The BTB-ZF gene Bm-mamo regulates pigmentation in silkworm caterpillars
Curation statements for this article:-
Curated by eLife

eLife assessment
This important study identifies the gene mamo as a new regulator of pigmentation in the silkworm Bombyx mori, a function that was previously unsuspected based on extensive work on Drosophila where the mamo gene is involved in gamete production. The evidence supporting the role of Bm-nano in pigmentation is convincing, including high-resolution linkage mapping of two mutant strains, expression profiling, and reproduction of the mutant phenotypes with state-of-the-art RNAi and CRISPR knock-out assays. The work will be of interest to evolutionary biologists and geneticists studying color patterns and evolution of gene networks.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The color pattern of insects is one of the most diverse adaptive evolutionary phenotypes. However, the molecular regulation of this color pattern is not fully understood. In this study, we found that the transcription factor Bm-mamo is responsible for black dilute ( bd ) allele mutations in the silkworm. Bm-mamo belongs to the BTB zinc finger family and is orthologous to mamo in Drosophila melanogaster . This gene has a conserved function in gamete production in Drosophila and silkworms and has evolved a pleiotropic function in the regulation of color patterns in caterpillars. Using RNAi and clustered regularly interspaced short palindromic repeats (CRISPR) technology, we showed that Bm-mamo is a repressor of dark melanin patterns in the larval epidermis. Using in vitro binding assays and gene expression profiling in wild-type and mutant larvae, we also showed that Bm-mamo likely regulates the expression of related pigment synthesis and cuticular protein genes in a coordinated manner to mediate its role in color pattern formation. This mechanism is consistent with the dual role of this transcription factor in regulating both the structure and shape of the cuticle and the pigments that are embedded within it. This study provides new insight into the regulation of color patterns as well as into the construction of more complex epidermal features in some insects.
Article activity feed
-

-
-
-

Author Response
The following is the authors’ response to the previous reviews.
eLife assessment
This important study identifies the gene mamo as a new regulator of pigmentation in the silkworm Bombyx mori, a function that was previously unsuspected based on extensive work on Drosophila where the mamo gene is involved in gamete production. The evidence supporting the role of Bm-nano in pigmentation is convincing, including high-resolution linkage mapping of two mutant strains, expression profiling, and reproduction of the mutant phenotypes with state-of-the-art RNAi and CRISPR knock-out assays. While the discussion about genetic changes being guided or accelerated by the environment is extremely speculative and has little relevance for the findings presented, the work will be of interest to evolutionary biologists and geneticists …
Author Response
The following is the authors’ response to the previous reviews.
eLife assessment
This important study identifies the gene mamo as a new regulator of pigmentation in the silkworm Bombyx mori, a function that was previously unsuspected based on extensive work on Drosophila where the mamo gene is involved in gamete production. The evidence supporting the role of Bm-nano in pigmentation is convincing, including high-resolution linkage mapping of two mutant strains, expression profiling, and reproduction of the mutant phenotypes with state-of-the-art RNAi and CRISPR knock-out assays. While the discussion about genetic changes being guided or accelerated by the environment is extremely speculative and has little relevance for the findings presented, the work will be of interest to evolutionary biologists and geneticists studying color patterns and evolution of gene networks.
Response: Thank you very much for your careful work. In the revised version, we conducted a comparative genomic analysis of the upstream regions of the Bm-mamo gene in 51 wild silkworms and 171 domesticated local silkworms. The analysis of nucleotide diversity (pi) and the fixation index (FSTs) of the Bm-mamo genome sequences in the wild and domesticated silkworm populations were also performed. The results showed that the Bm-mamo genome sequence of local silkworms was relatively conserved, while the upstream sequence of wild silkworms exhibited high nucleotide diversity. This finding suggested a high degree of variability in the regulatory region of the Bm-mamo gene, in wild strains. Additionally, the sequence in this region may have been fixed by domestication selection. We have optimized the description in the discussion section.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
This papers performs fine-mapping of the silkworm mutants bd and its fertile allelic version, bdf, narrowing down the causal intervals to a small interval of a handful of genes. In this region, the gene orthologous to mamo is impaired by a large indel, and its function is later confirmed using expression profiling, RNAi, and CRISPR KO. All these experiments are convincingly showing that mamo is necessary for the suppression of melanic pigmentation in the silkworm larval integument.
The authors also use in silico and in vitro assays to probe the potential effector genes that mamo may regulate.
Strengths:
The genotype-to-phenotype workflow, combining forward (mapping) and reverse genetics (RNAi and CRISPR loss-of-function assays) linking mamo to pigmentation are extremely convincing.
This revision is a much improved manuscript and I command the authors for many of their edits.
Response: Thank you very much for your careful work. With the help of reviewers and editors, we have revised the manuscript to improve its readability.
I find the last part of the discussion, starting at "It is generally believed that changes in gene expression patterns are the result of the evolution of CREs", to be confusing.
In this section, I believe the authors sequentially:
- emphasize the role of CRE in morphological evolution (I agree)
- emphasize that TF, and in particular their own CRE, are themselves important mutational targets of evolution (I agree, but the phrasing need to insist the authors are here talking about the CRE found at the TF locus, not the CRE bound by the TF).
- use the stickleback Pel enhancer as an example, which I think is a good case study, but the authors also then make an argument about DNA fragility sites, which is hard to connect with the present study.
- then continue on "DNA fragility" using the peppered moth and butterfly cortex locus. There is no evidence of DNA fragility at these loci, so the connection does not work. "The cortex gene locus is frequently mutated in Lepidoptera", the authors say. But a more accurate picture would be that the cortex locus is repeatedly involved in the generation of color pattern variants. Unlike for Pel fragile enhancer, we don't know if the causal mutations at this locus are repeatedly the same, and the haplotypes that have been described could be collateral rather than causal. Overall, it is important to clarify the idea that mutation bias is a possible factor explaining "genetic hotspots of evolution" (or genetic parallelism sensu 10.1038/nrg3483), but it is also possible that many genetic hotspots are repeated mutational targets because of their "optimal pleiotropy" (e.g. hub position in GRNs, such as mamo might be), or because of particularly modular CRE region that allow fine-tuning. Thus, I find the "fragility" argument misleading here. In fact the finding that "bd" and "bdf" alleles are different in nature is against the idea of a fragility bias (unless the authors can show increased mutation rates at this locus in a wild silkmoth species?). These alleles are also artificially-selected ie. they increased in frequency by breeding rather than natural selection in the wild, so while interesting for our understand of the genotype-phenotype map, they are not necessarily representative of the mutations that may underlie evolution in the wild.
Response: Thank you very much for your careful work. DNA fragility is an interesting topic, but some explanations for DNA fragility are confusing. One study measured the rate of DNA double-strand breaks (DSBs) in yeast artificial chromosomes (YACs), which are chromosomes containing marine Pel that broke ~25 to 50 times more frequently than did the control. These authors believe that the increase in the mutation rate is caused by DNA sequence characteristics, particularly TG-dinucleotide repeats. Moreover, they found that adding a replication origin on the opposite side of Pel did not cause the fungus to switch fragile, making the forward sequence stable and the reverse complement fragile. Thus, Pel fragility is also dependent on the direction of DNA replication. In summary, they suggested that the special DNA sequence is the cause of DNA fragility. In addition, the sequence features associated with DNA fragility in the Pel region are also found in thousands of other positions in the stickleback and human genomes (Xie KT et al, 2019, science).
In yeast artificial chromosomes (YACs), the characteristics of DNA sequences, such as TG-dinucleotide repeat sequences, may be important reasons for DNA fragility, and these breaks occur during DNA replication. However, the inserted sequence of YAC often undergoes deletion or recombination during cultivation and passage. In addition, yeast is a single-celled organism. Therefore, the results in yeast cannot represent the situation in multicellular organisms. If multicellular organisms are like this, there are several issues as follows:
(1) The DNA replication process occurs separately in different multicellular organisms. Because DNA breakage and repair are independent, they can lead to the presence of different alleles in different cells. This can potentially lead to the occurrence of extensive chimeric organisms. However, we have not found such a situation in the genome sequencing of many multicellular organisms.
(2) If the DNA sequence, TG-dinucleotide repeats, is the determining factor, the mutations near the sequence lose their strong correlation with environmental changes. The researchers conducted yeast artificial chromosome experiments in the same environment and found that the frequency of DNA breaks containing TG dinucleotide repeat sequences was 25 to 50 times greater than that of the control group. This means that, whether in the marine population or the lake population, this part of the sticklebacks’ genome has undergone frequent mutations. However, according to related research, populations of lake sticklebacks, rather than marine populations, often exhibit a decrease in the pelvic phenotype.
(3) Researchers have found thousands of loci in the genome of sticklebacks and humans that contain such sequences (TG-dinucleotide repeats). This means that thousands of sites undergo frequent mutations during DNA replication. Unless these sites do not possess functionality, they will have some impact on the organism, even causing damage. Even if they are not functional sequences, these sequences will gradually be discarded or replaced during frequent mutations rather than being present in large quantities in the genome.
Therefore, the study of DNA fragility in yeast cannot explain the situation in multicellular organisms.
As you noted, we want to express that the frequent variation in the cortex gene should be regulated by targeted regulation involving the GRN in Lepidoptera. In addition, studies on specific epigenetic modifications discovered through the referenced fragile DNA sites suggest that DNA fragility is not determined by the DNA sequence (Ji F, 2020, Cell Res) but rather by other factors, such as epigenetic factors. The sequence features discovered at fragile DNA sites are traces of frequent mutations, not causes.
