Salmonella-induced SIRT1 and SIRT3 are crucial for maintaining the metabolic switch in bacteria and host for successful pathogenesis
Curation statements for this article:-
Curated by eLife

eLife Assessment
This auhors present findings on the role of the sirtuins SIRT1 and SIRT3 during Salmonella Typhimurium infection. This valuable study increases our understanding of the mechanisms used by this pathogen to interact with its host and may have implications for other intracellular pathogens. The reviewers disagreed on the strength of the evidence to support the claims. Although one reviewer found the strength of the evidence convincing, the other found that it was incomplete, and that the main claims are only partially supported, as can be seen from the public reviews.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Sirtuins are the major players in host immunometabolic regulation. However, the role of sirtuins in the modulation of the immune metabolism pertaining to salmonellosis is largely unknown. Here, our investigation focussed on the role of two important sirtuins, SIRT1 and SIRT3, shedding light on their impact on intracellular Salmonella ’s metabolic switch and pathogenesis establishment. Our study indicated the ability of the live Salmonella Typhimurium to differentially regulate the levels of SIRT1 and SIRT3 for maintaining the high glycolytic metabolism and low fatty acid metabolism in Salmonella . Perturbing SIRT1 or SIRT3 through knockdown or inhibition resulted in a remarkable shift in the host metabolism to low fatty acid oxidation and high glycolysis. This switch led to decreased proliferation of Salmonella in the macrophages. Further, Salmonella -induced higher levels of SIRT1 and SIRT3 led to a skewed polarization state of the macrophages from a pro-inflammatory M1 state toward an immunosuppressive M2, making it more conducive for the intracellular life of Salmonella . Alongside, governing immunological functions by modulating p65 NF-κB acetylation, SIRT1, and SIRT3 also skew Salmonella- induced host metabolic switch by regulating the acetylation status of HIF-1α and PDHA1. Interestingly, though knockdown of SIRT1/3 attenuated Salmonella proliferation in macrophages, in in vivo mice model of infection, inhibition or knockdown of SIRT1/3 led to more dissemination and higher organ burden, which can be attributed to enhanced ROS and IL-6 production. Our study hence reports for the first time that Salmonella modulates SIRT1/3 levels to maintain its own metabolism for successful pathogenesis.
Article activity feed
-

-
-
-

eLife Assessment
This auhors present findings on the role of the sirtuins SIRT1 and SIRT3 during Salmonella Typhimurium infection. This valuable study increases our understanding of the mechanisms used by this pathogen to interact with its host and may have implications for other intracellular pathogens. The reviewers disagreed on the strength of the evidence to support the claims. Although one reviewer found the strength of the evidence convincing, the other found that it was incomplete, and that the main claims are only partially supported, as can be seen from the public reviews.
-
Reviewer #2 (Public review):
Dipasree Hajra et al demonstrated that Salmonella was able to modulate the expression of Sirtuins (Sirt1 and Sirt3) and regulate the metabolic switch in both host and Salmonella, promoting its pathogenesis. The authors found Salmonella infection induced high levels of Sirt1 and Sirt3 in macrophages, which were skewed toward the M2 phenotype allowing Salmonella to hyper-proliferate. Mechanistically, Sirt1 and Sirt3 regulated the acetylation of HIF-1alpha and PDHA1, therefore mediating Salmonella-induced host metabolic shift in the infected macrophages. Interestingly, Sirt1 and Sirt3-driven host metabolic switch also had an effect on the metabolic profile of Salmonella. Counterintuitively, inhibition of Sirt1/3 led to increased pathogen burdens in an in vivo mouse model. Overall, this is a well-designed study.
Reviewer #2 (Public review):
Dipasree Hajra et al demonstrated that Salmonella was able to modulate the expression of Sirtuins (Sirt1 and Sirt3) and regulate the metabolic switch in both host and Salmonella, promoting its pathogenesis. The authors found Salmonella infection induced high levels of Sirt1 and Sirt3 in macrophages, which were skewed toward the M2 phenotype allowing Salmonella to hyper-proliferate. Mechanistically, Sirt1 and Sirt3 regulated the acetylation of HIF-1alpha and PDHA1, therefore mediating Salmonella-induced host metabolic shift in the infected macrophages. Interestingly, Sirt1 and Sirt3-driven host metabolic switch also had an effect on the metabolic profile of Salmonella. Counterintuitively, inhibition of Sirt1/3 led to increased pathogen burdens in an in vivo mouse model. Overall, this is a well-designed study.
The revised manuscript has addressed all of the previous comments. The re-analysis of flow cytometry and WB data by authors makes the results and conclusion more complete and convincing.
-
Reviewer #3 (Public review):
Summary:
In this paper Hajra et al have attempted to identify the role of Sirt1 and Sirt3 in regulating metabolic reprogramming and macrophage host defense. They have performed gene knock down experiments in RAW macrophage cell line to show that depletion of Sirt1 or Sirt3 enhances the ability of macrophages to eliminate Salmonella Typhimurium. However, in mice inhibition of Sirt1 resulted in dissemination of the bacteria but the bacterial burden was still reduced in macrophages. They suggest that the effect they have observed is due to increased inflammation and ROS production by macrophages. They also try to establish a weak link with metabolism. They present data to show that the switch in metabolism from glycolysis to fatty acid oxidation is regulated by acetylation of Hif1a, and PDHA1.
Strengths:
The …
Reviewer #3 (Public review):
Summary:
In this paper Hajra et al have attempted to identify the role of Sirt1 and Sirt3 in regulating metabolic reprogramming and macrophage host defense. They have performed gene knock down experiments in RAW macrophage cell line to show that depletion of Sirt1 or Sirt3 enhances the ability of macrophages to eliminate Salmonella Typhimurium. However, in mice inhibition of Sirt1 resulted in dissemination of the bacteria but the bacterial burden was still reduced in macrophages. They suggest that the effect they have observed is due to increased inflammation and ROS production by macrophages. They also try to establish a weak link with metabolism. They present data to show that the switch in metabolism from glycolysis to fatty acid oxidation is regulated by acetylation of Hif1a, and PDHA1.
Strengths:
The strength of the manuscript is that the role of Sirtuins in host-pathogen interactions have not been previously explored in-depth making the study interesting. It is also interesting to see that depletion of either Sirt1 or Sirt3 result in a similar outcome.
Weaknesses:
The major weakness of the paper is the low quality of data, making it harder to substantiate the claims. Also, there are too many pathways and mechanisms being investigated. It would have been better if the authors had focussed on either Sirt1 or Sirt3 and elucidated how it reprograms metabolism to eventually modulate host response against Salmonella Typhimurium. Experimental evidences are also lacking to prove the proposed mechanisms. For instance they show correlative data that knock down of Sirt1 mediated shift in metabolism is due to HIF1a acetylation but this needs to be proven with further experiments.
-
Author response:
The following is the authors’ response to the current reviews.
Reviewer #2 (Public review):
Dipasree Hajra et al demonstrated that Salmonella was able to modulate the expression of Sirtuins (Sirt1 and Sirt3) and regulate the metabolic switch in both host and Salmonella, promoting its pathogenesis. The authors found Salmonella infection induced high levels of Sirt1 and Sirt3 in macrophages, which were skewed toward the M2 phenotype allowing Salmonella to hyper-proliferate. Mechanistically, Sirt1 and Sirt3 regulated the acetylation of HIF-1alpha and PDHA1, therefore mediating Salmonella-induced host metabolic shift in the infected macrophages. Interestingly, Sirt1 and Sirt3-driven host metabolic switch also had an effect on the metabolic profile of Salmonella. Counterintuitively, inhibition of Sirt1/3 led to increased …
Author response:
The following is the authors’ response to the current reviews.
Reviewer #2 (Public review):
Dipasree Hajra et al demonstrated that Salmonella was able to modulate the expression of Sirtuins (Sirt1 and Sirt3) and regulate the metabolic switch in both host and Salmonella, promoting its pathogenesis. The authors found Salmonella infection induced high levels of Sirt1 and Sirt3 in macrophages, which were skewed toward the M2 phenotype allowing Salmonella to hyper-proliferate. Mechanistically, Sirt1 and Sirt3 regulated the acetylation of HIF-1alpha and PDHA1, therefore mediating Salmonella-induced host metabolic shift in the infected macrophages. Interestingly, Sirt1 and Sirt3-driven host metabolic switch also had an effect on the metabolic profile of Salmonella. Counterintuitively, inhibition of Sirt1/3 led to increased pathogen burdens in an in vivo mouse model. Overall, this is a well-designed study.
The revised manuscript has addressed all of the previous comments. The re-analysis of flow cytometry and WB data by authors makes the results and conclusion more complete and convincing.We are immensely grateful to the reviewer for improving the strength of the manuscript by providing insightful comments and for appreciating the work.
Reviewer #3 (Public review):
Summary:
In this paper Hajra et al have attempted to identify the role of Sirt1 and Sirt3 in regulating metabolic reprogramming and macrophage host defense. They have performed gene knock down experiments in RAW macrophage cell line to show that depletion of Sirt1 or Sirt3 enhances the ability of macrophages to eliminate Salmonella Typhimurium. However, in mice inhibition of Sirt1 resulted in dissemination of the bacteria but the bacterial burden was still reduced in macrophages. They suggest that the effect they have observed is due to increased inflammation and ROS production by macrophages. They also try to establish a weak link with metabolism. They present data to show that the switch in metabolism from glycolysis to fatty acid oxidation is regulated by acetylation of Hif1a, and PDHA1.
Strengths:
The strength of the manuscript is that the role of Sirtuins in host-pathogen interactions have not been previously explored in-depth making the study interesting. It is also interesting to see that depletion of either Sirt1 or Sirt3 result in a similar outcome.
Weaknesses:
The major weakness of the paper is the low quality of data, making it harder to substantiate the claims. Also, there are too many pathways and mechanisms being investigated. It would have been better if the authors had focussed on either Sirt1 or Sirt3 and elucidated how it reprograms metabolism to eventually modulate host response against Salmonella Typhimurium. Experimental evidences are also lacking to prove the proposed mechanisms. For instance they show correlative data that knockdown of Sirt1 mediated shift in metabolism is due to HIF1a acetylation but this needs to be proven with further experiments.
