (see weaknesses).
Strengths:
• A single AMR recommender system could potentially facilitate the adoption of MALDI-TOF based antibiotic susceptibility profiling into clinical practices by reducing the number of models to be considered, and the efforts that may be required to periodically update them.
• Authors tested multiple combinations of embedders for the mass spectra and antibiotics while using different metrics to evaluate the performance of the resulting models. Models trained using different spectrum embedder-antibiotic embedder combinations had remarkably good performance for all tested metrics. The average ROC AUC scores for global and species-specific evaluations were above 0.8.
• Authors developed species-specific recommenders as an intermediate layer between the single recommender system and single species-antibiotic models. This intermediate approach achieved maximum performance (with one type of the species-specific recommender achieving a 0.9 ROC AUC), outlining the potential of this type of recommenders for frequent pathogens.
• Authors showed that data collected in one location can be leveraged to improve the performance of models generated using a smaller number of samples collected at a different location. This result may encourage researchers to optimize data integration to reduce the burden of data generation for institutions interested in testing this method.Weaknesses:
• Section 4.3 ("expert baseline model"): the authors need to explain how the probabilities defined as baselines were exactly used to predict individual patient susceptible profiles.
• Authors do not offer information about the model features associated with resistance. Although I understand the difficulty of mapping mass spectra to specific pathways or metabolites, mechanistic insights are much more important in the context of AMR than in the context of bacterial identification. For example, this information may offer additional antimicrobial targets. Thus, authors should at least identify mass spectra peaks highly associated with resistance profiles. Are those peaks consistent across species? This would be a key step towards a proteomic survey of mechanisms of AMR. See previous work on this topic: PMIDs: 35586072 and 23297261. -
Reviewer #2 (Public Review):
The authors frame the MS-spectrum-based prediction of antimicrobial resistance prediction as a drug recommendation task. Weis et al. introduced the dataset this model is tested on and benchmark models which take as input a single species and are trained to predict resistance to a single drug. Instead here, a pair of drugs and spectrum are fed to 2 neural network models to predict a resistance probability. In this manner, knowledge from different drugs and species can be shared through the model parameters. Questions asked: 1. what is the best way to encode the drugs? 2. does the dual NN outperform the single spectrum-drug?
Overall the paper is well-written and structured. It presents a novel framework for a relevant problem.
-
Author response:
The following is the authors’ response to the previous reviews.
Reviewer 1:
• Although ROC AUC is a widely used metric. Other metrics such as precision, recall, sensitivity, and specificity are not reported in this work. The last two metrics would help readers understand the model’s potential implications in the context of clinical research.
In response to this comment and related ones by Reviewer 2, we have overhauled how we evaluate our models. In the revised version, we have removed Micro ROC-AUC, as this evaluation metric is hard to interpret in the recommender system setting. Instead, the updated version fully focuses on two metrics: ROC-AUC and Precision at 1 of the negative class, both computed per spectrum and then averaged (equivalent to the instance-wise metrics in the previous version of the manuscript). We …
Author response:
The following is the authors’ response to the previous reviews.
Reviewer 1:
• Although ROC AUC is a widely used metric. Other metrics such as precision, recall, sensitivity, and specificity are not reported in this work. The last two metrics would help readers understand the model’s potential implications in the context of clinical research.
In response to this comment and related ones by Reviewer 2, we have overhauled how we evaluate our models. In the revised version, we have removed Micro ROC-AUC, as this evaluation metric is hard to interpret in the recommender system setting. Instead, the updated version fully focuses on two metrics: ROC-AUC and Precision at 1 of the negative class, both computed per spectrum and then averaged (equivalent to the instance-wise metrics in the previous version of the manuscript). We believe these metrics best reflect the use-case of AMR recommenders. In addition, we have kept (drug-)macro ROC-AUC as a complementary evaluation metric. As the ROC-AUC can be decomposed into sensitivity and specificity (at different prediction probability thresholds), we have added a ROC curve where sensitivity and specificity are indicated in Figure 8 (Appendices).