In this revision, we analyzed the nucleotide diversity of the mamo genome in 51 wild and 171 domestic silkworms. We found high nucleic acid diversity from the third exon to the upstream region of this gene in wild silkworms. We randomly selected 12 wild silkworms and 12 domestic silkworms and compared their upstream sequences to approximately 1 kb. In wild silkworms, there is significant diversity in their upstream sequences. In domestic silkworms, the sequences are highly conserved, but in some silkworms, a long interspersed nuclear element (LINE) is inserted. This finding suggested that there is frequent variation in the sequence of this region in wild silkworms, while fixation occurs in domesticated silkworms. These genomic data are sourced from the pangenome of silkworms (Tong X, 2022, Nat Commun.). In the pangenomic research, 1078 strains (205 local strains, 194 improved strains, 632 mutant strains, and 47 wild silkworms), which included 545 third-generation sequencing genomes, were obtained. An online website was built to utilize these data (http://silkmeta.org.cn/). We warmly welcome you to use these data.
In summary, for clearer expression, we have rewritten this section.
Xie KT, Wang G, Thompson AC, Wucherpfennig JI, Reimchen TE, MacColl ADC, Schluter D, Bell MA, Vasquez KM, Kingsley DM. DNA fragility in the parallel evolution of pelvic reduction in stickleback fish. Science. 2019 Jan 4;363(6422):81-84. doi: 10.1126/science.aan1425.
Ji F, Liao H, Pan S, Ouyang L, Jia F, Fu Z, Zhang F, Geng X, Wang X, Li T, Liu S, Syeda MZ, Chen H, Li W, Chen Z, Shen H, Ying S. Genome-wide high-resolution mapping of mitotic DNA synthesis sites and common fragile sites by direct sequencing. Cell Res. 2020 Nov;30(11):1009-1023. doi: 10.1038/s41422-020-0357-y.
Tong X, Han MJ, Lu K, Tai S, Liang S, Liu Y, Hu H, Shen J, Long A, Zhan C, Ding X, Liu S, Gao Q, Zhang B, Zhou L, Tan D, Yuan Y, Guo N, Li YH, Wu Z, Liu L, Li C, Lu Y, Gai T, Zhang Y, Yang R, Qian H, Liu Y, Luo J, Zheng L, Lou J, Peng Y, Zuo W, Song J, He S, Wu S, Zou Y, Zhou L, Cheng L, Tang Y, Cheng G, Yuan L, He W, Xu J, Fu T, Xiao Y, Lei T, Xu A, Yin Y, Wang J, Monteiro A, Westhof E, Lu C, Tian Z, Wang W, Xiang Z, Dai F. High-resolution silkworm pan-genome provides genetic insights into artificial selection and ecological adaptation. Nat Commun. 2022 Sep 24;13(1):5619. doi: 10.1038/s41467-022-33366-x.
Lu K, Pan Y, Shen J, Yang L, Zhan C, Liang S, Tai S, Wan L, Li T, Cheng T, Ma B, Pan G, He N, Lu C, Westhof E, Xiang Z, Han MJ, Tong X, Dai F. SilkMeta: a comprehensive platform for sharing and exploiting pan-genomic and multi-omic silkworm data. Nucleic Acids Res. 2024 Jan 5;52(D1):D1024-D1032. doi: 10.1093/nar/gkad956.
Curiously, the last paragraph ("Some research suggests that common fragile sites...") elaborate on the idea that some sites of the genome are prone to mutation. The connection with mamo and the current article are extremely thin. There is here an attempt to connect meiotic and mitotic breaks to Bm-mamo, but this is confusing: it seems to propose Bm-mamo as a recruiter of epigenetic modulators that may drive higher mutation rates elsewhere. Not only I am not convinced by this argument without actual data, but this would not explain how the mutations at the Bm-mamo itself evolved.
Response: Thank you very much for your careful work. This section mainly illustrates that DNA fragility is not determined by sequence but is regulated by other factors in animals. In fruit flies, they found that mamo is an important candidate gene for recombination hotspot setting in meiosis. First, we evaluated PRDM9, which plays an important role in setting recombination hotspots during meiosis. Our purpose in mentioning this information is to illustrate that chromosome recombination is a process of programmed double strand breaks and to answer another reviewer's question about programmed events in the genome. In summary, we suggest that some variations in DNA sequences are procedural results. We have optimized the description of this section in this version.
On a more positive note, I find it fascinating that the authors identified a TF that clearly articulates or orchestrate larval pattern development, and that when it is deleted, can generate healthy individuals. In other words, while it is a TF with many targets, it is not too pleiotropic. This idea, that the genetically causal modulators of developmental evolution are regulatory genes, has been described elsewhere (e.g. Fig 4c in 10.1038/s41576-020-0234-z, and associated refs). To me, the beautiful findings about Bm-mamo make sense in the general, existing framework that developmental processes and regulatory networks "shape" the evolutionary potential and trajectories of organisms. There is a degree of "programmability" in the genomes, because some loci are particularly prone to modulate a given type of trait. Here, Bm-mamo, as a potentially regulator of both CPs and melanin pathway genes, appear to be a potent modulator of epithelial traits. Claiming that there are inherent mutational biases behind this is unwarranted.
Response: Thank you very much for your careful work. I completely agree with your statement that the genome exhibits a certain degree of programmability. On the one hand, some transcription factors can precisely control the spatiotemporal expression levels of some structural genes (such as pigment synthesis genes). On the other hand, these transcription factors are also subject to strict expression regulation. Because the color pattern is complex, changes in single or minority structural genes result in incomplete or imprecise changes in coloring patterns. Nevertheless, several regulatory factors can regulate multiple downstream target genes. Changes in their expression patterns can lead to holistic and significant changes in color patterns. There are long intergenic regions upstream of many important transcription factors, dozens of kilobase pairs (Kb) to hundreds of Kb, which may contain many different regulatory elements for better control of their expression patterns. Therefore, gene regulatory networks can directly regulate transcription factors to modulate a given type of trait. Transcription factors and their downstream target genes can form a functional module, which is similar to a functional module in software or operating systems. This regulation of transcription factors is simpler in terms of steps, which are similar to a single click switch button. The gene regulatory network regulates these modules in response to environmental changes and is widely recognized.
Some people do not agree that genetic variations can also be regulated. They claim that this is completely random. The infinite monkey theorem (Félix-Édouard-Justin-Émile Borel, 1909) states that if an infinite number of monkeys were given typewriters and an infinite amount of time, they would eventually produce the complete works of Shakespeare. Although this theory advocates randomness on the surface, its conclusions are full of inevitability (tail event). In nature, some things we observe do not have obvious regularity because they involve relatively complex factors, and the underlying logic is obscure and difficult to understand. We often name them random. However, as we gradually understand the logic behind this complex event, we can also recognize the procedural nature of this randomness.
Previously, chromosomal recombination during meiosis was believed to be a random event. However, currently, it is believed that the process is procedural. The occurrence of meiotic recombination mentioned earlier indicates that the genome has the ability to self-set the position of double-strand breaks to form new allelic forms. Because meiotic recombination is programmed, transcription factors that recognize DNA sites, enzymes that cleave double strands, and DNA repair systems exist, programming can also introduce genetic variation. A study in plants has provided insights into this programmed mutation (Monroe JG, 2023, nature). Frequent changes in the expression patterns of some transcription factors occur between and/or within species. In this article, we only discuss the possible reasons for variations in the expression patterns of some transcription factors in a general manner and simple reasoning. We have added an analysis of the response of wild silkworms and improved the relevance of the discussion.
Monroe JG, Srikant T, Carbonell-Bejerano P, Becker C, Lensink M, Exposito-Alonso M, Klein M, Hildebrandt J, Neumann M, Kliebenstein D, Weng ML, Imbert E, Ågren J, Rutter MT, Fenster CB, Weigel D. Mutation bias reflects natural selection in Arabidopsis thaliana. Nature. 2022 Feb;602(7895):101-105. doi: 10.1038/s41586-021-04269-6. Epub 2022 Jan 12. Erratum in: Nature. 2023 Aug;620(7973):
Recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
- Please structure your Discussion with section headers.
Response: Thank you very much for your careful work. We have added relevant section headers.
- As explained in my public review, I found the two last sections of the Discussion to be dispersed and confusing. I also must say that I carefully read the Response to Reviewers on this, which helped me to better understand the authors' intentions here. Please consider the revision of this Discussion as this feels extremely speculative difficult to connect with Bm-mamo.
Response: Thank you very much for your careful work. We have rewritten this part of the content.
- typo: were found near the TTS of yellow --> TSS
Response: Thank you very much for your careful work. We have made these modifications.
- l. 234 :"expression level of the 18 CP genes in the integument". Consider adding a mention of Figure 7 here, as only Fig. S10 is cited here.
Response: Thank you very much for your careful work. We have made these modifications.
- Editorial comment on the second half of the Abstract:
Wu et al : "We found that Bm-mamo can comprehensively regulate the expression of related pigment synthesis and cuticular protein genes to form color patterns. This indicates that insects have a genetic basis for coordinate regulation of the structure and shape of the cuticle, as well as color patterns. This genetic basis provides the possibility for constructing the complex appearances of some insects. This study provides new insight into the regulation of color patterns."
I respectfully suggest a more accurate rephrasing, where the methods are mentioned, and where the logical argument is more straightforward. For example
"Using RNAi and CRISPR we show that Bm-mamo is a repressor or dark melanin patterns in the larval epithelium. Using in-vitro binding assays and gene expression profiling in wild-type and mutant larvae, we also show that Bm-mamo likely regulate the expression of related pigment synthesis and cuticular protein genes in a coordinated manner to mediate its role in color pattern formation. This mechanism is consistent with a dual role of this transcription factor in regulating both the structure and shape of the cuticle and pigments that are embedded within it. This study provides new insight into the regulation of color patterns as well as in the construction more complex epithelial features in some insects."
I hope this let the ideas of the original version transpire as the authors intended.
Response: Thank you very much for your careful work. We have made these modifications.
-
eLife assessment
This important study identifies the gene mamo as a new regulator of pigmentation in the silkworm Bombyx mori, a function that was previously unsuspected based on extensive work on Drosophila where the mamo gene is involved in gamete production. The evidence supporting the role of Bm-nano in pigmentation is convincing, including high-resolution linkage mapping of two mutant strains, expression profiling, and reproduction of the mutant phenotypes with state-of-the-art RNAi and CRISPR knock-out assays. The work will be of interest to evolutionary biologists and geneticists studying color patterns and evolution of gene networks.