As the public review of the reviewer remains unaltered as the previous version without further recommendations for authors, we are sticking to our former author’s response. We respect the reviewer’s opinion and thank the reviewer for the critical analysis of our work.
---------
The following is the authors’ response to the previous reviews.
Reviewer #2 (Public Review):
Dipasree Hajra et al demonstrated that Salmonella was able to modulate the expression of Sirtuins (Sirt1 and Sirt3) and regulate the metabolic switch in both host and Salmonella, promoting its pathogenesis. The authors found Salmonella infection induced high levels of Sirt1 and Sirt3 in macrophages, which were skewed toward the M2 phenotype allowing Salmonella to hyper-proliferate. Mechanistically, Sirt1 and Sirt3 regulated the acetylation of HIF-1alpha and PDHA1, therefore mediating Salmonella-induced host metabolic shift in the infected macrophages. Interestingly, Sirt1 and Sirt3-driven host metabolic switch also had an effect on the metabolic profile of Salmonella. Counterintuitively, inhibition of Sirt1/3 led to increased pathogen burdens in an in vivo mouse model. Overall, this is a well-designed study.
Comments on revised version:
The authors have performed additional experiments to address the discrepancy between in vitro and in vivo data. While this offers some potential insights into the in vivo role of Sirt1/3 in different cell types and how this affects bacterial growth/dissemination, I still believe that Sirt1/3 inhibitors could have some effect on the gut microbiota contributing to increased pathogen counts. This possibility can be discussed briefly to give a better scenario of how Sirt1/3 inhibitors work in vivo. Additionally, the manuscript would improve significantly if some of the flow cytometry analysis and WB data could be better analyzed.
We are highly grateful for your valuable and insightful comments. Thank you for appreciating the merit of our manuscript. As rightly pointed out by the eminent reviewer, we acknowledge the probable link of Sirtuin on gut microbiota and its effect on increased bacterial loads as indicated by previous literature studies (PMID: 22115311, PMID: 19228061). These reports suggested that a low dose of Sirt1 activator, resveratrol treatment in rats for 25 days treatment under 5% DSS induced colitis condition led to alterations in gut microbiota profile with increased lactobacilli and bifidobacteria alongside reduced abundance of enterobacteria. This study correlates with our study wherein we have detected enhanced Salmonella (belonging to Enterobacteriaceae family) loads under both Sirt1/3 in vivo knockdown condition or inhibitor-treated condition in C57BL/6 mice and reduced burden under Sirt-1 activator treatment SRT1720.
As per your valid suggestion, we have discussed this possibility in our discussion section. (Line- 541-548).
We have incorporated the suggestions for the improvement in the analysis of WB data and flow cytometry.
Reviewer #3 (Public Review):
Summary:
In this paper Hajra et al have attempted to identify the role of Sirt1 and Sirt3 in regulating metabolic reprogramming and macrophage host defense. They have performed gene knock down experiments in RAW macrophage cell line to show that depletion of Sirt1 or Sirt3 enhances the ability of macrophages to eliminate Salmonella Typhimurium. However, in mice inhibition of Sirt1 resulted in dissemination of the bacteria but the bacterial burden was still reduced in macrophages. They suggest that the effect they have observed is due to increased inflammation and ROS production by macrophages. They also try to establish a weak link with metabolism. They present data to show that the switch in metabolism from glycolysis to fatty acid oxidation is regulated by acetylation of Hif1a, and PDHA1.
Strengths:
The strength of the manuscript is that the role of Sirtuins in host-pathogen interactions has not been previously explored in-depth making the study interesting. It is also interesting to see that depletion of either Sirt1 or Sirt3 results in a similar outcome.
Weaknesses:
The major weakness of the paper is the low quality of data, making it harder to substantiate the claims. Also, there are too many pathways and mechanisms being investigated. It would have been better if the authors had focussed on either Sirt1 or Sirt3 and elucidated how it reprograms metabolism to eventually modulate host response against Salmonella Typhimurium. Experimental evidence is also lacking to prove the proposed mechanisms. For instance they show correlative data that knock down of Sirt1 mediated shift in metabolism is due to HIF1a acetylation but this needs to be proven with further experiments.
We appreciate the reviewer’s critical analysis of our work. In the revised manuscript, we aimed to eliminate the low-quality data sets and have tried to substantiate them with better and conclusive ones, as directed in the recommendations for the author section. We agree with the reviewer that the inclusion of both Sirtuins 1 and 3 has resulted in too many pathways and mechanisms and focusing on one SIRT and its mechanism of metabolic reprogramming and immune modulation would have been a less complicated alternative approach. However, as rightly pointed out, our work demonstrated the shared and few overlapping roles of the two sirtuins, SIRT1 and SIRT3, together mediating the immune-metabolic switch upon Salmonella infection. As per the reviewer’s suggestion, we have performed additional experiments with HIF-1α inhibitor treatment in our revised manuscript to substantiate our correlative findings on SIRT1-mediated regulation of host glycolysis (Fig.7G). We wanted to clarify our claim in this regard. Our results suggested that loss of SIRT1 function triggered increased host glycolysis alongside hyperacetylation of HIF-1α. HIF-1α is reported to be one of the important players in glycolysis regulation (Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol. 2021;599(1):23-37. doi:10.1113/JP280572.) and additionally, SIRT1 has been shown to regulate HIF-1α acetylation status (Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1 alpha. Mol Cell. 2010;38(6):864-878. doi:10.1016/j.molcel.2010.05.023.) Further, ectopic expression of SIRT1 has been demonstrated to reduce glycolysis by negatively regulating HIF-1α. (Wang Y, Bi Y, Chen X, et al. Histone Deacetylase SIRT1 Negatively Regulates the Differentiation of Interleukin-9-Producing CD4(+) T Cells. Immunity. 2016;44(6):1337-1349. doi:10.1016/j.immuni.2016.05.009). We have subsequently shown in Fig. 7G, that the increase in host glycolysis upon SIRT knockdown in the infected macrophages gets lowered upon HIF-1α inhibitor treatment, suggesting that one of the mechanisms of SIRT-mediated regulation of host glycolysis is via regulation of HIF-1α. However, this warrants further future mechanistic research.
Recommendations for the authors:
Reviewer #2 (Recommendations For The Authors):
(1) Figures 8I-S: are only viable cells used for analysis? Please provide gating strategy used for these analyses.
(2) Many changes seen in WB seem to be marginal. Since the authors used densitometric plot to quantify the band intensities, I expect these experiments were repeated at least three times. Please indicate the number of repeats. For instance, Figures 7C, 7I (UI SCR vs UI shSIRT3), 7J, show marginal changes or no changes. What do other WB images look like? Are they more convincing than the ones currently shown? Please provide them in the response letter.
(3) Figure 7C: label is a bit misleading. Please relabel the figure title to Acetylated HIF vs total levels
(4) Figure 7J: which band is AcPDHA1?
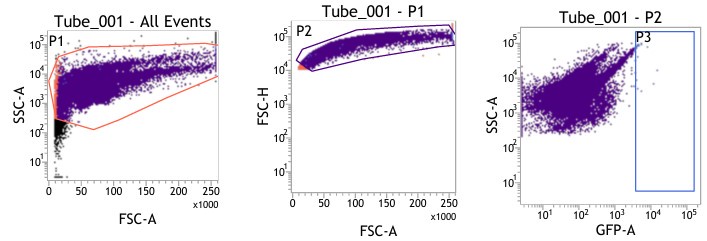
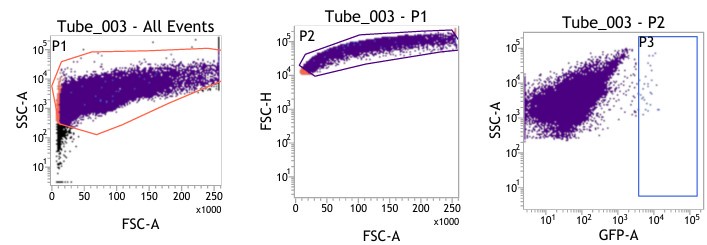
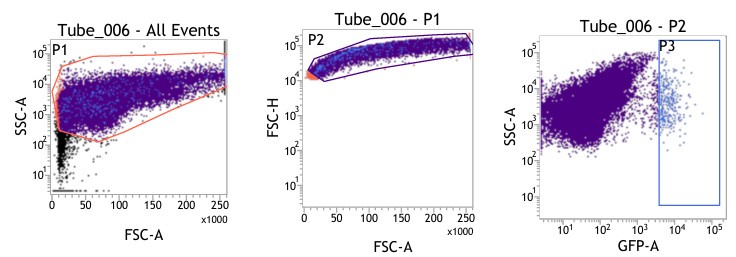
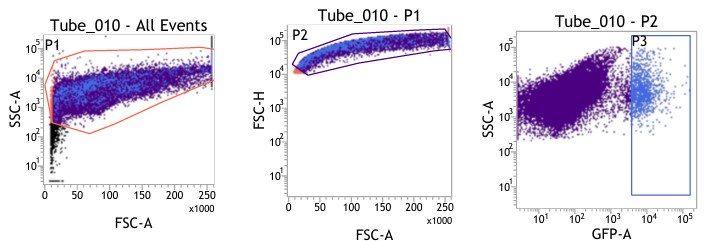
(1) We are highly apologetic for not clarifying our gating strategy for the analysis.
We initially gated the viable splenocyte population based on Forward scatter (FSC) and Side Scatter (SSC). This gated population was further subjected to gating based on cell FSC-H (height) versus FSC-A (area). Subsequently, the population was gated as per SSC-A and GFP (expressed by intracellular bacteria) based on the autofluorescence exhibited by the uninfected control (Fig. 8I-J).
Author response image 1.
UNINFECTED
Author response image 2.