• The authors did not hypothesize or describe in any way what an acceptable performance of their recommender system should be in order to be adopted by clinicians.
In Section 4.3, we have extended our experiments to include a baseline that represents a “simulated expert”. In short, given a species, an expert can already make some best guesses as to what drugs will be effective or not. To simulate this, we count resistance frequencies per species and per drug in the training set, and use this as predictions of a “simulated expert”.
We now mention in our manuscript that any performance above this level results in a real-world information gain for clinical diagnostic labs.
• Related to the previous comment, this work would strongly benefit from the inclusion of 1-2 real-life applications of their method that could showcase the benefits of their strategy for designing antibiotic treatment in a clinical setting.
While we think this would be valuable to try out, we are an in silico research lab, and the study we propose is an initial proof-of-concept focusing on the methodology. Because of this, we feel a real-life application of the model is out-of-scope for the present study.
• The authors do not offer information about the model features associated with resistance. This information may offer insights about mechanisms of antimicrobial resistance and how conserved they are across species.
In general, MALDI-TOF mass spectra are somewhat hard to interpret. Because of a limited body of work analyzing resistance mechanisms with MALDI-TOF MS, it is hard to link peaks back to specific pathways. For this reason, we have chosen to forego such an analysis. After all, as far as we know, typical MALDI-TOF MS manufacturers’ software for bacterial identification also does not provide interpretability results or insights into peaks, but merely gives an identification and confidence score.
However, we do feel that the whole topic revolving around “the degree of biological insight a data modality might give versus actual performance and usability” merits further discussion. We have ultimately decided not to include a segment in our discussion section as it is hard to discuss this matter concisely.
• Comparison of AUC values across models lacks information regarding statistical significance. Without this information it is hard for a reader to figure out which differences are marginal and which ones are meaningful (for example, it is unclear if a difference in average AUC of 0.02 is significant). This applied to Figure 2, Figure 3, and Table 2 (and the associated supplementary figures).
To make trends a bit more clear and easier to discern, in our revised manuscript, all models are run for 5 replicates (as opposed to 3 in the previous version).
There is an ongoing debate in the ML community whether statistical tests are useful for comparing machine learning models. A simple argument against them is that model runs are typically not independent from each other, as they are all trained on the same data. The assumptions of traditional statistical tests are therefore violated (t-test, Wilcoxon test, etc.). With such tests statistical significance of the smallest differences can simply be achieved by increasing the number of replicates (i.e. training the same models more times).
More complicated but more appropriate statistical tests also exist, such as the 5x2 cross-validated t-test of Dietterich: “Approximate statistical tests for comparing supervised classification learning algorithms”, Neural computation 1998. However, these tests are typically not considered in deep learning, because only 10% of the data can be used for training, which is practically not desirable. The Friedman test of Demšar "On the appropriateness of statistical tests in machine learning." Workshop on Evaluation Methods for Machine Learning in conjunction with ICML. 2008., in combination with posthoc pairwise tests, is still frequently used in machine learning, but that test is only applicable in studies where many datasets are tested.
For those reasons, most deep learning papers that only analyse a few datasets typically do not consider any statistical tests. For the same reasons, we are also not convinced of the added value of statistical tests in our study.
• One key claim of this work was that their single recommender system outperformed specialist (single species-antibiotic) models. However, in its current status, it is not possible to determine that in fact that is the case (see comment above). Moreover, comparisons to species-level models (that combine all data and antibiotic susceptibility profiles for a given species) would help to illustrate the putative advantages of the dual branch neural network model over species-based models. This analysis will also inform the species (and perhaps datasets) for which specialist models would be useful to consider.