-
Joint Public Review:
This papers performs fine-mapping of the silkworm mutants bd and its fertile allelic version, bdf, narrowing down the causal intervals to a small interval of a handful of genes. In this region, the gene orthologous to mamo is impaired by a large indel, and its function is later confirmed using expression profiling, RNAi, and CRISPR KO. All these experiments are convincingly showing that mamo is necessary for the suppression of melanic pigmentation in the silkworm larval integument.
The authors also use in silico and in vitro assays to probe the potential effector genes that mamo may regulate.
The genotype-to-phenotype workflow, combining forward (mapping) and reverse genetics (RNAi and CRISPR loss-of-function assays) linking mamo to pigmentation are extremely convincing.
Comments on latest version:
This second …
Joint Public Review:
This papers performs fine-mapping of the silkworm mutants bd and its fertile allelic version, bdf, narrowing down the causal intervals to a small interval of a handful of genes. In this region, the gene orthologous to mamo is impaired by a large indel, and its function is later confirmed using expression profiling, RNAi, and CRISPR KO. All these experiments are convincingly showing that mamo is necessary for the suppression of melanic pigmentation in the silkworm larval integument.
The authors also use in silico and in vitro assays to probe the potential effector genes that mamo may regulate.
The genotype-to-phenotype workflow, combining forward (mapping) and reverse genetics (RNAi and CRISPR loss-of-function assays) linking mamo to pigmentation are extremely convincing.
Comments on latest version:
This second revision took into account all the reviewers' comments. The authors added an interesting analysis of nucleotide diversity at the Bm-mamo locus, using available sequence data from 51 wild silkworms and 171 domesticated silkworms.
The last paragraph added to the discussion, starting with "It has often been believed that changes in CREs are caused by random mutations", is speculative. There is currently no evidence that the mutation rate is biased at the Bm-mamo locus. -
-
Author Response
The following is the authors’ response to the original reviews.
Reviewer #1:
Summary:
This paper performs fine-mapping of the silkworm mutants bd and its fertile allelic version, bdf, narrowing down the causal intervals to a small interval of a handful of genes. In this region, the gene orthologous to mamo is impaired by a large indel, and its function is later confirmed using expression profiling, RNAi, and CRISPR KO. All these experiments are convincingly showing that mamo is necessary for the suppression of melanic pigmentation in the silkworm larval integument. The authors also use in silico and in vitro assays to probe the potential effector genes that mamo may regulate. Strengths: The genotype-to-phenotype workflow, combining forward (mapping) and reverse genetics (RNAi and CRISPR loss-of-function assays) …
Author Response
The following is the authors’ response to the original reviews.
Reviewer #1:
Summary:
This paper performs fine-mapping of the silkworm mutants bd and its fertile allelic version, bdf, narrowing down the causal intervals to a small interval of a handful of genes. In this region, the gene orthologous to mamo is impaired by a large indel, and its function is later confirmed using expression profiling, RNAi, and CRISPR KO. All these experiments are convincingly showing that mamo is necessary for the suppression of melanic pigmentation in the silkworm larval integument. The authors also use in silico and in vitro assays to probe the potential effector genes that mamo may regulate. Strengths: The genotype-to-phenotype workflow, combining forward (mapping) and reverse genetics (RNAi and CRISPR loss-of-function assays) linking mamo to pigmentation are extremely convincing.
Response: Thank you very much for your affirmation of our work. The reviewer discussed the parts of our manuscript that involve evolution sentence by sentence. We have further refined the description in this regard and improved the logical flow. Thank you again for your help.
Weaknesses:
- The last section of the results, entitled "Downstream target gene analysis" is primarily based on in silico genome-wide binding motif predictions.
While the authors identify a potential binding site using EMSA, it is unclear how much this general approach over-predicted potential targets. While I think this work is interesting, its potential caveats are not mentioned. In fact the Discussion section seems to trust the high number of target genes as a reliable result. Specifically, the authors correctly say: "even if there are some transcription factor-binding sites in a gene, the gene is not necessarily regulated by these factors in a specific tissue and period", but then propose a biological explanation that not all binding sites are relevant to expression control. This makes a radical short-cut that predicted binding sites are actual in vivo binding sites. This may not be true, as I'd expect that only a subset of binding motifs predicted by Positional Weight Matrices (PWM) are real in vivo binding sites with a ChIP-seq or Cut-and-Run signal. This is particularly problematic for PWM that feature only 5-nt signature motifs, as inferred here for mamo-S and mamo-L, simply because we can expect many predicted sites by chance.
Response: Thank you very much for your careful work. The analysis and identification of transcription factor-binding sites is an important issue in gene regulation research. Techniques such as ChIP-seq can be used to experimentally identify the binding sites of transcription factors (TFs). However, reports using these techniques often only detect specific cell types and developmental stages, resulting in a limited number of downstream target genes for some TFs. Interestingly, TFs may regulate different downstream target genes in different cell types and developmental stages.
Previous research has suggested that the ZF-DNA binding interface can be understood as a “canonical binding model”, in which each finger contacts DNA in an antiparallel manner. The binding sequence of the C2H2-ZF motif is determined by the amino acid residue sequence of its α-helical component. Considering the first amino acid residue in the α-helical region of the C2H2-ZF domain as position 1, positions -1, 2, 3, and 6 are key amino acids for recognizing and binding DNA. The residues at positions -1, 3, and 6 specifically interact with base 3, base 2, and base 1 of the DNA sense sequence, respectively, while the residue at position 2 interacts with the complementary DNA strand (Wolfe SA et al., 2000; Pabo CO et al., 2001). Based on this principle, the binding sites of C2H2-ZF have good reference value. For the 5-nt PWM sequence, we referred to the study of D. melanogaster, which was identified by EMSA (Shoichi Nakamura et al., 2019). In the new version, we have rewritten this section.
Pabo CO, Peisach E, Grant RA. Design and selection of novel Cys2His2 zinc finger proteins. Annu Rev Biochem. 2001;70:313-340.
Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct. 2000;29:183-212.
Nakamura S, Hira S, Fujiwara M, et al. A truncated form of a transcription factor Mamo activates vasa in Drosophila embryos. Commun Biol. 2019;2:422. Published 2019 Nov 20.
- The last part of the current discussion ("Notably, the industrial melanism event, in a short period of several decades ... a more advanced self-regulation program") is flawed with important logical shortcuts that assign "agency" to the evolutionary process. For instance, this section conveys the idea that phenotypically relevant mutations may not be random. I believe some of this is due to translation issues in English, as I understand that the authors want to express the idea that some parts of the genome are paths of least resistance for evolutionary change (e.g. the regulatory regions of developmental regulators are likely to articulate morphological change). But the language and tone is made worst by the mention that in another system, a mechanism involving photoreception drives adaptive plasticity, making it sound like the authors want to make a Lamarckian argument here (inheritance of acquired characteristics), or a point about orthogenesis (e.g. the idea that the environment may guide non-random mutations).
Because this last part of the current discussion suffers from confused statements on modes and tempo of regulatory evolution and is rather out of topic, I would suggest removing it.
In any case, it is important to highlight here that while this manuscript is an excellent genotype-to-phenotype study, it has very few comparative insights on the evolutionary process. The finding that mamo is a pattern or pigment regulatory factor is interesting and will deserve many more studies to decipher the full evolutionary study behind this Gene Regulatory Network.
Response: Thank you very much for your careful work. In this part of the manuscript, we introduced some assumptions that make the statement slightly unconventional. The color pattern of insects is an adaptive trait. The bd and bdf mutants used in the study are formed spontaneously. As a frequent variation and readily observable phenotype, color patterns have been used as models for evolutionary research (Wittkopp PJ et al., 2011). Darwin's theory of natural selection has epoch-making significance. I deeply believe in the theory that species strive to evolve through natural selection. However, with the development of molecular genetics, Darwinism’s theory of undirected random mutations and slow accumulation of micromutations resulting in phenotype evolution has been increasingly challenged.
The prerequisite for undirected random mutations and micromutations is excessive reproduction to generate a sufficiently large population. A sufficiently large population can contain sufficient genotypes to face various survival challenges. However, it is difficult to explain how some small groups and species with relatively low fertility rates have survived thus far. More importantly, the theory cannot explain the currently observed genomic mutation bias. In scientific research, every theory is constantly being modified to adapt to current discoveries. The most famous example is the debate over whether light is a particle or a wave, which has lasted for hundreds of years. However, in the 20th century, both sides seemed to compromise with each other, believing that light has a wave‒particle duality.
In summary, we have rewritten this section to reduce unnecessary assumptions.
Wittkopp PJ, Kalay G. Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat Rev Genet. 2011;13(1):59-69.
Minor Comment:
The gene models presented in Figure 1 are obsolete, as there are more recent annotations of the Bm-mamo gene that feature more complete intron-exon structures, including for the neighboring genes in the bd/bdf intervals. It remains true that the mamo locus encodes two protein isoforms.
An example of the Bm-mamo locus annotation, can be found at: https://www.ncbi.nlm.nih.gov/gene/101738295 RNAseq expression tracks (including from larval epidermis) can be displayed in the embedded genome browser from the link above using the "Configure Tracks" tool.
Based on these more recent annotations, I would say that most of the work on the two isoforms remains valid, but FigS2, and particularly Fig.S2C, need to be revised.
Response: Thank you very much for your careful work. In this study, we referred to the predicted genes of SilkDB, NCBI and Silkbase. In different databases, there are varying degrees of differences in the number of predicted genes and the length of gene mRNA. Because the SilkDB database is based on the first silkworm genome, it has been used for the longest time and has a relatively large number of users. In the revised manuscript, we have added the predicted genes of NCBI and Silkbase in Figure S1.