VEHICLE CONTROL INFECTED
Author response image 3.
EX-527 INFECTED
Author response image 4.
3TYP INFECTED
Author response image 5.
SRT 1720 INFECTED
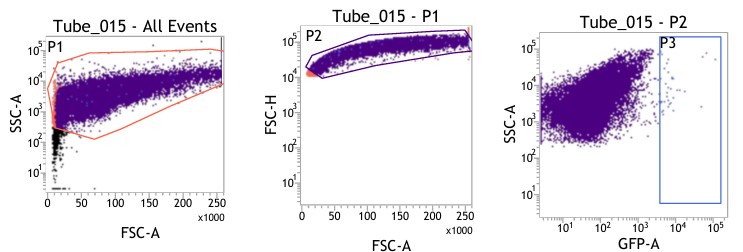
For gating different cell types such as F4/80 (PE) positive population in Fig. 8K-L, the viable cell population was gated based on SSC-A versus PE-A to gate the macrophage population. These macrophage populations were gated further based on GFP (Salmonella) + population to obtain the percentage of macrophage population harboring GFP+ bacteria. Similar strategies were followed for other cell types as depicted in Fig. 8M-S, Fig. S8.
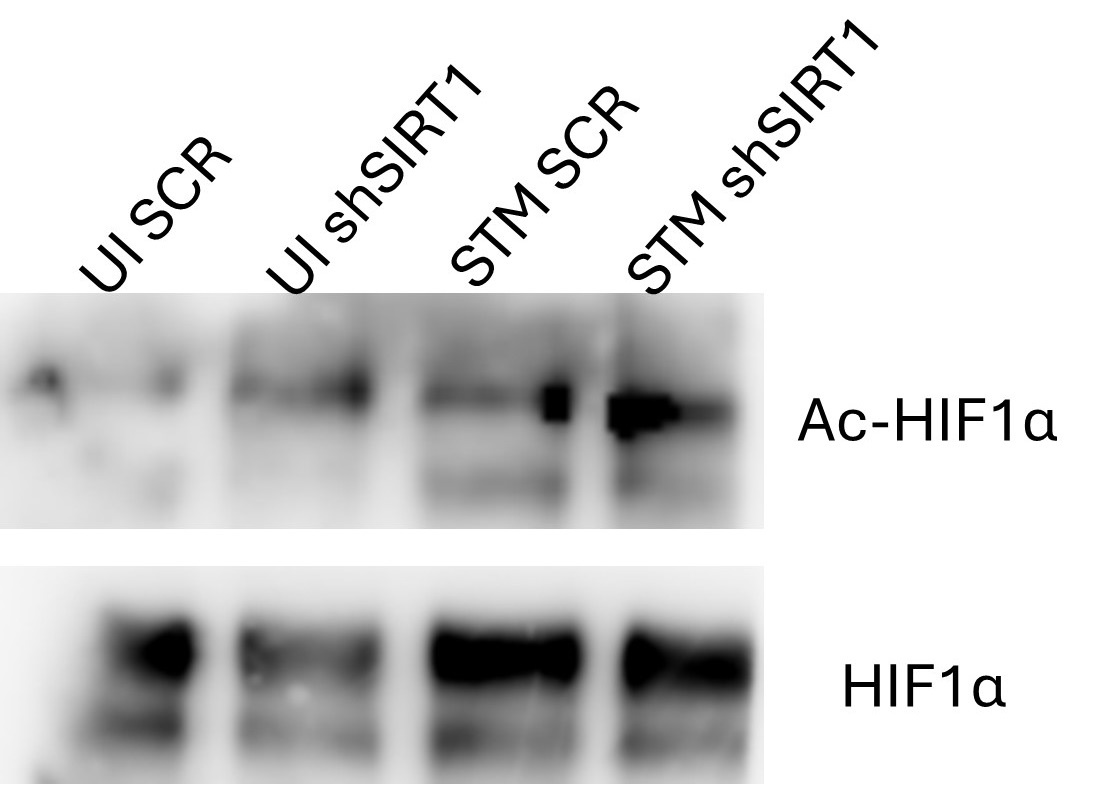
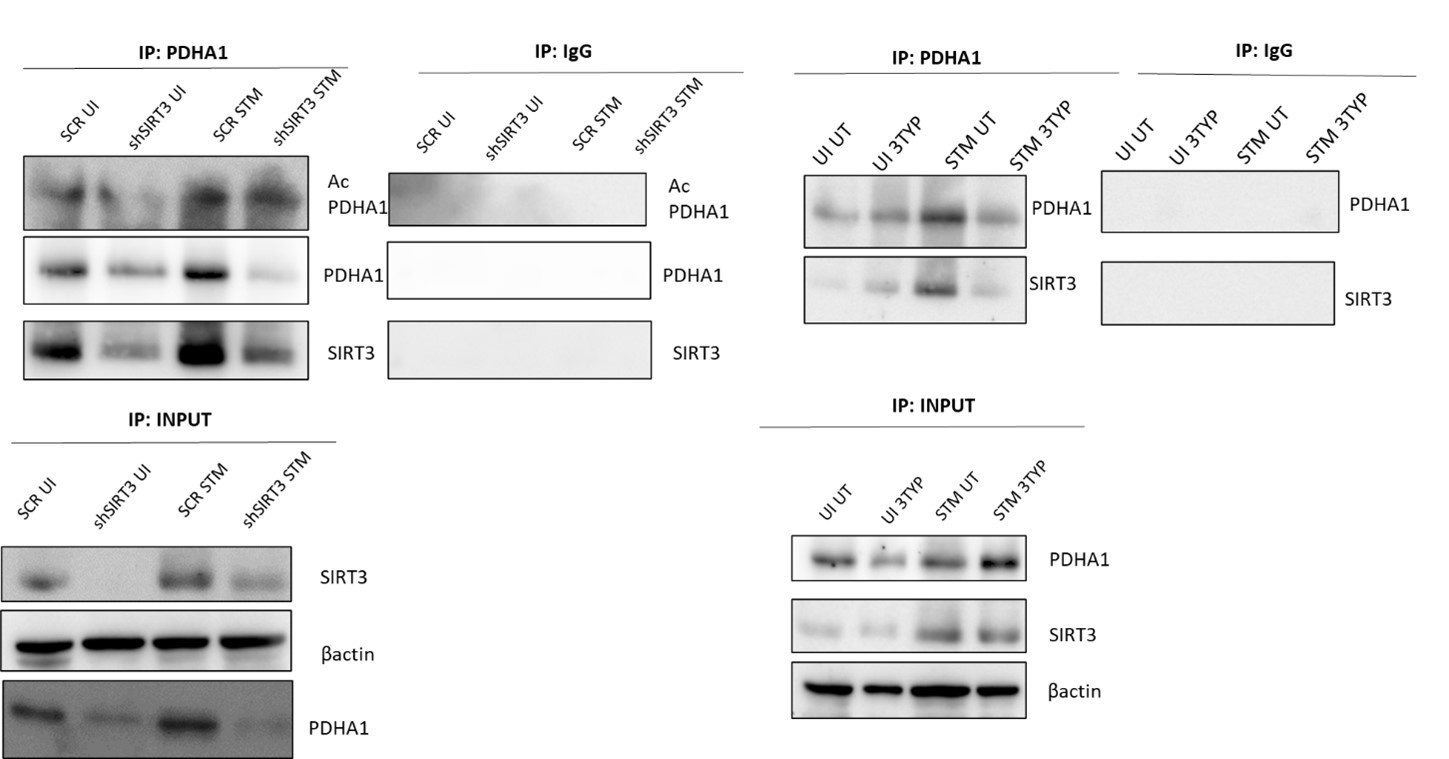
(2) We agree with the reviewer’s concern with the marginal changes in the western blots (Figures 7C, 7I (UI SCR vs UI shSIRT3), 7J). As per the suggestions, we have provided the alternate blot images and have indicated the number of repeats in the manuscript. The alternate blot images are provided herewith:
Author response image 6.
Alternate blot images for Fig. 7B-C
Author response image 7.
Alternate blot images for Fig. 7I, J
(1) We are highly thankful to the reviewer for recommending this suggestion. We have made the necessary modifications of relabelling Fig. C to Acetylated HIF-1α over total HIF-1α as per the suggestion.
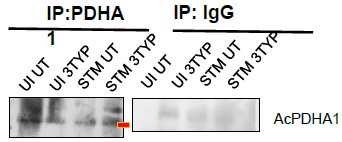
(2) 7J Acetylated PDHA1 has been duly pointed as per the suggestion. We are extremely apologetic for the inconvenience caused.
Author response image 8.
Reviewer #3 (Recommendations For The Authors):
The authors have done some work to improve the manuscript. However, the data presented lacks clarity.
Fig 4B: I still do not see a change in Ac p65 in the less saturated blot. It looks reduced as the band is distorted. I am not sure how this could be quantified.
Fig S2 b-actin bands are hyper saturated, and it is not possible to decipher the knockdown efficiency. It is probably better to provide a ponceau staining similar to S2C. The band intensity values are out of place.
Fig 5F HADHA blot: Lane 1 expression appears to be significantly higher than lane 3, but the values mentioned do not match the intensity of the bands.
It is hard to interpret the authors' claim that the shift in metabolism is HIF1a-dependent.
Fig 7B: I would expect HIF1a acetylation to be increased in UI ShSIRT1 compared to UI SCR. The blot shows reduced HIF1a acetylation.
Fig 7D: SIRT1 immunoprecipitates with HIF1a equally under all conditions. Is this what the authors expect? Labelling of the blots are not clear. It looks like the bottom SIRT1 blot is from Beads IgG control.
Fig 7H: How does PDHA1 interact with SIRT3 so strongly in shSIRT3 cells (lane 2)?
Authors have mentioned in their response that a knockdown of 40% has been achieved in the uninfected but the blot does not reflect that. SIRT3 expression seems to be more in the knockdown.
Blots are also not labelled properly especially Input. The lanes are not marked.
We thank the reviewer for acknowledging the improvements in the revised version and for suggesting further clarifications and improvements.