We thank the reviewer for this excellent suggestion. In our new manuscript, we have dedicated an entire section of experiments to testing such species-specific recommender models (Section 4.2). We find that species-specific recommender systems generally outperform the models trained globally across all species. As a result, our manuscript has been majorly reworked.
• Taking into account that the clustering of spectra embeddings seemed to be species-driven (Figure 4), one may hypothesize that there is limited transfer of information between species, and therefore the neural network model may be working as an ensemble of species models. Thus, this work would deeply benefit from a comparison between the authors' general model and an ensemble model in which the species is first identified and then the relevant species recommender is applied. If authors had identified cases to illustrate how data from one species positively influence the results for another species, they should include some of those examples.
See the answer to the remark above.
• The authors should check that all abbreviations are properly introduced in the text so readers understand exactly what they mean. For example, the Prec@1 metric is a little confusing.
See the answer to a remark above for how we have overhauled our evaluation metrics in the revised version. In addition, in the revised version, we have bundled our explanations on evaluation metrics together in Section 3.2. We feel that having these explanations in a separate section will improve overall comprehensibility of the manuscript.
• The authors should include information about statistical significance in figures and tables that compare performance across models.
See answer above.
• An extra panel showing species labels would help readers understand Figure 11.
We have tried to play around with including species labels in these plots, but could not make it work without overcrowding the figure. Instead, we have added a reminder in the caption that readers should refer back to an earlier figure for species labels.
• The authors initially stated that molecular structure information is not informative. However, in a second analysis, the authors stated that molecular structures are useful for less common drugs. Please explain in more detail with specific examples what you mean.
In the previous version of our manuscript, we found that one-hot embedding-based models were superior to structure-based drug embedders for general performance. The latter however, delivered better transfer learning performance.
In our new experiments however, we perform early stopping on “spectrum-macro” ROC-AUC (as opposed to micro ROC-AUC in the previous version). As a consequence, our results are different. In the new version of our manuscript, Morgan Fingerprints-based drug embedders generally outperform others both “in general” and for transfer learning. Hence, our previously conflicting statements are not applicable to our new results.
• The authors may want to consider adding a few sentences that summarize the 'Related work' section into the introduction, and converting the 'Related work' section into an appendix.
While we acknowledge that such a section is uncommon in biology, in machine learning research, a “related work” section is very common. As this research lies on the intersection of the two, we have decided to keep the section as such.
Reviewer 2:
• Are the specialist models re-trained on the whole set of spectra? It was shown by Weis et al. that pooling spectra from different species hinders performance. It would then be better to compare directly to the models developed by Weis et al, using their splitting logic since it could be that the decay in performance from specialists comes from the pooling. See the section "Species-stratified learning yields superior predictions" in https://doi.org/10.1038/s41591-021-01619-9.
We train our “specialist” (or now-called “species-drug classifiers”) just as described in Weis et al.: All labels for a drug are taken, and then subsetted for a single species. We have clarified this a bit better in our new manuscript. The text now reads:
“Previous studies have studied AMR prediction in specific species-drug combinations. For this reason, it is useful to compare how the dual-branch setup weighs up against training separate models for separate species and drugs. In Weis et al. (2020b), for example, binary AMR classifiers are trained for the following three combinations: (1) E. coli with Ceftriaxone, (2) K. pneumoniae with Ceftriaxone, and (3) S. aureus with Oxacillin. Here, such "species-drug-specific classifiers" are trained for the 200 most-common combinations of species and drugs in the training dataset.
• Going back to Weis et al. a high variance in performance between species/drug pairs was observed. The metrics in Table 2 do not offer any measurement of variance or statistical testing. Indeed, some values are quite close e.g. Macro AUROC of Specialist MLP-XL vs One-hot M.
See our answer to a remark of Reviewer 1 for our viewpoint on statistical significance testing in machine learning.
• Since this is a recommendation task, why were no recommendation system metrics used, e.g. mAP@K, mRR, and so (apart from precision@1 for the negative class)? Additionally, since there is a high label imbalance in this task (~80% negatives) a simple model would achieve a very high precision@1.