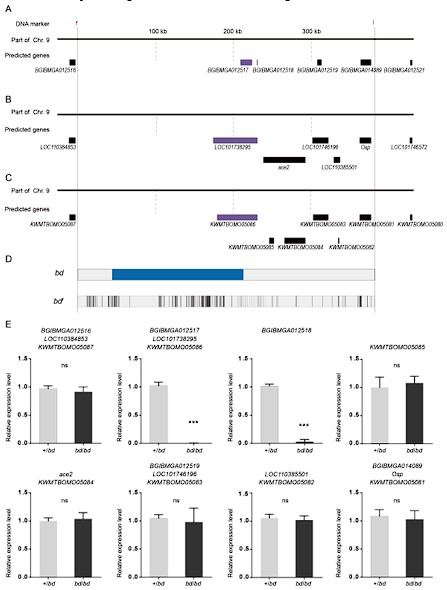
Author response image 1.
The predicted genes and qPCR analysis of candidate genes in the responsible genomic region for bd mutant. (A) The predicted genes in SilkDB;(B) the predicted genes in Genbak;(C) the predicted genes in Silkbase;(D) analysis of nucleotide differences in the responsible region of bd;(E) investigation of the expression level of candidate genes.
Reviewer #2 (Public Review):
Summary:
The authors tried to identify new genes involved in melanin metabolism and its spatial distribution in the silkworm Bombyx mori. They identified the gene Bm-mamo as playing a role in caterpillar pigmentation. By functional genetic and in silico approaches, they identified putative target genes of the Bm-mamo protein. They showed that numerous cuticular proteins are regulated by Bm-mamo during larval development.
Strengths:
- preliminary data about the role of cuticular proteins to pattern the localization of pigments
- timely question
- challenging question because it requires the development of future genetic and cell biology tools at the nanoscale
Response: Thank you very much for your affirmation of our work. The reviewer's familiarity with the color patterns of Lepidoptera is helpful, and the recommendation raised has provided us with very important assistance. This has allowed us to make significant progress with our manuscript.
Weaknesses:
- statistical sampling limited
- the discussion would gain in being shorter and refocused on a few points, especially the link between cuticular proteins and pigmentation. The article would be better if the last evolutionary-themed section of the discussion is removed.
A recent paper has been published on the same gene in Bombyx mori (https://www.sciencedirect.com/science/article/abs/pii/S0965174823000760) in August 2023. The authors must discuss and refer to this published paper through the present manuscript.
Response: Thank you very much for your careful work. First, we believe that competitive research is sometimes coincidental and sometimes intentional. Our research began in 2009, when we began to configure the recombinant population. In 2016, we published an article on comparative transcriptomics (Wu et al. 2016). The article mentioned above has a strong interest in our research and is based on our transcriptome analysis for further research, with the aim of making a preemptive publication. To discourage such behavior, we cannot cite it and do not want to discuss it in our paper.
Songyuan Wu et al. Comparative analysis of the integument transcriptomes of the black dilute mutant and the wild-type silkworm Bombyx mori. Sci Rep. 2016 May 19:6:26114. doi: 10.1038/srep26114.
Reviewer #1 (Recommendations For The Authors):
- please consider using a more recent annotation model of the B. mori genome to revise your Result Section 1, Fig.1, and Fig. S2. https://www.ncbi.nlm.nih.gov/gene/101738295
Specifically, you used BGIM____ gene models, while the current annotation such as the one above featured in the NCBI database provides more accurate intron-exon structures without splitting mamo into tow genes. I believe this can be done with minor revisions of the figures, and you could keep the BGIM____ gene names for the text.
Response: Thank you very much for your careful work. The GenBank of NCBI (National Center for Biotechnology Information) is a very good database that we often use and refer to in this research process. Our research started in 2009, so we mainly referred to the SilkDB database (Jun Duan et al., 2010), although other databases also have references, such as NCBI and Silkbase (https://silkbase.ab.a.u-tokyo.ac.jp/cgi-bin/index.cgi). Because the SilkDB database was constructed based on the first published silkworm genome data, it has been used for the longest time and has a relatively large number of users. Recently, researchers are still using these data (Kejie Li et al., 2023).
The problem with predicting the mamo gene as two genes (BGIBMGA012517 and BGIBMGA012518) in SilkDB is mainly due to the presence of alternative splicing of the mamo gene. BGIBMGA012517 corresponds to the shorter transcript (mamo-s) of the mamo gene. Due to the differences in sequencing individuals, sequencing methods, and methods of gene prediction, there are differences in the number and sequence of predicted genes in different databases. We added the pattern diagram of predicted genes from NCBI and Silkbase, and the expression levels of new predicted genes are shown in Supplemental Figure S1.
Jun Duan et al., SilkDB v2.0: a platform for silkworm (Bombyx mori) genome biology. Nucleic Acids Res. 2010 Jan;38(Database issue): D453-6. doi: 10.1093/nar/gkp801. Kejie Li et al., Transcriptome analysis reveals that knocking out BmNPV iap2 induces apoptosis by inhibiting the oxidative phosphorylation pathway. Int J Biol Macromol. 2023 Apr 1;233:123482. doi: 10.1016/j.ijbiomac.2023.123482. Epub 2023 Jan 31.
Author response image 2.
The predicted genes and qPCR analysis of candidate genes in the responsible genomic region for bd mutant. (A) The predicted genes in SilkDB;(B) the predicted genes in Genbak;(C) the predicted genes in Silkbase;(D) analysis of nucleotide differences in the responsible region of bd;(E) investigation of the expression level of candidate genes.
- As I mentioned in my public review, I strongly believe the interpretation of the PWM binding analyses require much more conservative statements taking into account the idea that short 5-nt motifs are expected by chance. The work in this section is interesting, but the manuscript would benefit from a quite significant rewrite of the corresponding Discussion section, making it that the in silico approach is prone to the identification of many sites in the genomes, and that very few of those sites are probably relevant for probabilistic reasons. I would recommend statements such as "Future experiments assessing the in vivo binding profile of Bm-mamo (eg. ChIP-seq or Cut&Run), will be required to further understand the GRNs controlled by mamo in various tissues".
Response: Thank you very much for your careful work. Previous research has suggested that the ZF-DNA binding interface can be understood as a “canonical binding model”, in which each finger contacts DNA in an antiparallel manner. The binding sequence of the C2H2-ZF motif is determined by the amino acid residue sequence of its α-helical component. Considering the first amino acid residue in the α-helical region of the C2H2-ZF domain as position 1, positions -1, 2, 3, and 6 are key amino acids for recognizing and binding DNA. The residues at positions -1, 3, and 6 specifically interact with base 3, base 2, and base 1 of the DNA sense sequence, respectively, while the residue at position 2 interacts with the complementary DNA strand (Wolfe SA et al., 2000; Pabo CO et al., 2001). Based on this principle, the prediction of DNA recognition motifs of C2H2-type zinc finger proteins currently has good accuracy.
The predicted DNA binding sequence (GTGCGTGGC) of the mamo protein in Drosophila melanogaster was highly consistent with that of silkworms. In addition, in D. melanogaster, the predicted DNA binding sequence of mamo, the bases at positions 1 to 7 (GTGCGTG), was highly similar to the DNA binding sequence obtained from EMSA experiments (Seiji Hira et al., 2013). Furthermore, in another study on the mamo protein of Drosophila melanogaster, five bases (TGCGT) were used as the DNA recognition core sequence of the mamo protein (Shoichi Nakamura et al., 2019). In the JASPAR database (https://jaspar.genereg.net), there are also some shorter (4-6 nt) DNA recognition sequences; for example, the DNA binding sequence of Ubx is TAAT (ID MA0094.1) in Drosophila melanogaster. However, we used longer DNA binding motifs (9 nt and 15 nt) of mamo to study the 2 kb genomic regions near the predicted gene. Over 70% of predicted genes were found to have these feature sequences near them. This analysis method is carried out with common software and processes. Due to sufficient target proteins, the accessibility of DNA, the absence of suppressors, the suitability of ion environments, etc., zinc finger protein transcription factors are more likely to bind to specific DNA sequences in vitro than in vivo. Using ChIP-seq or Cut&Run techniques to analyze various tissues and developmental stages in silkworms can yield one comprehensive DNA-binding map of mamo, and some false positives generated by predictions can be excluded. Thank you for your suggestion. We will conduct this work in the next research step. In addition, for brevity, we deleted the predicted data (Supplemental Tables S7 and S8) that used shorter motifs.
Pabo CO, Peisach E, Grant RA. Design and selection of novel Cys2His2 zinc finger proteins. Annu Rev Biochem. 2001;70:313-340.
Wolfe SA, Nekludova L, Pabo CO. DNA recognition by Cys2His2 zinc finger proteins. Annu Rev Biophys Biomol Struct. 2000;29:183-212.
Anton V Persikov et al., De novo prediction of DNA-binding specificities for Cys2His2 zinc finger proteins. Nucleic Acids Res. 2014 Jan;42(1):97-108. doi: 10.1093/nar/gkt890. Epub 2013 Oct 3.
Seiji Hira et al., Binding of Drosophila maternal Mamo protein to chromatin and specific DNA sequences. Biochem Biophys Res Commun. 2013 Aug 16;438(1):156-60. doi: 10.1016/j.bbrc.2013.07.045. Epub 2013 Jul 20.
Shoichi Nakamura et al., A truncated form of a transcription factor Mamo activates vasa in Drosophila embryos. Commun Biol. 2019 Nov 20;2: 422. doi: 10.1038/s42003-019-0663-4. eCollection 2019.
- In my opinion, the last section of the Discussion needs to be completely removed ("Notably, the industrial melanism event, in a short period of several decades ... a more advanced self-regulation program"), as it is over-extending the data into evolutionary interpretations without any support. I would suggest instead writing a short paragraph asking whether the pigmentary role of mamo is a Lepidoptera novelty, or if it could have been lost in the fly lineage.
Below, I tried to comment point-by-point on the main issues I had.