We have tried to incorporate the specified modifications to the best of our abilities in the revised manuscript.
We are highly apologetic for the inconclusive blot image in the figure 4B. We have provided an alternative blot image with better clarity for Fig.4B used for quantification analysis.
Author response image 9.
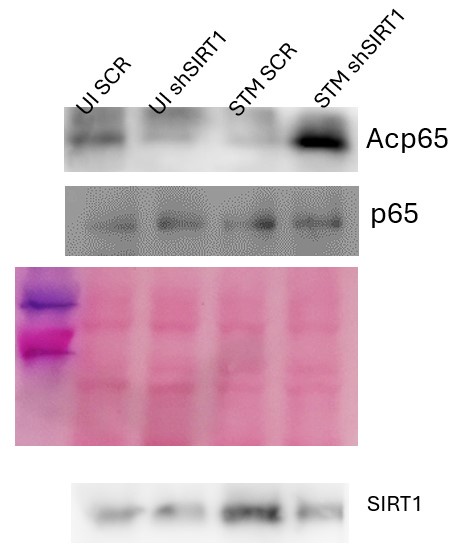
As per the reviewer’s valuable suggestions, we have provided the ponceau image in the Fig. S2B.
We thank the reviewers for rightly pointing out the discrepancy in the band intensity quantification in the Fig. 5F. We have re-evaluated the intensities on imageJ and have provided with the correct band intensities. We are highly apologetic for the inaccuracies.
As per the reviewer’s previous suggestion, we have performed additional experiments with HIF-1α inhibitor treatment in our revised manuscript to substantiate our correlative findings on SIRT1-mediated regulation of host glycolysis (Fig.7G). We wanted to clarify our claim in this regard. Our results suggested that loss of SIRT1 function triggered increased host glycolysis alongside hyperacetylation of HIF-1α. HIF-1α is reported to be one of the important players of glycolysis regulation (Kierans SJ, Taylor CT. Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol. 2021;599(1):23-37. doi:10.1113/JP280572.) and additionally, SIRT1 has been shown to regulate HIF-1α acetylation status (Lim JH, Lee YM, Chun YS, Chen J, Kim JE, Park JW. Sirtuin 1 modulates cellular responses to hypoxia by deacetylating hypoxia-inducible factor 1alpha. Mol Cell. 2010;38(6):864-878. doi:10.1016/j.molcel.2010.05.023.) Further, ectopic expression of SIRT1 has been demonstrated to reduce glycolysis by negatively regulating HIF-1α. (Wang Y, Bi Y, Chen X, et al. Histone Deacetylase SIRT1 Negatively Regulates the Differentiation of Interleukin-9-Producing CD4(+) T Cells. Immunity. 2016;44(6):1337-1349. doi:10.1016/j.immuni.2016.05.009). We have subsequently shown in Fig. 7G, that the increase in host glycolysis upon SIRT knockdown in the infected macrophages gets lowered upon HIF-1α inhibitor treatment, suggesting that one of the mechanisms of SIRT-mediated regulation of host glycolysis is via regulation of HIF-1α. However, this warrants further future mechanistic research.
We agree with the reviewer’s claim of increased HIF-1α acetylation in the UI sh1 versus UI SCR. The apparent reduced acetylation depicted in UI sh1 in Fig. 7B could be attributed to lower HIF-1α levels in the UI sh1 compared to UI SCR. Therefore, we have provided an alternate blot image that been used for quantification in Fig. 7C (Author response image 6).
To answer the reviewer’s question in Fig. 7D, we have noticed more or less equal degree of immunoprecipitation of HIF-1α under pull down of HIF-1α in all the sample cohorts under conditions of SIRT1 inhibitor treatment. However, we have observed reduced interaction of HIF-1α with SIRT1 in the infected sample upon SIRT1 inhibitor treatment.
We thank the reviewers for suggesting improvements in the blot labelling and for raising this concern. We have corrected the blot labelling to avoid the previous confusion.
We appreciate the reviewer’s concern and therefore we have provided an alternate blot image for Fig. 7H which might address the previous stated concern wherein we have achieved an enhanced SIRT3 knockdown percentage.
We are extremely apologetic for the improper labelling of the Input blot with unmarked lanes. We have addressed this issue by labelling the lanes in the input section of the blots.
-
-
eLife assessment
This study presents valuable findings on the role of the sirtuins SIRT1 and SIRT3 during Salmonella Typhimurium infection. Although the work increases our understanding of the mechanisms used by this pathogen to interact with its host and may have implications for other intracellular pathogens, the reviewers found that the evidence to support the claims is incomplete. In particular, the discrepancy between results obtained using cultured cell lines and the animal model of infection, as well as potential indirect effects through the microbiome stand out.
-
Reviewer #2 (Public Review):
Dipasree Hajra et al demonstrated that Salmonella was able to modulate the expression of Sirtuins (Sirt1 and Sirt3) and regulate the metabolic switch in both host and Salmonella, promoting its pathogenesis. The authors found Salmonella infection induced high levels of Sirt1 and Sirt3 in macrophages, which were skewed toward the M2 phenotype allowing Salmonella to hyper-proliferate. Mechanistically, Sirt1 and Sirt3 regulated the acetylation of HIF-1alpha and PDHA1, therefore mediating Salmonella-induced host metabolic shift in the infected macrophages. Interestingly, Sirt1 and Sirt3-driven host metabolic switch also had an effect on the metabolic profile of Salmonella. Counterintuitively, inhibition of Sirt1/3 led to increased pathogen burdens in an in vivo mouse model. Overall, this is a well-designed study.
Reviewer #2 (Public Review):
Dipasree Hajra et al demonstrated that Salmonella was able to modulate the expression of Sirtuins (Sirt1 and Sirt3) and regulate the metabolic switch in both host and Salmonella, promoting its pathogenesis. The authors found Salmonella infection induced high levels of Sirt1 and Sirt3 in macrophages, which were skewed toward the M2 phenotype allowing Salmonella to hyper-proliferate. Mechanistically, Sirt1 and Sirt3 regulated the acetylation of HIF-1alpha and PDHA1, therefore mediating Salmonella-induced host metabolic shift in the infected macrophages. Interestingly, Sirt1 and Sirt3-driven host metabolic switch also had an effect on the metabolic profile of Salmonella. Counterintuitively, inhibition of Sirt1/3 led to increased pathogen burdens in an in vivo mouse model. Overall, this is a well-designed study.
Comments on revised version:
The authors have performed additional experiments to address the discrepancy between in vitro and in vivo data. While this offers some potential insights into the in vivo role of Sirt1/3 in different cell types and how this affects bacterial growth/dissemination, I still believe that Sirt1/3 inhibitors could have some effect on the gut microbiota contributing to increased pathogen counts. This possibility can be discussed briefly to give a better scenario of how Sirt1/3 inhibitors work in vivo. Additionally, the manuscript would improve significantly if some of the flow cytometry analysis and WB data could be better analyzed.
-
Reviewer #3 (Public Review):
Summary:
In this paper Hajra et al have attempted to identify the role of Sirt1 and Sirt3 in regulating metabolic reprogramming and macrophage host defense. They have performed gene knock down experiments in RAW macrophage cell line to show that depletion of Sirt1 or Sirt3 enhances the ability of macrophages to eliminate Salmonella Typhimurium. However, in mice inhibition of Sirt1 resulted in dissemination of the bacteria but the bacterial burden was still reduced in macrophages. They suggest that the effect they have observed is due to increased inflammation and ROS production by macrophages. They also try to establish a weak link with metabolism. They present data to show that the switch in metabolism from glycolysis to fatty acid oxidation is regulated by acetylation of Hif1a, and PDHA1.
Strengths:
The …
Reviewer #3 (Public Review):
Summary:
In this paper Hajra et al have attempted to identify the role of Sirt1 and Sirt3 in regulating metabolic reprogramming and macrophage host defense. They have performed gene knock down experiments in RAW macrophage cell line to show that depletion of Sirt1 or Sirt3 enhances the ability of macrophages to eliminate Salmonella Typhimurium. However, in mice inhibition of Sirt1 resulted in dissemination of the bacteria but the bacterial burden was still reduced in macrophages. They suggest that the effect they have observed is due to increased inflammation and ROS production by macrophages. They also try to establish a weak link with metabolism. They present data to show that the switch in metabolism from glycolysis to fatty acid oxidation is regulated by acetylation of Hif1a, and PDHA1.
Strengths:
The strength of the manuscript is that the role of Sirtuins in host-pathogen interactions has not been previously explored in-depth making the study interesting. It is also interesting to see that depletion of either Sirt1 or Sirt3 results in a similar outcome.
Weaknesses:
The major weakness of the paper is the low quality of data, making it harder to substantiate the claims. Also, there are too many pathways and mechanisms being investigated. It would have been better if the authors had focussed on either Sirt1 or Sirt3 and elucidated how it reprograms metabolism to eventually modulate host response against Salmonella Typhimurium. Experimental evidence is also lacking to prove the proposed mechanisms. For instance they show correlative data that knock down of Sirt1 mediated shift in metabolism is due to HIF1a acetylation but this needs to be proven with further experiments.
-
Author response:
The following is the authors’ response to the original reviews.
Reviewer #1 (Public Review):
Summary:
The current manuscript by Hajra et al deals with the role of the prominent Sirtuins SIRT1 and -3 during infection of macrophages with Salmonella Typhimurium (ST). Apparently, ST infection induces upregulation of host cell SRTs to aid its own metabolism during the intracellular lifestyle and to help reprogramming macrophage polarization. The manuscript has two parts, namely one part that deals with Salmonella infection in cells, where RAW 264.7 murine macrophage-like cells, sharing some features with primary macrophages, were employed. Infected RAW cells displayed a tendency to polarize towards wound-healing M2 and not inflammatory M1 macrophages, which was dependent on SRT. Consequently, the inflammatory response in …
Author response:
The following is the authors’ response to the original reviews.