See the answer to a remark above for how we have overhauled our evaluation metrics in the revised version. In addition, in choosing our metrics, we wanted metrics that are both (1) appropriate (i.e. recommender system metrics), but also (2) easy to interpret for clinicians. For this reason, we have not included metrics such as mAP@K or mRR. We feel that “spectrum-macro” ROC-AUC and precision@1 cover a sufficiently broad evaluation set of metrics but are easy enough to interpret.
• A highly similar approach was recently published (https://doi.org/10.1093/bioinformatics/btad717). Since it is quite close to the publication date of this paper, it could be discussed as concurrent work.
We thank the reviewer for bringing our attention to this study. We have added a paragraph in our revised version discussing this paper as concurrent work.
• It is difficult to observe a general trend from Figure 2. A statistical test would be advised here.
See our answer to a remark of Reviewer 1 for our viewpoint on statistical significance testing in machine learning.
• Figure 5. UMAPs generally don't lead to robust quantitative conclusions. However, the analysis of the embedding space is indeed interesting. Here I would recommend some quantitative measures directly using embedding distances to accompany the UMAP visualizations. E.g. clustering coefficients, distribution of pairwise distances, etc.
In accordance with this recommendation, we have computed many statistics on the MALDI-TOF spectra embedding spaces. However, we could not come up with any statistic that illuminated us more than the visualization itself. For this reason, we have kept this section as is, and let the figure speak for itself.
• Weis et al. also perform a transfer learning analysis. How does the transfer learning capacity of the proposed models differ from those in Weis et al?
Weis et al. perform experiments towards “transferability”, not actual transfer learning. In essence, they use a model trained on data from one diagnostic lab towards prediction on data from another. However, they do not conduct experiments to learn how much data such a pre-trained classifier needs to fine-tune it for adequate performance on the new diagnostic lab, as we do. The end of Section 4.4 discusses how our proposed models specifically shine in transfer learning. The paragraph reads:
“Lowering the amount of data required is paramount to expedite the uptake of AMR models in clinical diagnostics. The transfer learning qualities of dual-branch models may be ascribed to multiple properties. First of all, since different hospitals use much of the same drugs, transferred drug embedders allow for expressively representing drugs out of the box. Secondly, owing to multi-task learning, even with a limited number of spectra, a considerable fine-tuning dataset may be obtained, as all available data is "thrown on one pile".”
-
-
eLife assessment
This valuable study presents a machine learning model to recommend effective antimicrobial drugs from patients' samples analysed with mass spectrometry. While the proposed approach of training a single model across different bacterial species and drugs seems promising, the comparison with baselines and related work is incomplete. With the evaluation part strengthened, this paper would be of interest to computational biologists, microbiologist, and clinicians.
-
Reviewer #1 (Public Review):
Summary:
De Waele et al. reported a dual-branch neural network model for predicting antibiotic resistance profiles using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry data. Neural networks were trained on the recently available DRIAMS database of MALDI-TOF mass spectrometry data and their associated antibiotic susceptibility profiles. The authors used a dual branch neural network approach to simultaneously represent information about mass spectra and antibiotics for a wide range of species and antibiotic combinations. The authors showed consistent performance of their strategy to predict antibiotic susceptibility for different spectrums and antibiotic representations (i.e., embedders). Remarkably, the authors showed how small datasets collected at one location can …
Reviewer #1 (Public Review):
Summary:
De Waele et al. reported a dual-branch neural network model for predicting antibiotic resistance profiles using matrix-assisted laser desorption/ionization time-of-flight (MALDI-TOF) mass spectrometry data. Neural networks were trained on the recently available DRIAMS database of MALDI-TOF mass spectrometry data and their associated antibiotic susceptibility profiles. The authors used a dual branch neural network approach to simultaneously represent information about mass spectra and antibiotics for a wide range of species and antibiotic combinations. The authors showed consistent performance of their strategy to predict antibiotic susceptibility for different spectrums and antibiotic representations (i.e., embedders). Remarkably, the authors showed how small datasets collected at one location can improve the performance of a model trained with limited data collected at a second location. Despite these promising results, there are several analyses that the authors could incorporate to offer additional support to some of their claims (see weaknesses). In particular, this work would benefit from a more comprehensive comparison of the author's single recommender model vs an ensemble of specialist models, and the inclusion of 1-2 examples that showcase how their model could be translated into the clinic.