Wu et al: Notably, the industrial melanism event, in a short period of several decades, resulted in significant changes in the body color of multiple Lepidoptera species(46). Industrial melanism events, such as changes in the body color of pepper moths, are heritable and caused by genomic mutations(47).
Yes, but the selective episode was brief, and the relevant "carbonaria" mutations may have existed for a long time at low-frequency in the population.
Response: Thank you very much for your careful work. Moth species often have melanic variants at low frequencies outside industrial regions. Recent molecular work on genetics has revealed that the melanic (carbonaria) allele of the peppered moth had a single origin in Britain. Further research indicated that the mutation event causing industrial melanism of peppered moth (Biston betularia) in the UK is the insertion of a transposon element into the first intron of the cortex gene. Interestingly, statistical inference based on the distribution of recombined carbonaria haplotypes indicates that this transposition event occurred in approximately 1819, a date highly consistent with a detectable frequency being achieved in the mid-1840s (Arjen E Van't Hof, et al., 2016). From molecular research, it is suggested that this single origin melanized mutant (carbonaria) was generated near the industrial development period, rather than the ancient genotype, in the UK. We have rewritten this part of the manuscript.
Arjen E Van't Hof, et al., The industrial melanism mutation in British peppered moths is a transposable element. Nature. 2016 Jun 2;534(7605):102-5. doi: 10.1038/nature17951.
Wu et al: If relying solely on random mutations in the genome, which have a time unit of millions of years, to explain the evolution of the phenotype is not enough.
What you imply here is problematic for several reasons.
First, as you point out later, some large-effect mutations (e.g. transpositions) can happen quickly.
Second, it's unclear what "the time units of million of years" means here... mutations occur, segregate in populations, and are selected. The speed of this process depends on the context and genetic architectures.
Third, I think I understand what you mean with "to explain the evolution of the phenotype is not enough", but this would probably need a reformulation and I don't think it's relevant to bring it here. After all, you used loss-of-function mutants to explain the evolution of artificially selected mutants. The evolutionary insights from these mutants are limited. Random mutations at the mamo locus are perfectly sufficient here to explain the bd and bdf phenotypes and larval traits.
Response: Thank you very much for your careful work. Charles Darwin himself, who argued that “natural selection can act only by taking advantage of slight successive variations; she can never take a leap, but must advance by the shortest and slowest steps” (Darwin, C. R. 1859). This ‘micromutational’ view of adaptation proved extraordinarily influential. However, the accumulation of micromutations is a lengthy process, which requires a very long time to evolve a significant phenotype. This may be only a proportion of the cases. Interestingly, recent molecular biology studies have shown that the evolution of some morphological traits involves a modest number of genetic changes (H Allen Orr. 2005).
One example is the genetic basis analysis of armor-plate reduction and pelvic reduction of the three-spined stickleback (Gasterosteus aculeatus) in postglacial lakes. Although the marine form of this species has thick armor, the lake population (which was recently derived from the marine form) does not. The repeated independent evolution of lake morphology has resulted in reduced armor plate and pelvic structures, and there is no doubt that these morphological changes are adaptive. Research has shown that pelvic loss in different natural populations of three-spined stickleback fish occurs by regulatory mutations deleting a tissue-specific enhancer (Pel) of the pituitary homeobox transcription factor 1 (Pitx1) gene. The researchers genotyped 13 pelvic-reduced populations of three-spined stickleback from disparate geographic locations. Nine of the 13 pelvic-reduced stickleback populations had sequence deletions of varying lengths, all of which were located at the Pel enhancer. Relying solely on random mutations in the genome cannot lead to such similar mutation forms among different populations. The author suggested that the Pitx1 locus of the stickleback genome may be prone to double-stranded DNA breaks that are subsequently repaired by NHEJ (Yingguang Frank Chan et al., 2010).
The bd and bdf mutants used in the study are formed spontaneously. Natural mutation is one of the driving forces of evolution. Nevertheless, we have rewritten the content of this section.
Darwin, C. R. The Origin of Species (J. Murray, London, 1859).
H Allen Orr. The genetic theory of adaptation: a brief history. Nat Rev Genet. 2005 Feb;6(2):119-27. doi: 10.1038/nrg1523.
Yingguang Frank Chan et al., Adaptive evolution of pelvic reduction in sticklebacks by recurrent deletion of a Pitx1 enhancer. Science. 2010 Jan 15;327(5963):302-5. doi: 10.1126/science.1182213. Epub 2009 Dec 10.
Wu et al: Interestingly, the larva of peppered moths has multiple visual factors encoded by visual genes, which are conserved in multiple Lepidoptera, in the skin. Even when its compound eyes are covered, it can rely on the skin to feel the color of the environment to change its body color and adapt to the environment(48). Therefore, caterpillars/insects can distinguish the light wave frequency of the background. We suppose that perceptual signals can stimulate the GRN, the GRN guides the expression of some transcription factors and epigenetic factors, and the interaction of epigenetic factors and transcription factors can open or close the chromatin of corresponding downstream genes, which can guide downstream target gene expression.
This is extremely confusing because you are bringing in a plastic trait here. It's possible there is a connection between the sensory stimulus and the regulation of mamo in peppered moths, but this is a mere hypothesis. Here, by mentioning a plastic trait, this paragraph sounds as if it was making a statement about directed evolution, especially after implying in the previous sentence that (paraphrasing) "random mutations are not enough". To be perfectly honest, the current writing could be misinterpreted and co-opted by defenders of the Intelligent Design doctrine. I believe and trust this is not your intention.
Response: Thank you very much for your careful work. The plasticity of the body color of peppered moth larvae is very interesting, but we mainly wanted to emphasize that their skin shows the products of visual genes that can sense the color of the environment by perceiving light. Moreover, these genes are conserved in many insects. Human skin can also perceive light by opsins, suggesting that they might initiate light–induced signaling pathways (Haltaufderhyde K et al., 2015). This indicates that the perception of environmental light by the skin of animals and the induction of feedback through signaling pathways is a common phenomenon. For clarity, we have rewritten this section of the manuscript.
Haltaufderhyde K, Ozdeslik RN, Wicks NL, Najera JA, Oancea E. Opsin expression in human epidermal skin. Photochem Photobiol. 2015;91(1):117-123.
Wu et al: In addition, during the opening of chromatin, the probability of mutation of exposed genomic DNA sequences will increase (49).
Here again, this is veering towards a strongly Lamarckian view with the environment guiding specific mutation. I simply cannot see how this would apply to mamo, nothing in the current article indicates this could be the case here. Among many issues with this, it's unclear how chromatin opening in the larval integument may result in heritable mutations in the germline.
Response: Thank you very much for your careful work. Previous studies have shown that there is a mutation bias in the genome; compared with the intergenic region, the mutation frequency is reduced by half inside gene bodies and by two-thirds in essential genes. In addition, they compared the mutation rates of genes with different functions. The mutation rate in the coding region of essential genes (such as translation) is the lowest, and the mutation rates in the coding region of specialized functional genes (such as environmental response) are the highest. These patterns are mainly affected by the traits of the epigenome (J Grey Monroe et al., 2022).
In eukaryotes, chromatin is organized as repeating units of nucleosomes, each consisting of a histone octamer and the surrounding DNA. This structure can protect DNA. When one gene is activated, the chromatin region of this gene is locally opened, becoming an accessible region. Research has found that DNA accessibility can lead to a higher mutation rate in the region (Radhakrishnan Sabarinathan et al., 2016; Schuster-Böckler B et al., 2012; Lawrence MS et al., 2013; Polak P et al., 2015). In addition, the BTB-ZF protein mamo belongs to this family and can recruit histone modification factors such as DNA methyltransferase 1 (DMNT1), cullin3 (CUL3), histone deacetylase 1 (HDAC1), and histone acetyltransferase 1 (HAT1) to perform chromatin remodeling at specific genomic sites. Although mutations can be predicted by the characteristics of apparent chromatin, the forms of mutations are diverse and random. Therefore, this does not violate randomness. For clarity, we have rewritten this section of the manuscript.
J Grey Monroe, Mutation bias reflects natural selection in Arabidopsis thaliana. Nature. 2022 Feb;602(7895):101-105.
Sabarinathan R, Mularoni L, Deu-Pons J, Gonzalez-Perez A, López-Bigas N. Nucleotide excision repair is impaired by binding of transcription factors to DNA. Nature. 2016;532(7598):264-267.
Schuster-Böckler B, Lehner B. Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature. 2012;488(7412):504-507.
Lawrence MS, Stojanov P, Polak P, et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature. 2013;499(7457):214-218.
Polak P, Karlić R, Koren A, et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature. 2015;518(7539):360-364.
Mathew R, Seiler MP, Scanlon ST, et al. BTB-ZF factors recruit the E3 ligase cullin 3 to regulate lymphoid effector programs. Nature. 2012;491(7425):618-621.
Wu et al: Transposon insertion occurs in a timely manner upstream of the cortex gene in melanic pepper moths (47), which may be caused by the similar binding of transcription factors and opening of chromatin.
No, we do not think that the peppered moth mutation is Lamarckian at all, as seems to be inferred here (notice that by mentioning the peppered moth twice, you are juxtaposing a larval plastic trait and then a purely genetic wing trait, making it even more confusing). Also, the "in a timely manner" is superfluous, because all the data are consistent with a chance mutation being eventually picked up by strong directional mutation. The mutation and selection did NOT occur at the same time.