Reviewer #1 (Public Review):
Summary:
The current manuscript by Hajra et al deals with the role of the prominent Sirtuins SIRT1 and -3 during infection of macrophages with Salmonella Typhimurium (ST). Apparently, ST infection induces upregulation of host cell SRTs to aid its own metabolism during the intracellular lifestyle and to help reprogramming macrophage polarization. The manuscript has two parts, namely one part that deals with Salmonella infection in cells, where RAW 264.7 murine macrophage-like cells, sharing some features with primary macrophages, were employed. Infected RAW cells displayed a tendency to polarize towards wound-healing M2 and not inflammatory M1 macrophages, which was dependent on SRT. Consequently, the inflammatory response in RAW was more robust in the absence of SRT. Moreover, loss of SRTs leads to impaired bacterial proliferation in these cells, which was attributed to defects in metabolic adaption of the bacteria in the absence of SRT-activity and to the increased M1 inflammatory response.
Unfortunately, the line of argumentation remains incomplete because corresponding assays in mice showed the opposite result as compared to the experiments using RAW 264.7 cells. i.e. loss of SRTs leads to increased bacterial load in animals (versus impaired proliferation in RAW 264.7 cells). The authors cannot explain this discrepancy.
Strengths:
Extensive analysis of Salmonella infection in RAW macrophage-like cells and mice in the context of SRT1/3 function.
Weaknesses:
Lack of connection between the cell-based and organismic data, which are not supportive of each other.
We are highly grateful for your valuable and insightful comments. Thank you for appreciating the merit of our manuscript. We agree with the opposing phenotypes among the RAW264.7 cell line (Fig. 2A), primary peritoneal macrophages (ex vivo) (Fig.2B), and in vivo mouse model (Fig.8) findings. Both RAW264.7 macrophage and peritoneal macrophage infection show attenuated intracellular bacterial proliferation owing to the heightened proinflammatory burst. This is in sharp contrast to our in vivo mouse model of infection which shows increased organ burden and bacterial dissemination. The higher bacterial load in the organs including the spleen (Fig.8B) is attributed to increased pro-inflammatory cytokine burst and ROS production (Fig.8F-H, Fig.S9) triggering bacterial dissemination. The pro-inflammatory arsenals like IL-6, IL-1β and ROS that limit bacterial proliferation within the macrophages (F4/80+ macrophages within the spleen or in RAW264.7 macrophages or primary peritoneal macrophages) are facilitating bacterial dissemination in blood and to the other organs (Fig. 8I-L, Fig.S3F-G). This is in line with the following previous findings-
Klebsiella pneumoniae infection triggers an inflammatory response via secretion of IL-6 upon HIF-1α activation that induces bacterial dissemination (Holden VI, Breen P, Houle S, Dozois CM, Bachman MA. Klebsiella pneumoniae Siderophores Induce Inflammation, Bacterial Dissemination, and HIF-1α Stabilization during Pneumonia. mBio. 2016 Sep 13;7(5):e01397-16. doi: 10.1128/mBio.01397-16. PMID: 27624128; PMCID: PMC5021805.).
Correlation analysis of immune responses to Salmonella infection revealed that increased innate immune “cassette” opposes the adaptive immune arm leading to increased bacterial load in mice (Hotson AN, Gopinath S, Nicolau M, Khasanova A, Finck R, Monack D, et al. Coordinate actions of innate immune responses oppose those of the adaptive immune system during Salmonella infection of mice. Science signaling. 2016;9(410):ra4).
In our revised manuscript, we have assessed additional splenic populations including CD45+, Ly6C+, and CD11c+ populations. Our results show that the CD45+ splenic population depicts increased bacterial loads like that of the total splenic population within the SIRT1/3 inhibited cohorts. However, CD45+ monocytes and Ly6C positive splenic population exhibit compromised burden within the SIRT1/3 inhibited cohorts. Moreover, within the CD11c+ population, CD45+ granulocytes or lymphocytes show comparable organ loads to that of the vehicle control or SIRT1 activator-treated mice group (Fig. M-S, Fig.S8). Overall, our data suggest heterogeneous bacterial burden in diverse splenic populations.
Reviewer #2 (Public Review):
Dipasree Hajra et al demonstrated that Salmonella was able to modulate the expression of Sirtuins (Sirt1 and Sirt3) and regulate the metabolic switch in both host and Salmonella, promoting its pathogenesis. The authors found Salmonella infection induced high levels of Sirt1 and Sirt3 in macrophages, which were skewed toward the M2 phenotype allowing Salmonella to hyper-proliferate. Mechanistically, Sirt1 and Sirt3 regulated the acetylation of HIF-1alpha and PDHA1, therefore mediating Salmonella-induced host metabolic shift in the infected macrophages. Interestingly, Sirt1 and Sirt3-driven host metabolic switch also had an effect on the metabolic profile of Salmonella. Counterintuitively, inhibition of Sirt1/3 led to increased pathogen burdens in an in vivo mouse model. Overall, this is a well-designed study. There are a few comments below that would further strengthen the current study.
Major comments:
In the in vivo study (lines 436-446) - the authors noticed increased pathogen burden in the EX-527 or the 3TYP-treated mice cohorts but decreased pathogen burden within the F4/80+ macrophage population. What are the other cell types that have increased pathogen burden in splenocytes from EX-527 or the 3TYP treated? Can this be further explored and explained?
While the authors indicated that IL-6 cytokine storm and elevated ROS production could result in bacterial dissemination in vivo, one could also argue that Sirt1/3 inhibitors might have an impact on gut function and/or gut microbiota (PMID: 22115311). Did Sirt1/3 inhibitors also lead to increased pathogen burdens in the gut? If so, the potential effect of these in vivo treatments on gut microbiota/colonization resistance should be discussed.
Minor comment:
Sirt1 has been shown to be degraded during Salmonella infection (PMID: 28192515), which is different from the current study. An explanation should be provided for this.
We thank you for your encouraging and gracious comments. We deeply appreciate your time and efforts in providing constructive feedback for the betterment of our work. As per your precious suggestions, we have assessed additional splenic populations including CD45+, Ly6C+, and CD11c+ populations apart from F4/80+ macrophage populations. Our analysis suggests that the CD45+ splenic population show increased bacterial loads similar to the total splenic population within the SIRT1/3 inhibited cohorts. However, CD45+ monocytes and Ly6C positive splenic population exhibit compromised burden within the SIRT1/3 inhibited cohorts. Moreover, CD11c+ population, CD45+ granulocytes or lymphocytes show comparable organ loads to that of the vehicle control or SIRT1 activator treated mice group (Fig. 8M-S). Overall, our data suggest heterogeneous bacterial burden in diverse splenic populations.
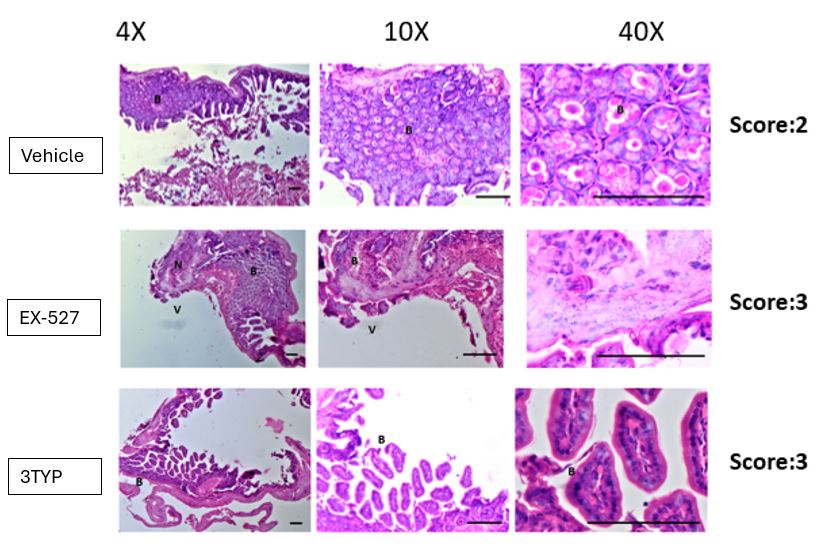
We immensely appreciate the reviewer for this insightful question about the effect of SIRT1/3 on the gut per se. To answer your question, we observed increased pathogen loads within the mesenteric lymph nodes of the gut in the SIRT1/3 inhibitor-treated mice groups (Fig.8B). In our revised manuscript, we evaluated gut inflammation via IL1-β estimation in the mice's ileal tissues and have observed heightened IL-1β production in the inhibitor-treated mice cohorts in comparison to the vehicle control (Fig. S3G). We have also examined gut epithelial pathology via Haematoxylin-Eosin (H&E) staining of the ileal sections to address the effect of in vivo treatment on gut microbiota and colonization resistance which is appended here. However, the gut microbiota crosstalk and their effect on colonization resistance is a part of another current study and it is being examined in detail there. Therefore, this appended H&E has not been incorporated in the revised manuscript.
Author response image 1.
In line with the reference PMID: 28192515, where Sirt1 has been shown to be degraded during Salmonella infection at later time points of infection, our study also has shown that both SIRT1 mRNA (Fig. 1A) and protein levels (Fig. S1A) show an elevated expression at 2h and 6h post-infection and show a downregulation at 16h in comparison to the 6h time point. However, SIRT3 expression levels remain elevated even at later time points of infection. Therefore, we speculate that there is a shared role between SIRT1 and SIRT3 that facilitates the phenotypes reported in our study.
Reviewer #3 (Public Review):
Summary:
In this paper, Hajra et al have attempted to identify the role of Sirt1 and Sirt3 in regulating metabolic reprogramming and macrophage host defense. They have performed gene knockdown experiments in RAW macrophage cell lines to show that depletion of Sirt1 or Sirt3 enhances the ability of macrophages to eliminate Salmonella Typhimurium. However, in mice, inhibition of Sirt1 resulted in dissemination of the bacteria but the bacterial burden was still reduced in macrophages. They suggest that the effect they have observed is due to increased inflammation and ROS production by macrophages. They also try to establish a weak link with metabolism. They present data to show that the switch in metabolism from glycolysis to fatty acid oxidation is regulated by acetylation of Hif1a, and PDHA1.