Strengths:
• A single AMR recommender system could potentially facilitate the adoption of MALDI-TOF-based antibiotic susceptibility profiling into clinical practices by reducing the number of models to be considered, and the efforts that may be required to periodically update them.
• Authors tested multiple combinations of embedders for the mass spectra and antibiotics while using different metrics to evaluate the performance of the resulting models. Models trained using different spectrum embedder-antibiotic embedder combinations had remarkably good performance for all tested metrics. The average ROC AUC scores for global and spectrum-level evaluations were above 0.9. Average ROC AUC scores for antibiotic-level evaluations were greater than 0.75.
• Authors showed that data collected in one location can be leveraged to improve the performance of models generated using a smaller number of samples collected at a different location. This result may encourage researchers to optimize data integration to reduce the burden of data generation for institutions interested in testing this method.
Weaknesses:
• Although ROC AUC is a widely used metric. Other metrics such as precision, recall, sensitivity, and specificity are not reported in this work. The last two metrics would help readers understand the model's potential implications in the context of clinical research.
• The authors did not hypothesize or describe in any way what an acceptable performance of their recommender system should be in order to be adopted by clinicians.
• Related to the previous comment, this work would strongly benefit from the inclusion of 1-2 real-life applications of their method that could showcase the benefits of their strategy for designing antibiotic treatment in a clinical setting.
• The authors do not offer information about the model features associated with resistance. This information may offer insights about mechanisms of antimicrobial resistance and how conserved they are across species.
• Comparison of AUC values across models lacks information regarding statistical significance. Without this information it is hard for a reader to figure out which differences are marginal and which ones are meaningful (for example, it is unclear if a difference in average AUC of 0.02 is significant). This applied to Figure 2, Figure 3, and Table 2 (and the associated supplementary figures).
• One key claim of this work was that their single recommender system outperformed specialist (single species-antibiotic) models. However, in its current status, it is not possible to determine that in fact that is the case (see comment above). Moreover, comparisons to species-level models (that combine all data and antibiotic susceptibility profiles for a given species) would help to illustrate the putative advantages of the dual branch neural network model over species-based models. This analysis will also inform the species (and perhaps datasets) for which specialist models would be useful to consider.
• Taking into account that the clustering of spectra embeddings seemed to be species-driven (Figure 4), one may hypothesize that there is limited transfer of information between species, and therefore the neural network model may be working as an ensemble of species models. Thus, this work would deeply benefit from a comparison between the authors' general model and an ensemble model in which the species is first identified and then the relevant species recommender is applied. If authors had identified cases to illustrate how data from one species positively influence the results for another species, they should include some of those examples.
-
Reviewer #2 (Public Review):
The authors frame the MS-spectrum-based prediction of antimicrobial resistance prediction as a drug recommendation task. Weis et al introduced the dataset this model is tested on and benchmark models which take as input a single species and are trained to predict resistance to a single drug. Instead here, a pair of drug and spectrum are fed to 2 neural network models to predict a resistance probability. In this manner, knowledge from different drugs and species can be shared through the model parameters. Three questions are asked: 1. what is the best way to encode the drugs? 2. does the dual NN outperform the single-spectrum drug?
Overall the paper is well-written and structured. It presents a novel framework for a relevant problem. The work would benefit from more work on evaluation.
-