Response: Thank you very much for your careful work. The insertion of one transposon into the first intron of the cortex gene of industrial melanism in peppered moth occurred in approximately 1819, which is similar to the time of industrial development in the UK (Arjen E Van't Hof, et al., 2016). In multiple species of Heliconius, the cortex gene is the shared genetic basis for the regulation of wing coloring patterns. Interestingly, the SNP of the cortex, associated with the wing color pattern, does not overlap among different Heliconius species, such as H. erato dephoon and H. erato favorinus, which suggests that the mutations of this cortex gene have different origins (Nadeau NJ et al., 2016). In addition, in Junonia coenia (van der Burg KRL et al., 2020) and Bombyx mori (Ito K et al., 2016), the cortex gene is a candidate for regulating changes in wing coloring patterns. Overall, the cortex gene is an evolutionary hotspot for the variation of multiple butterfly and moth wing coloring patterns. In addition, it was observed that the variations in the cortex are diverse in these species, including SNPs, indels, transposon insertions, inversions, etc. This indicates that although there are evolutionary hotspots in the insect genome, this variation is random. Therefore, this is not completely detached from randomness.
Arjen E Van't Hof, et al., The industrial melanism mutation in British peppered moths is a transposable element. Nature. 2016 Jun 2;534(7605):102-5. doi: 10.1038/nature17951.
Nadeau NJ, Pardo-Diaz C, Whibley A, et al. The gene cortex controls mimicry and crypsis in butterflies and moths. Nature. 2016;534(7605):106-110.
van der Burg KRL, Lewis JJ, Brack BJ, Fandino RA, Mazo-Vargas A, Reed RD. Genomic architecture of a genetically assimilated seasonal color pattern. Science. 2020;370(6517):721-725.
Ito K, Katsuma S, Kuwazaki S, et al. Mapping and recombination analysis of two moth colour mutations, Black moth and Wild wing spot, in the silkworm Bombyx mori. Heredity (Edinb). 2016;116(1):52-59.
Wu et al: Therefore, we proposed that the genetic basis of color pattern evolution may mainly be system-guided programmed events that induce mutations in specific genomic regions of key genes rather than just random mutations of the genome.
While the mutational target of pigment evolution may involve a handful of developmental regulator genes, you do not have the data to infer such a strong conclusion at the moment.
The current formulation is also quite strong and teleological: "system-guided programmed events" imply intentionality or agency, an idea generally assigned to the anti-scientific Intelligent Design movement. There are a few examples of guided mutations, such as the adaptation phase of gRNA motifs in bacterial CRISPR assays, where I could see the term ""system-guided programmed events" to be applicable. But it is irrelevant here.
Response: Thank you very much for your careful work. The CRISPR-CAS9 system is indeed very well known. In addition, recent studies have found the existence of a Cas9-like gene editing system in eukaryotes, such as Fanzor. Fanzor (Fz) was reported in 2013 as a eukaryotic TnpB-IS200/IS605 protein encoded by the transposon origin, and it was initially thought that the Fz protein (and prokaryotic TnpBs) might regulate transposon activity through methyltransferase activity (Saito M et al., 2023). Fz has recently been found to be a eukaryotic CRISPR‒Cas system. Although this system is found in fungi and mollusks, it raises hopes for scholars to find similar systems in other higher animals. However, before these gene-editing systems became popular, zinc finger nucleases (ZFNs) were already being studied as a gene-editing system in many species. The mechanism by which ZFN recognizes DNA depends on its zinc finger motif (Urnov FD et al., 2005). This is consistent with the mechanism by which transcription factors recognize DNA-binding sites.
Furthermore, a very important evolutionary event in sexual reproduction is chromosome recombination during meiosis, which helps to produce more abundant alleles. Current research has found that this recombination event is not random. In mice and humans, the PRDM9 transcription factors are able to plan the sites of double-stranded breaks (DSBs) in meiosis recombination. PRDM9 is a histone methyltransferase consisting of three main regions: an amino-terminal region resembling the family of synovial sarcoma X (SSX) breakpoint proteins, which contains a Krüppel-associated box (KRAB) domain and an SSX repression domain (SSXRD); a PR/SET domain (a subclass of SET domains), surrounded by a pre-SET zinc knuckle and a post-SET zinc finger; and a long carboxy-terminal C2H2 zinc finger array. In most mammalian species, during early meiotic prophase, PRDM9 can determine recombination hotspots by H3K4 and H3K36 trimethylation (H3K4me3 and H3K36me3) of nucleosomes near its DNA-binding site. Subsequently, meiotic DNA DSBs are formed at hotspots through the combined action of SPO11 and TOPOVIBL. In addition, some proteins (such as RAD51) are involved in repairing the break point. In summary, programmed events of induced and repaired DSBs are widely present in organisms (Bhattacharyya T et al., 2019).
These studies indicate that on the basis of randomness, the genome also exhibits programmability.
Saito M, Xu P, Faure G, et al. Fanzor is a eukaryotic programmable RNA-guided endonuclease. Nature. 2023;620(7974):660-668.
Urnov FD, Miller JC, Lee YL, et al. Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 2005;435(7042):646-651.
Bhattacharyya T, Walker M, Powers NR, et al. Prdm9 and Meiotic Cohesin Proteins Cooperatively Promote DNA Double-Strand Break Formation in Mammalian Spermatocytes [published correction appears in Curr Biol. 2021 Mar 22;31(6):1351]. Curr Biol. 2019;29(6):1002-1018.e7.
Wu et al: Based on this assumption, animals can undergo phenotypic changes more quickly and more accurately to cope with environmental changes. Thus, seemingly complex phenotypes such as cryptic coloring and mimicry that are highly similar to the background may have formed in a short period. However, the binding sites of some transcription factors widely distributed in the genome may be reserved regulatory interfaces to cope with potential environmental changes. In summary, the regulation of genes is smarter than imagined, and they resemble a more advanced self-regulation program.
Here again, I can agree with the idea that certain genetic architectures can evolve quickly, but I cannot support the concept that the genetic changes are guided or accelerated by the environment. And again, none of this is relevant to the current findings about Bm-mamo.
Response: Thank you very much for your careful work. Darwin's theory of natural selection has epoch-making significance. I deeply believe in the theory that species strive to evolve through natural selection. However, with the development of molecular genetics, Darwinism’s theory of undirected random mutations and slow accumulation of micromutations resulting in phenotype evolution has been increasingly challenged.
The prerequisite for undirected random mutations and micromutations is excessive reproduction to generate a sufficiently large population. A sufficiently large population can contain sufficient genotypes to face various survival challenges. However, it is difficult to explain how some small groups and species with relatively low fertility rates have survived thus far. More importantly, the theory cannot explain the currently observed genomic mutation bias. In scientific research, every theory is constantly being modified to adapt to current discoveries. The most famous example is the debate over whether light is a particle or a wave, which has lasted for hundreds of years. However, in the 20th century, both sides seemed to compromise with each other, believing that light has a wave‒particle duality.
Epigenetics has developed rapidly since 1987. Epigenetics has been widely accepted, defined as stable inheritance caused by chromosomal conformational changes without altering the DNA sequence, which differs from genetic research on variations in gene sequences. However, an increasing number of studies have found that histone modifications can affect gene sequence variation. In addition, both histones and epigenetic factors are essentially encoded by genes in the genome. Therefore, genetics and epigenetics should be interactive rather than parallel. However, some transcription factors play an important role in epigenetic modifications. Meiotic recombination is a key process that ensures the correct separation of homologous chromosomes through DNA double-stranded break repair mechanisms. The transcription factor PRDM9 can determine recombination hotspots by H3K4 and H3K36 trimethylation (H3K4me3 and H3K36me3) of nucleosomes near its DNA-binding site (Bhattacharyya T et al., 2019). Interestingly, mamo has been identified as an important candidate factor for meiosis hotspot setting in Drosophila (Winbush A et al., 2021).
Bhattacharyya T, Walker M, Powers NR, et al. Prdm9 and Meiotic Cohesin Proteins Cooperatively Promote DNA Double-Strand Break Formation in Mammalian Spermatocytes [published correction appears in Curr Biol. 2021 Mar 22;31(6):1351]. Curr Biol. 2019;29(6):1002-1018.e7.
Winbush A, Singh ND. Genomics of Recombination Rate Variation in Temperature-Evolved Drosophila melanogaster Populations. Genome Biol Evol. 2021;13(1): evaa252.
Reviewer #2 (Recommendations For The Authors):
Major comments
- A recent paper has been published on the same gene in Bombyx mori (https://www.sciencedirect.com/science/article/abs/pii/S0965174823000760) in August 2023. The authors must discuss and refer to this published paper through the present manuscript.
Response: Thank you very much for your careful work. First, we believe that competitive research is sometimes coincidental and sometimes intentional. Our research began in 2009, when we began to configure the recombinant population. In 2016, we published an article on comparative transcriptomics (Wu et al. 2016). The article mentioned above has a strong interest in our research and is based on our transcriptome analysis for further research, with the aim of making a preemptive publication.
To discourage such behavior, we cannot cite it and do not want to discuss it in our paper.
Songyuan Wu et al. Comparative analysis of the integument transcriptomes of the black dilute mutant and the wild-type silkworm Bombyx mori. Sci Rep. 2016 May 19:6:26114. doi: 10.1038/srep26114.
- line 52-54. The numerous biological functions of insect coloration have been thoroughly investigated. It is reasonable to expect more references for each function.
Response: Thank you very much for your careful work. We have made the appropriate modifications.
Sword GA, Simpson SJ, El Hadi OT, Wilps H. Density-dependent aposematism in the desert locust. Proc Biol Sci. 2000;267(1438):63-68. … Behavior.
Barnes AI, Siva-Jothy MT. Density-dependent prophylaxis in the mealworm beetle Tenebrio molitor L. (Coleoptera: Tenebrionidae): cuticular melanization is an indicator of investment in immunity. Proc Biol Sci. 2000;267(1439):177-182. … Immunity.
N. F. Hadley, A. Savill, T. D. Schultz, Coloration and Its Thermal Consequences in the New-Zealand Tiger Beetle Neocicindela-Perhispida. J Therm Biol. 1992;17, 55-61…. Thermoregulation.
Y. G. Hu, Y. H. Shen, Z. Zhang, G. Q. Shi, Melanin and urate act to prevent ultraviolet damage in the integument of the silkworm, Bombyx mori. Arch Insect Biochem. 2013; 83, 41-55…. UV protection.