Strengths:
The strength of the manuscript is that the role of Sirtuins in host-pathogen interactions has not been previously explored in-depth making the study interesting. It is also interesting to see that depletion of either Sirt1 or Sirt3 results in a similar outcome.
Weaknesses:
The major weakness of the paper is the low quality of data, making it harder to substantiate the claims. Also, there are too many pathways and mechanisms being investigated. It would have been better if the authors had focussed on either Sirt1 or Sirt3 and elucidated how it reprograms metabolism to eventually modulate host response against Salmonella Typhimurium. Experimental evidence is also lacking to prove the proposed mechanisms. For instance, they show correlative data that the knockdown of Sirt1-mediated shift in metabolism is due to HIF1a acetylation but this needs to be proven with further experiments.
We appreciate the reviewer’s critical analysis of our work. In the revised manuscript, we aimed to eliminate the low-quality data sets and have tried to substantiate them with better and conclusive ones, as directed in the recommendations for the author section. We agree with the reviewer that the inclusion of both Sirtuins 1 and 3 has resulted in too many pathways and mechanisms and focusing on one SIRT and its mechanism of metabolic reprogramming and immune modulation would have been a less complicated alternative approach. However, as rightly pointed out, our work demonstrated the shared and few overlapping roles of the two sirtuins, SIRT1 and SIRT3, together mediating the immune-metabolic switch upon Salmonella infection. As per the reviewer’s suggestion, we have performed additional experiments with HIF-1α inhibitor treatment in our revised manuscript to substantiate our correlative findings on SIRT1-mediated regulation of host glycolysis (Fig.7G).
Reviewer #1 (Recommendations For The Authors):
The authors state "SIRT1 and SIRT3 inhibition resulted in increased pathogen loads in organs and triggered enhanced bacterial dissemination, together leading to increased susceptibility of the mice to S. Typhimurium infection owing to increased ROS and IL-6 production." How can this be reconciled? To the reviewer, this is not a convincing explanation. The reviewer is not a mouse pathologist, so maybe did not understand the argument in full.
However, in order to clarify whether these phenomena can be brought into context and explained by for instance cell-autonomous (in (RAW) macrophages) versus non-autonomous (in mice) mechanisms, it would be required to bring in context the organismic phenotype with a cellular phenotype, using more physiologic primary macrophages.
(1) The authors show in Figure 8 that in general SRT inhibition leads to increased infection whereas SRT activation results in decreased infection. This is even true for e the spleen (e.g. Figure 8B), which should be full of macrophages upon infection.
(2) Only Figure 8L implies that endogenous primary, splenic macrophages show a higher infection rate upon pharmacologic SRT activation, which would potentially mirror the RAW results. This is however not supportive of their own explanation: Who would now produce more ROS and IL6 if these macrophages are more supportive of intracellular ST? Is there a difference in the roles or SRTs between different types of macrophages and/or neutrophils? And between macrophages and somatic cells concerning ST infection? The reviewer tends to believe that RAW cells display a defective killing response (such as ROS production) as they are highly transformed cells. Therefore, the authors should use cultured peritoneal macrophages or BMDMs in addition to RAW264.7 cells.
The literature cited by the authors also implies that the inflammatory response in mice is higher in the absence of SRTs. This is in line with a role for SRTs in (negatively) regulating M1 inflammatory polarization but probably not with increased bacterial burden in mice. If it was, then increased dissemination could be explained by increased tissue damage. However, the flow cytometry experiments from infected organs then do not confirm that, as the infection of individual cells is higher upon SRT inhibition. Thus there seems a broad gap between the role of SRTs in ST infection in RAW264.7 cells versus non-transformed cells.
I would not discard the RAW results, as I am convinced that they contain valuable data. However, it needs to be clarified what aspect of the host response RAW 264.7 cells represent. Primary macrophages might likely be more aggressive towards the bacteria. Finally, the question arises: what is the role of the metabolic switch in the in vivo setting?
The reviewer recommends repeating some key experiments by in-vitro-infecting BMDMs or isolated peritoneal macrophages (after some days of culturing) to bridge between the present RAW-derived data and the mouse data. How is the bacterial load with and without SRT inhibitor/activator in primary macrophages, when infected outside of the body? Can ex-vivo infection also affect polarization of e.g. peritoneal macrophages or the metabolic switch? If it is possible to find a conclusive explanation for their data, then this story might really add to our understanding of another aspect of how ST manipulates the host to survive.
In case the reviewer understands the mouse experiments correctly, all assays on peritoneal cells were performed after in-vivo-infection and/or treatment.
Together, RAW 264.7 murine macrophage-like cells might not be the right model to understand the phenotypes in full. As far as the reviewer knows, these cells are not capable of killing bacteria as effectively as activated primary macrophages or neutrophils.
A few of the key findings of RAW264.7 macrophages have been replicated in primary peritoneal macrophages (Fig. 2B, S3E-F, S6B, S7B-D). We wanted to clarify that the peritoneal macrophage experiments were performed ex vivo, wherein peritoneal macrophages were isolated from mice were then subjected to SIRT1/3 inhibitor treatments and Salmonella infection and not after in vivo treatment or infection. In ex vivo setting, we have examined the effect of SIRTs on the metabolic switch during Salmonella infection (Fig. S7B-D) which resembled our RAW264.7 macrophage data. Additionally, in in vivo setting, we have analyzed the transcript level expression of host metabolic genes and corresponding bacterial metabolic genes in infected mice liver and spleen tissue under SIRT1/3 inhibitor treatment (Fig.S7E-F, Fig.6C-D). Our primary peritoneal macrophage data exactly mirrors the RAW264.7 macrophage findings showing attenuated intracellular bacterial proliferation owing to the heightened proinflammatory burst upon SIRT1/3 knockdown or inhibition (Fig.2A-B). This is opposite to our in vivo mouse model of infection which shows increased organ burden and bacterial dissemination (Fig.8A-H). The pro-inflammatory arsenals that limit bacterial proliferation within the macrophages (F4/80+ macrophages within the spleen or in RAW264.7 macrophages or primary peritoneal macrophages) are facilitating bacterial dissemination in blood and to the other organs owing to tissue damage (Fig.8E-L). This is in line with the following previous findings-
Klebsiella pneumoniae infection triggers an inflammatory response via secretion of IL-6 upon HIF-1α activation that induces bacterial dissemination (Holden VI, Breen P, Houle S, Dozois CM, Bachman MA. Klebsiella pneumoniae Siderophores Induce Inflammation, Bacterial Dissemination, and HIF-1α Stabilization during Pneumonia. mBio. 2016 Sep 13;7(5):e01397-16. doi: 10.1128/mBio.01397-16. PMID: 27624128; PMCID: PMC5021805.).
Correlation analysis of immune responses to Salmonella infection revealed that increased innate immune “cassette” opposes the adaptive immune arm leading to increased bacterial load in mice (Hotson AN, Gopinath S, Nicolau M, Khasanova A, Finck R, Monack D, et al. Coordinate actions of innate immune responses oppose those of the adaptive immune system during Salmonella infection of mice. Science Signaling. 2016;9(410):ra4).
As per the reviewer’s suggestions, we have analyzed other populations apart from F4/80+ macrophages and have observed that the CD45+ splenic population depicts increased bacterial loads like that of the total splenic population within the SIRT1/3 inhibited cohorts. However, CD45+ monocytes and Ly6C positive splenic population exhibit compromised burden within the SIRT1/3 inhibited cohorts. Moreover, the CD1c+ population, CD45+ granulocytes, or lymphocytes show comparable organ loads to that of the vehicle control or SIRT1 activator-treated mice group (Fig.8M-S, Fig.S8). Overall, our data suggest heterogeneous bacterial burden in diverse splenic populations.
Reviewer #3 (Recommendations For The Authors):
Abstract
The authors state that perturbing Sirt1 and Sirt3 results in a shift in Salmonella's metabolism. On the contrary, the data reflects the metabolism in the host cell and not the bacteria. This statement is wrong. They only show increased expression of some of the glycolytic genes in Salmonella, which is not sufficient to make the claim that the switch to fatty acid oxidation in macrophages is due to utilisation of glucose by the bacteria.
We value the reviewer’s response and have accordingly reframed our sentence in the abstract (Line 24-25).
Fig 1: Expression of Sirt1 - The data needs to be supported with a western blot for Sirt1 and Sirt3 but the Western blots shown in the supplementary figure are of very poor quality and do not support the authors' claim.
We have repeated the western blot and have supplemented the previous blot with an alternate blot in Fig. S1A as per your precious input.
Why haven't the authors shown any representative blots for Sirt1 and Sirt3 upon infection with Salmonella mutants? They need to italicize the genes when they describe mRNA expression.
Previously we had only performed transcript-level expression of Sirt1 and Sirt3 upon infection with Salmonella mutants and therefore representative blot image was absent. The gene names have been duly italicized while describing mRNA expression (Line 126-154). We regret the inconvenience caused. We have performed the western blotting to assess the protein expression profile upon infection with Salmonella mutants as per the reviewer’s suggestion and the representative blot image has been duly appended in the revised manuscript (Fig. S1B).
What is the rationale for examining Sirt1 and Sirt3 mRNA in M1 and M2 macrophages? Salmonella infection on its own will polarise the macrophages towards M1. How long were these macrophages infected? The time points are missing.
The rationale behind the examination of Sirt1 and Sirt3 mRNA in M1 and M2 polarized was to ascertain whether indeed M1 polarized macrophages exhibit decreased expression of Sirt1 or Sirt3 and polarization of macrophages toward M2 state show upregulation of Sirt1 and Sirt3 upon Salmonella infection. After confirming these above-mentioned findings through this preliminary experiment, we then hypothesized whether Salmonella infection on its own will polarise the macrophages toward an immunosuppressive M2 state at a later time course of infection as infection drives the induction of SIRT expression and whether this is mediated by Sirt1 and Sirt3 (Fig. 3). We are extremely apologetic for not mentioning the 16h time-point in the figure and the missing time point has been duly documented in the revised manuscript (Line 155).