M. Stevens, G. D. Ruxton, Linking the evolution and form of warning coloration in nature. P Roy Soc B-Biol Sci. 2012; 279, 417-426…. Aposematism.
K. K. Dasmahapatra et al., Butterfly genome reveals promiscuous exchange of mimicry adaptations among species. Nature.2012; 487, 94-98…. Mimicry.
Gaitonde N, Joshi J, Kunte K. Evolution of ontogenic change in color defenses of swallowtail butterflies. Ecol Evol. 2018;8(19):9751-9763. Published 2018 Sep 3. …Crypsis.
B. S. Tullberg, S. Merilaita, C. Wiklund, Aposematism and crypsis combined as a result of distance dependence: functional versatility of the colour pattern in the swallowtail butterfly larva. P Roy Soc B-Biol Sci.2005; 272, 1315-1321…. Aposematism and crypsis combined.
- line 59-60. This general statement needs to be rephrased. I suggest remaining simple by indicating that insect coloration can be pigmentary, structural, or bioluminescent. About the structural coloration and associated nanostructures, the authors could cite recent reviews, such as: Seago et al., Interface 2009 + Lloyd and Nadeau, Current Opinion in Genetics & Development 2021 + "Light as matter: natural structural colour in art" by Finet C. 2023. I suggest doing the same for recent reviews that cover pigmentary and bioluminescent coloration in insects. The very recent paper by Nishida et al. in Cell Reports 2023 on butterfly wing color made of pigmented liquid is also unique and worth to consider.
Response: Thank you very much for your careful work. We have made the appropriate modifications.
Insect coloration can be pigmentary, structural, or bioluminescent. Pigments are mainly synthesized by the insects themselves and form solid particles that are deposited in the cuticle of the body surface and the scales of the wings (10, 11). Interestingly, recent studies have found that bile pigments and carotenoid pigments synthesized through biological synthesis are incorporated into body fluids and passed through the wing membranes of two butterflies (Siproeta stelenes and Philaethria diatonica) via hemolymph circulation, providing color in the form of liquid pigments (12). The pigments form colors by selective absorption and/or scattering of light depending on their physical properties (13). However, structural color refers to colors, such as metallic colors and iridescence, generated by optical interference and grating diffraction of the microstructure/nanostructure of the body surface or appendages (such as scales) (14, 15). Pigment color and structural color are widely distributed in insects and can only be observed by the naked eye in illuminated environments. However, some insects, such as fireflies, exhibit colors (green to orange) in the dark due to bioluminescence (16). Bioluminescence occurs when luciferase catalyzes the oxidation of small molecules of luciferin (17). In conclusion, the color patterns of insects have evolved to be highly sophisticated and are closely related to their living environments. For example, cryptic color can deceive animals via high similarity to the surrounding environment. However, the molecular mechanism by which insects form precise color patterns to match their living environment is still unknown.
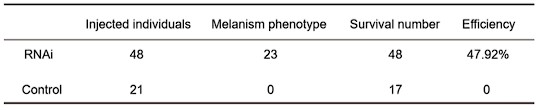
- RNAi approach. I have no doubt that obtaining phenocopies by electroporation might be difficult. However, I find the final sampling a bit limited to draw conclusions from the RT-PCR (n=5 and n=3 for phenocopies and controls). Three control individuals is a very low number. Moreover, it would nice to see the variability on the plot, using for example violin plots.
Response: Thank you very much for your careful work. In the RNAi experiment, we injected more than 20 individuals in the experimental group and control group. We have added the RNAi data in Figure 4.
Author response table 1.
- Figure 6. Higher magnification images of Dazao and Bm-mamo knockout are needed, as shown in Figure 5 on RNAi.
Response: Thank you very much for your careful work. We have added enlarged images.
Author response image 3.
- Phylogenetic analysis/Figure S6. I am not sure to what extent the sampling is biased or not, but if not, it is noteworthy that mamo does not show duplicated copies (negative selection?). It might be interesting to discuss this point in the manuscript.
Response: Thank you very much for your careful work. mamo belongs to the BTB/POZ zinc finger family. The members of this family exhibit significant expansion in vertebrates. For example, there are 3 members in C. elegans, 13 in D. melanogaster, 16 in Bombyx mori, 58 in M. musculus and 63 in H. sapiens (Wu et al, 2019). These members contain conserved BTB/POZ domains but vary in number and amino acid residue compositions of the zinc finger motifs. Due to the zinc finger motifs that bind to different DNA recognition sequences, there may be differences in their downstream target genes. Therefore, when searching for orthologous genes from different species, we required high conservation of their zinc finger motif sequences. Due to these strict conditions, only one orthologous gene was found in these species.
- Differentially-expressed genes and CP candidate genes (line 189-191). The manuscript would gain in clarity if the authors explain more in details their procedure. For instance, they moved from a list of 191 genes to CP genes only. Can they say a little bit more about the non-CP genes that are differentially expressed? Maybe quantify the number of CPs among the total number of differentially-expressed genes to show that CPs are the main class?
Response: Thank you very much for your careful work. The nr (Nonredundant Protein Sequence Database) annotations for 191 differentially expressed genes in Supplemental Table S3 were added. Among them, there were 19 cuticular proteins, 17 antibacterial peptide genes, 6 transporter genes, 5 transcription factor genes, 5 cytochrome genes, 53 enzyme-encoding genes and others. Because CP genes were significantly enriched in differentially expressed genes (DEGs), previous studies have found that BmorCPH24 can affect pigmentation. Therefore, we first conducted an investigation into CP genes.
- Interaction between Bm-mamo. It is not clear why the authors chose to investigate the physical interaction of Bm-mamo protein with the putative binding site of yellow, and not with the sites upstream of tan and DDC. Do the authors test one interaction and assume the conclusion stands for the y, tan and DDC?
Response: Thank you very much for your careful work. In D. melanogaster, the yellow gene is the most studied pigment gene. The upstream and intron sequences of the yellow gene have been identified as containing multiple cis-regulatory elements. Due to the important pigmentation role of the yellow gene and its variable cis-regulatory sequence among different species, it has been considered a research model for cis-regulatory elements (Laurent Arnoult et al. 2013, Gizem Kalay et al. 2019, Yaqun Xin et al. 2020, Yann Le Poul et al. 2020). We use yellow as an example to illustrate the regulation of the mamo gene. We added this description to the discussion.
Laurent Arnoult et al. Emergence and diversification of fly pigmentation through evolution of a gene regulatory module. Science. 2013 Mar 22;339(6126):1423-6. doi: 10.1126/science.1233749.
Gizem Kalay et al. Redundant and Cryptic Enhancer Activities of the Drosophila yellow Gene. Genetics. 2019 May;212(1):343-360. doi: 10.1534/genetics.119.301985. Epub 2019 Mar 6.
Yaqun Xin et al. Enhancer evolutionary co-option through shared chromatin accessibility input. Proc Natl Acad Sci U S A. 2020 Aug 25;117(34):20636-20644. doi: 10.1073/pnas.2004003117. Epub 2020 Aug 10.
Yann Le Poul et al. Regulatory encoding of quantitative variation in spatial activity of a Drosophila enhancer. Sci Adv. 2020 Dec 2;6(49):eabe2955. doi: 10.1126/sciadv.abe2955. Print 2020 Dec.
- Please note that some controls are missing for the EMSA experiments. For instance, the putative binding-sites should be mutated and it should be shown that the interaction is lost.
Response: Thank you very much for your careful work. In this study, we found that the DNA recognition sequence of mamo is highly conserved across multiple species. In D. melanogaster, studies have found that mamo can directly bind to the intron of the vasa gene to activate its expression. The DNA recognition sequence they use is TGCGT (Shoichi Nakamura et al. 2019). We chose a longer sequence, GTGCGTGGC, to detect the binding of mamo. This binding mechanism is consistent across species.
- Figure 7 and supplementary data. How did the name of CPs attributed? According to automatic genome annotation of Bm genes and proteins? Based on Drosophila genome and associated gene names? Did the authors perform phylogenetic analyses to name the different CP genes?
Response: Thank you very much for your careful work. The naming of CPs is based on their conserved motif and their arrangement order on the chromosome. In previous reports, sequence identification and phylogenetic analysis of CPs have been carried out in silkworms (Zhengwen Yan et al. 2022, Ryo Futahashi et al. 2008). The members of the same family have sequence similarity between different species, and their functions may be similar. We have completed the names of these genes in the text, for example, changing CPR2 to BmorCPR2.
Zhengwen Yan et al. A Blueprint of Microstructures and Stage-Specific Transcriptome Dynamics of Cuticle Formation in Bombyx mori. Int J Mol Sci. 2022 May 5;23(9):5155.
Ningjia He et al. Proteomic analysis of cast cuticles from Anopheles gambiae by tandem mass spectrometry. Insect Biochem Mol Biol. 2007 Feb;37(2):135-46.
Maria V Karouzou et al. Drosophila cuticular proteins with the R&R Consensus: annotation and classification with a new tool for discriminating RR-1 and RR-2 sequences. Insect Biochem Mol Biol. 2007 Aug;37(8):754-60.
Ryo Futahashi et al. Genome-wide identification of cuticular protein genes in the silkworm, Bombyx mori. Insect Biochem Mol Biol. 2008 Dec;38(12):1138-46.
- Discussion. I think the discussion would gain in being shorter and refocused on the understudied role of CPs. Another non-canonical aspect of the discussion is the reference to additional experiments (e.g., parthogenesis line 290-302, figure S14). This is not the place to introduce more results, and it breaks the flow of the discussion. I encourage the authors to reshuffle the discussion: 1) summary of their findings on mamo and CPs, 2) link between pigmentation mutant phenotypes, pigmentation pattern and CPs, 3) general discussion about the (evo-)devo importance of CPs and link between pigment deposition and coloration. Three important papers should be mentioned here:
- Matsuoka Y and A Monteiro (2018) Melanin pathway genes regulate color and morphology of butterfly wing scales. Cell Reports 24: 56-65... Yellow has a pleiotropic role in cuticle deposition and pigmentation.