Fig S2 knockdown of Sirt1 and Sirt3 are not convincing.
We are extremely sorry for the inconclusive knockdown blot. An alternative blot has been substantiated in the revised manuscript (Fig. S2,C-D).
Fig 2A and 2B the time point post infection has not been mentioned. Although it is stated that 2h and 16h post-infection samples were analysed. Only one time point has been shown.
We are sorry for the confusion. We wanted to clarify that Fig.2A and Fig. 2B show the fold proliferation where fold proliferation was calculated as CFU at 16hr divided by CFU at 2hr as mentioned in the materials and methods section under the heading of Intracellular proliferation or gentamicin protection assay.
Fold Proliferation= [CFU at 16h]/[CFU at 2h]
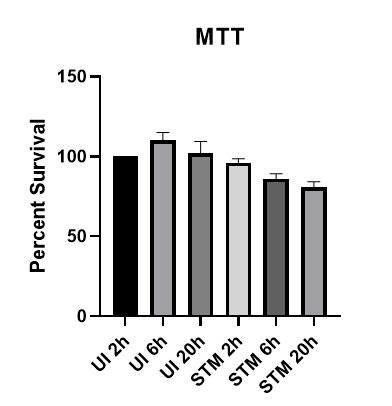
The cytokines data are intriguing in that the increase in IL-6 relative to control is seen only at 2h and 20h but not at 6h. Il-6 at 20h in untransfected cells is comparable to uninfected cells. Did the authors investigate cell death? Salmonella induces various forms of cell death which could account for the decreased cytokine production at later time points.
We have investigated the cell death upon Salmonella infection via MTT assay. At later time points of infection, we indeed observed around 16 percent decrease in cell survival compared to the initial time point of 2h. The results have been appended here and it supports our eminent reviewer’s reasoning for the decreased cytokine production at later time points.
Author response image 2.
Additional cytokines such as IL-1b would be helpful. Also, not sure how uninfected macrophages produce nearly 200pg of IL-10.
As per the author’s critical suggestion, we have assessed the IL-1b cytokine production at 16h post-infection in RAW264.7 macrophages and peritoneal macrophages and mice serum samples at 5th day post-infection (Fig.S3C, S3E-F). Our results indicate increased production of IL-b in the infected SIRT1/3 knockdown RAW264.7 macrophages, SIRT1/3 inhibitor-treated peritoneal macrophages and in mice serum samples under SIRT1/3 inhibitor treatment in comparison to the vehicle control. Additionally, we have quantified IL-1b in mice ileal tissues under SIRT1/3 inhibitor treatment (Fig.S3G) and have obtained heightened intestinal IL-1b production in the inhibitor-treated cohorts. We thank the reviewer for raising the concern for 200pg of IL-10 in the uninfected macrophages. We have repeated the experiment and have provided an alternative representative graph for the experiment wherein the IL-10 levels in the uninfected cohorts range between 20-40pg/ml (Fig. S3B).
It is surprising that the authors have found increased Sirt1 binding to NFkB, however there is no change in acetylated NFkB upon infection (Fig 4B). Acetylated p65 is equally high in uninfected Scrambled siRNA, UI shSirt1, STM Scr, and STM shSirt1. Furthermore, increased binding of Sirt1 with NFkb would mean decreased acetylation hence decreased inflammation. However, Salmonella induces profound inflammation.
We thank the reviewers for their insightful and critical questioning. We truly acknowledge that due to oversaturation there was no apparent change in the acetylated p65 among the different sample sets. Therefore, in the revised manuscript we have provided an image at lower exposure where the changes in the acetylation of the p65 subunit are apparent. Salmonella induces inflammation upon challenge similar to any other pathogens and induces acute inflammatory responses. This heightened acute inflammation at the initial phases of infection subsides at a later phase of infection. Here, we have performed the Sirt1 interaction with NFκB at 16hr post-infection where increased binding of Sirt1 with NFκB facilitates the resolution of the _Salmonella-_induced acute inflammation. This is in line with previous reports that suggest SIRT1 suppresses acute inflammation through the promotion of p65 acetylation and inhibition of NFκB activity. (Yang H, Zhang W, Pan H, et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-κB activity. PLoS One. 2012;7(9):e46364. doi:10.1371/journal.pone.0046364, Liu TF, Yoza BK, El Gazzar M, Vachharajani VT, McCall CE. NAD+-dependent SIRT1 deacetylase participates in epigenetic reprogramming during endotoxin tolerance. J Biol Chem. 2011;286(11):9856–64., Liu TF, Vachharajani V, Millet P, Bharadwaj MS, Molina AJ, McCall CE. Sequential actions of SIRT1-RELB-SIRT3 coordinate nuclear-mitochondrial communication during immunometabolic adaptation to acute inflammation and sepsis. J Biol Chem. 2015;290(1):396–408.)
Please explain how the acetylated p65 was analysed.
Total endogenous p65 subunit was immunoprecipitated using Anti-NFκB p65 antibody and the immunoprecipitated fraction was probed with Anti-Acetylated Lysine antibody to assess acetylated p65.
An increase in ROS production is seen in a relatively small percentage of cells- not more than 4% of cells. How does this contribute to such a significant difference in intracellular bacterial burden? Also, it is not clear how the authors calculated the fold change in proliferation. It is better to show the actual bacterial burden logarithmically.
We strongly agree with the reviewer’s concerns, and we have reanalyzed the flow cytometric data set. The revised data have been presented in Fig. S5 which shows a considerable increase in DCFDA positive population. For instance, the infected scrambled control shows around 2.44% of ROS-producing cells, however knockdown of SIRT1 and SIRT3 increases the ROS-producing cells to 27.34% and 28.64% respectively.
Fold proliferation was calculated as CFU at 16hr divided by CFU at 2hr as mentioned in the materials and methods section under the heading of Intracellular proliferation or gentamicin protection assay. Fold proliferation has been calculated as opposed to absolute CFU values to nullify the differential phagocytosis of bacteria to the macrophages among the samples.
Fold Proliferation= [CFU at 16h]/[CFU at 2h]
An increase in metabolic genes is not sufficient to show that the macrophages are metabolically reprogrammed.
We thank the reviewer for the valuable comment. We agree that an increase in metabolic gene profile is not sufficient to claim metabolic reprogramming. Therefore, in addition to the metabolic gene profile, we have estimated lactate production (end-product of glycolysis) as an indicator of glycolysis (Fig. 5 C-E) and have performed the fatty acid β oxidation activity (Fig. 5G-H) to support our claims.
Figure 5F the band intensities do not visually match the bands shown for PFK. For instance, shSIRT1 STM (1.00) and shSIRT3 STM (0.81).
We are extremely sorry for the erroneous band intensity for shSIRT3. Upon reanalysis of the band intensities, we have corrected the band intensity for shSIRT3 to 2.28 (Fig.5F).
It is surprising that HADHA is not expressed in uninfected samples.
We are extremely apologetic for the inappropriate representative blot. We feel that the discrepancy might have arisen due to the usage of old antibodies. We have provided an alternate blot for the HADHA gene where fresh antibody staining solution was used for probing which shows expression even in the uninfected samples (Fig.5F).
Figure 6A - What is the significance of PFA fixed samples (PI) compared to SI samples? This has not been discussed.
PFA-fixed samples are paraformaldehyde-treated bacterial samples that harbor the immune signals or Pattern Associated Molecular Patterns (PAMPs). The rationale for using PI in addition to SI samples was to show whether the phenomena is driven by live metabolically active pathogens or is mediated by PAMPs.
I understand that the hypothesis is that during the later phase of infection, there is an increase in fatty acid oxidation which correlates with a decrease in inflammation. However, at 6h there is no increase in genes regulating fatty acid oxidation. Why did the authors choose 6h when the previous experiments have been done at 16h?
We indeed agree with the reviewer’s understanding of our hypothesis that there is an increase in fatty acid oxidation along the progression of infection which correlates with a decrease in inflammation. The Salmonella intracellular replication has been reported to commence at 6h post-internalization when SPI-2 effector expression is fully established (Helaine S, Thompson JA, Watson KG, Liu M, Boyle C, Holden DW. Dynamics of intracellular bacterial replication at the single cell level. Proc Natl Acad Sci U S A. 2010;107(8):3746-3751. doi:10.1073/pnas.1000041107). Therefore, we have assessed the 6h timepoint post-infection in addition to the initial and later timepoints of 2h and 16h respectively. Additionally, the nanostring gene profiling data of both host and bacterial genes indicate the onset of both metabolic (Fig. 5A, 6A) and immune genes (Fig. 3A) modulation at 6h post-infection. We have validated these results via qPCR studies and have observed an upregulation in the transcript level of fatty acid oxidation genes as depicted in Fig. S7A in RAW264.7 macrophages.
Line 355 it is mentioned that Sirt1 and Sirt3 abrogate metabolic shift by reducing glycolytic flux. This is incorrect as experiments such as carbon chase assays have not been performed to investigate glycolytic flux.
As per the reviewer’s valuable suggestion, we have removed the word ‘flux’ from the above-mentioned statement(Line 351, Line 353).
Lines 392-393: "We immunoprecipitated PDHA1 and checked for its interaction with SIRT3 or SIRT1 under knockdown condition of SIRT3 or upon SIRT3 inhibitor treatment (Fig.7 G-H)"
What is the rationale for checking PDHA1 interaction with Sirt under Sirt knockdown conditions?
We are thankful to the reviewer for the critical comments. The rationale for checking PDHA1 interaction with Sirt was to ascertain that indeed Sirt interacted with PDHA1 under S. Typhimurium infection and abrogation of either protein expression (knockdown) or their enzymatic activity (inhibitor treatment) diminished the interaction.
Moreover, the blots are very confusing and do not represent the authors' claims.
(1) In the input blot I do not see Sirt3 depletion in shSirt3 knockdown sample.
The knockdown has been quantified in the input blot as per your suggestion. A knockdown of 40% has been obtained in the uninfected dataset whereas a knockdown of 47.1% has been obtained in the infected data set at 16h post-infection (Fig.7H).
(2) Why does Sirt1 interact with PDHA1 similar to Sirt3. Do both the proteins bind to PDHA1 at the same time/ competitively? If so do they both deacetylate?
In literature, Sirt3 has been shown to interact with PDHA1 and deacetylate PDHA1. However, the interaction of Sirt1 with PDHA1 has not been reported previously and therefore we are unable to comment on the exact dynamics of the interaction. Future studies need to be performed to explore these phenomena in depth. However, SIRT1 agonist SRT1720 has been shown to impact PDH phosphorylation and its activity (Han Y, Sun W, Ren D, Zhang J, He Z, Fedorova J, Sun X, Han F, Li J. SIRT1 agonism modulates cardiac NLRP3 inflammasome through pyruvate dehydrogenase during ischemia and reperfusion. Redox Biol. 2020 Jul;34:101538).
(3) Figure 7I in the IP: IgG samples Sirt3 seem to bind to IgG non-specifically, which questions the specificity of Sirt3 binding to PDHA1.
We appreciate the reviewer for pointing out this concern. The immunoprecipitation experiment has been repeated and the same has been appended in the revised manuscript and we observe no non-specific binding of Sirt3 antibody to IgG.
(4) In Figure 7I all the bands Ac PDHA1, PDHA1, and Sirt3 look similar with double bands, which has not been seen in other blots. How is this possible?
This cannot explain the increase in beta-oxidation observed.
We thank the reviewer for raising this concern. We have repeated the experiment and provided the alternative blot as per the reviewer’s suggestion.
The rationale for performing this experiment was to show that SIRT plays an important role in the activation of downstream TCA cycle pathways via PDHA1 deacetylation during Salmonella infection. The deacetylation of PDHA1 has been previously reported to cause transcriptional activation of the downstream TCA cycle and oxidative phosphorylation (Zhang Y, Wen P, Luo J, et al., Cell Death Dis.,2021). Additionally, PDHA1 hyperacetylation has been reported to cause lactate overproduction (An, S., Yao, Y., Hu, H. et al. PDHA1 hyperacetylation-mediated lactate overproduction promotes sepsis-induced acute kidney injury via Fis1 lactylation. Cell Death Dis 14, 457 (2023)). In our study, increased lactate production and PDHA1 hyperacetylation have been observed during SIRT3 inhibition conditions upon Salmonella infection.
-
-
eLife assessment
This study presents valuable findings on the role of the sirtuins SIRT1 and SIRT3 during Salmonella Typhimurium infection. Although the work increases our understanding of the mechanisms used by this pathogen to interact with its host and may have implications for other intracellular pathogens, the reviewers found that the evidence to support the claims is incomplete. In particular, the discrepancy between results obtained using cultured cell lines and the animal model of infection stands out.
-
Reviewer #1 (Public Review):
Summary:
The current manuscript by Hajra et al deals with the role of the prominent Sirtuins SIRT1 and -3 during infection of macrophages with Salmonella Typhimurium (ST). Apparently, ST infection induces upregulation of host cell SRTs to aid its own metabolism during the intracellular lifestyle and to help reprogramming macrophage polarization. The manuscript has two parts, namely one part that deals with Salmonella infection in cells, where RAW 264.7 murine macrophage-like cells, sharing some features with primary macrophages, were employed. Infected RAW cells displayed a tendency to polarize towards wound-healing M2 and not inflammatory M1 macrophages, which was dependent on SRT. Consequently, the inflammatory response in RAW was more robust in the absence of SRT. Moreover, loss of SRTs leads to impaired …Reviewer #1 (Public Review):
Summary:
The current manuscript by Hajra et al deals with the role of the prominent Sirtuins SIRT1 and -3 during infection of macrophages with Salmonella Typhimurium (ST). Apparently, ST infection induces upregulation of host cell SRTs to aid its own metabolism during the intracellular lifestyle and to help reprogramming macrophage polarization. The manuscript has two parts, namely one part that deals with Salmonella infection in cells, where RAW 264.7 murine macrophage-like cells, sharing some features with primary macrophages, were employed. Infected RAW cells displayed a tendency to polarize towards wound-healing M2 and not inflammatory M1 macrophages, which was dependent on SRT. Consequently, the inflammatory response in RAW was more robust in the absence of SRT. Moreover, loss of SRTs leads to impaired bacterial proliferation in these cells, which was attributed to defects in metabolic adaption of the bacteria in the absence of SRT-activity and to the increased M1 inflammatory response.Unfortunately, the line of argumentation remains incomplete because corresponding assays in mice showed the opposite result as compared to the experiments using RAW 264.7 cells. i.e. loss of SRTs leads to increased bacterial load in animals (versus impaired proliferation in RAW 264.7 cells). The authors cannot explain this discrepancy.
Strengths:
Extensive analysis of Salmonella infection in RAW macrophage-like cells and mice in the context of SRT1/3 function.Weaknesses:
Lack of connection between the cell-based and organismic data, which are not supportive of each other. -
Reviewer #2 (Public Review):
Dipasree Hajra et al demonstrated that Salmonella was able to modulate the expression of Sirtuins (Sirt1 and Sirt3) and regulate the metabolic switch in both host and Salmonella, promoting its pathogenesis. The authors found Salmonella infection induced high levels of Sirt1 and Sirt3 in macrophages, which were skewed toward the M2 phenotype allowing Salmonella to hyper-proliferate. Mechanistically, Sirt1 and Sirt3 regulated the acetylation of HIF-1alpha and PDHA1, therefore mediating Salmonella-induced host metabolic shift in the infected macrophages. Interestingly, Sirt1 and Sirt3-driven host metabolic switch also had an effect on the metabolic profile of Salmonella. Counterintuitively, inhibition of Sirt1/3 led to increased pathogen burdens in an in vivo mouse model. Overall, this is a well-designed study. …
Reviewer #2 (Public Review):
Dipasree Hajra et al demonstrated that Salmonella was able to modulate the expression of Sirtuins (Sirt1 and Sirt3) and regulate the metabolic switch in both host and Salmonella, promoting its pathogenesis. The authors found Salmonella infection induced high levels of Sirt1 and Sirt3 in macrophages, which were skewed toward the M2 phenotype allowing Salmonella to hyper-proliferate. Mechanistically, Sirt1 and Sirt3 regulated the acetylation of HIF-1alpha and PDHA1, therefore mediating Salmonella-induced host metabolic shift in the infected macrophages. Interestingly, Sirt1 and Sirt3-driven host metabolic switch also had an effect on the metabolic profile of Salmonella. Counterintuitively, inhibition of Sirt1/3 led to increased pathogen burdens in an in vivo mouse model. Overall, this is a well-designed study. There are a few comments below that would further strengthen the current study.
Major comments:
In the in vivo study (lines 436-446) - the authors noticed increased pathogen burden in the EX-527 or the 3TYP-treated mice cohorts but decreased pathogen burden within the F4/80+ macrophage population. What are the other cell types that have increased pathogen burden in splenocytes from EX-527 or the 3TYP treated? Can this be further explored and explained?While the authors indicated that IL-6 cytokine storm and elevated ROS production could result in bacterial dissemination in vivo, one could also argue that Sirt1/3 inhibitors might have an impact on gut function and/or gut microbiota (PMID: 22115311). Did Sirt1/3 inhibitors also lead to increased pathogen burdens in the gut? If so, the potential effect of these in vivo treatments on gut microbiota/colonization resistance should be discussed.
Minor comment:
Sirt1 has been shown to be degraded during Salmonella infection (PMID: 28192515), which is different from the current study. An explanation should be provided for this. -
Reviewer #3 (Public Review):
Summary:
In this paper, Hajra et al have attempted to identify the role of Sirt1 and Sirt3 in regulating metabolic reprogramming and macrophage host defense. They have performed gene knockdown experiments in RAW macrophage cell lines to show that depletion of Sirt1 or Sirt3 enhances the ability of macrophages to eliminate Salmonella Typhimurium. However, in mice, inhibition of Sirt1 resulted in dissemination of the bacteria but the bacterial burden was still reduced in macrophages. They suggest that the effect they have observed is due to increased inflammation and ROS production by macrophages. They also try to establish a weak link with metabolism. They present data to show that the switch in metabolism from glycolysis to fatty acid oxidation is regulated by acetylation of Hif1a, and PDHA1.Strengths:
The …Reviewer #3 (Public Review):
Summary:
In this paper, Hajra et al have attempted to identify the role of Sirt1 and Sirt3 in regulating metabolic reprogramming and macrophage host defense. They have performed gene knockdown experiments in RAW macrophage cell lines to show that depletion of Sirt1 or Sirt3 enhances the ability of macrophages to eliminate Salmonella Typhimurium. However, in mice, inhibition of Sirt1 resulted in dissemination of the bacteria but the bacterial burden was still reduced in macrophages. They suggest that the effect they have observed is due to increased inflammation and ROS production by macrophages. They also try to establish a weak link with metabolism. They present data to show that the switch in metabolism from glycolysis to fatty acid oxidation is regulated by acetylation of Hif1a, and PDHA1.Strengths:
The strength of the manuscript is that the role of Sirtuins in host-pathogen interactions has not been previously explored in-depth making the study interesting. It is also interesting to see that depletion of either Sirt1 or Sirt3 results in a similar outcome.Weaknesses:
The major weakness of the paper is the low quality of data, making it harder to substantiate the claims. Also, there are too many pathways and mechanisms being investigated. It would have been better if the authors had focussed on either Sirt1 or Sirt3 and elucidated how it reprograms metabolism to eventually modulate host response against Salmonella Typhimurium. Experimental evidence is also lacking to prove the proposed mechanisms. For instance, they show correlative data that the knockdown of Sirt1-mediated shift in metabolism is due to HIF1a acetylation but this needs to be proven with further experiments. -