- https://arxiv.org/abs/2305.16628... Link between nanoscale cuticle density and pigmentation
- https://www.cell.com/cell-reports/pdf/S2211-1247(23)00831-8.pdf... Variation in pigmentation and implication of endosomal maturation (gene red).
Response: Thank you very much for your careful work. We have rewritten the discussion section.
- We have summarized our findings.
Bm-mamo may affect the synthesis of melanin in epidermis cells by regulating yellow, DDC, and tan; regulate the maturation of melanin granules in epidermis cells through BmMFS; and affect the deposition of melanin granules in the cuticle by regulating CP genes, thereby comprehensively regulating the color pattern in caterpillars.
- We describe the relationship among the pigmentation mutation phenotype, pigmentation pattern, and CP.
Previous studies have shown that the lack of expression of BmorCPH24, which encodes important components of the endocuticle, can lead to dramatic changes in body shape and a significant reduction in the pigmentation of caterpillars (53). We crossed Bo (BmorCPH24 null mutation) and bd to obtain F1(Bo/+Bo, bd/+), then self-crossed F1 and observed the phenotype of F2. The lunar spots and star spots decreased, and light-colored stripes appeared on the body segments, but the other areas still had significant melanin pigmentation in double mutation (Bo, bd) individuals (Fig. S13). However, in previous studies, introduction of Bo into L (ectopic expression of wnt1 results in lunar stripes generated on each body segment) (24) and U (overexpression of SoxD results in excessive melanin pigmentation of the epidermis) (58) strains by genetic crosses can remarkably reduce the pigmentation of L and U (53). Interestingly, there was a more significant decrease in pigmentation in the double mutants (Bo, L) and (Bo, U) than in (Bo, bd). This suggests that Bm-mamo has a stronger ability than wnt1 and SoxD to regulate pigmentation. On the one hand, mamo may be a stronger regulator of the melanin metabolic pathway, and on the other hand, mamo may regulate other CP genes to reduce the impact of BmorCPH24 deficiency.
- We discussed the importance of (evo-) devo in CPs and the relationship between pigment deposition and coloring.
CP genes usually account for over 1% of the total genes in an insect genome and can be categorized into several families, including CPR, CPG, CPH, CPAP1, CPAP3, CPT, CPF and CPFL (68). The CPR family is the largest group of CPs, containing a chitin-binding domain called the Rebers and Riddiford motif (R&R) (69). The variation in the R&R consensus sequence allows subdivision into three subfamilies (RR-1, RR-2, and RR-3) (70). Among the 28 CPs, 11 RR-1 genes, 6 RR-2 genes, 4 hypothetical cuticular protein (CPH) genes, 3 glycine-rich cuticular protein (CPG) genes, 3 cuticular protein Tweedle motif (CPT) genes, and 1 CPFL (like the CPFs in a conserved C-terminal region) gene were identified. The RR-1 consensus among species is usually more variable than RR-2, which suggests that RR-1 may have a species-specific function. RR-2 often clustered into several branches, which may be due to gene duplication events in co-orthologous groups and may result in conserved functions between species (71). The classification of CPH is due to their lack of known motifs. In the epidermis of Lepidoptera, the CPH genes often have high expression levels. For example, BmorCPH24 had a highest expression level, in silkworm larvae epidermis (72). The CPG protein is rich in glycine. The CPH and CPG genes are less commonly found in insects outside the order Lepidoptera (73). This suggests that they may provide species specific functions for the Lepidoptera. CPT contains a Tweedle motif, and the TweedleD1 mutation has a dramatic effect on body shape in D. melanogaster (74). The CPFL members are relatively conserved in species and may be involved in the synthesis of larval cuticles (75). CPT and CPFL may have relatively conserved functions among insects. The CP genes are a group of rapidly evolving genes, and their copy numbers may undergo significant changes in different species. In addition, RNAi experiments on 135 CP genes in brown planthopper (Nilaparvata lugens) showed that deficiency of 32 CP genes leads to significant defective phenotypes, such as lethal, developmental retardation, etc. It is suggested that the 32 CP genes are indispensable, and other CP genes may have redundant and complementary functions (76). In previous studies, it was found that the construction of the larval cuticle of silkworms requires the precise expression of over two hundred CP genes (22). The production, interaction, and deposition of CPs and pigments are complex and precise processes, and our research shows that Bm-mamo plays an important regulatory role in this process in silkworm caterpillars. For further understanding of the role of CPs, future work should aim to identify the function of important cuticular protein genes and the deposition mechanism in the cuticle.
Minor comments
- Title. At this stage, there is no evidence that Bm-mamo regulates caterpillar pigmentation outside of Bombyx mori. I suggest to precise 'silkworm caterpillars' in the title.
Response: Thank you very much for your careful work. We have modified the title.
- Abstract, line 29. Because the knowledge on pigmentation pathway(s) is advanced, I would suggest writing 'color pattern is not fully understood' instead of 'color pattern is not clear'.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 29. I suggest 'the transcription factor' rather than 'a transcription factor'.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 30. If you want to mention the protein, the name 'Bm-mamo' should not be italicized.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 30. 'in the silkworm'.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 31. 'mamo' should not be italicized.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 31. 'in Drosophila' rather 'of Drosophila'.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 32. Bring detail if the gamete function is conserved in insects? In all animals?
Response: Thank you very much for your careful work. The sentence was changed to “This gene has a conserved function in gamete production in Drosophila and silkworms and evolved a pleiotropic function in the regulation of color patterns in caterpillars.”
- Introduction, line 51. I am not sure what the authors mean by 'under natural light'. Please rephrase.
Response: Thank you very much for your careful work. We have deleted “under natural light”.
- line 43. I find that the sentence 'In some studies, it has been proven that epidermal proteins can affect the body shape and appendage development of insects' is not necessary here. Furthermore, this sentence breaks the flow of the teaser.
Response: Thank you very much for your careful work. We have deleted this sentence.
- line 51-52. 'Greatly benefit them' should be rephrased in a more neutral way. For example, 'colours pattern have been shown to be involved in...'.
Response: Thank you very much for your careful work. We have modified to “and the color patterns have been shown to be involved in…”
- line 62. CPs are secreted by the epidermis, but I would say that CPs play their structural role in the cuticle, not directly in the epidermis. I suggest rephrasing this sentence and adding references.
Response: Thank you very much for your careful work. We have modified “epidermis” to “cuticle”.
- line 67. Please indicate that pathways have been identified/reported in Lepidoptera (11). Otherwise, the reader does not understand if you refer to previous biochemical in Drosophila for example.
Response: Thank you very much for your careful work. We have modified this sentence. “Moreover, the biochemical metabolic pathways of pigments used for color patterning in Lepidoptera…have been reported.”
- line 69. Missing examples of pleiotropic factors and associated references. For example, I suggest adding: engrailed (Dufour, Koshikawa and Finet, PNAS 2020) + antennapedia (Prakash et al., Cell Reports 2022) + optix (Reed et al., Science 2011), etc. Need to add references for clawless, abdominal-A.
Response: Thank you very much for your careful work. We have made modifications.
- line 76. The simpler term moth might be enough (instead of Lepidoptera).
Response: Thank you very much for your careful work. We have modified this to “insect”.
- line 96. I would simplify the text by writing "Then, quantitative RT-PCR was performed..."
Response: Thank you very much for your careful work. We have modified this sentence.
- line 112. 'Predict' instead of 'estimate'?
Response: Thank you very much for your careful work. We have modified this sentence.
- line 113. I would rather indicate the full name first, then indicate mamo between brackets.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 144. The Perl script needs to be made accessible on public repository.
Response: Thank you very much for your careful work.
- line 147-150. Too many technical details here. The details are already indicated in the material and methods section. Furthermore, the details break the flow of the paragraph.
Response: Thank you very much for your careful work. We have modified this section.
- line 152. Needs to make the link with the observed phenotypes in Figure 1. Just needs to state that RNAi phenocopies mimic the mutant alleles.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 153-157. Too many technical details here. The details are already indicated in the material and methods section. Furthermore, the details break the flow of the paragraph.
Response: Thank you very much for your careful work. We have simplified this paragraph.
- line 170. Please rephrase 'conserved in 30 species' because it might be understood as conserved in 30 species only, and not in other species.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 182. Maybe explain the rationale behind restricting the analysis to +/- 2kb. Can you cite a paper that shows that most of binding sites are within 2kb from the start codon?
Response: Thank you very much for your careful work. We have modified this sentence.
- line 182. '14,623 predicted genes'.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 183. '10,622 genes'
Response: Thank you very much for your careful work. We have modified this sentence.
- line 183. Redundancy. Please remove 'silkworm' or 'B. mori'.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 187. '10,072 genes'
Response: Thank you very much for your careful work. We have modified this sentence.
- line 188. '9,853 genes'
Response: Thank you very much for your careful work. We have modified this sentence.
- line 200. "Therefore, the differential...in caterpillars" is a strong statement.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 204. Remove "The" in front of eight key genes. Also, needs a reference... maybe a recent review on the biochemical pathway of melanin in insects.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 220. This sentence is too general and vague. Please explicit what you mean by "in terms of evolution". Number of insect species? Diversity of niche occupancy? Morphological, physiological diversity?
Response: Thank you very much for your careful work. We have modified this sentence.
- line 285. The verb "believe" should be replaced by a more neutral one.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 354-355. This sentence needs to be rephrased in a more objective way.
Response: Thank you very much for your careful work. We have rewritten this sentence.
- line 378. Missing reference for MUSCLE.
Response: Thank you very much for your careful work. We have modified this sentence.
- line 379. Pearson model?
Response: Thank you very much for your careful work. We have modified this sentence.
- line 408. "The CRISPRdirect online software was used...".
Response: Thank you very much for your careful work. We have modified this sentence.
- Figure 1. In the title, I suggest indicating Dazao, bd, bdf as it appears in the figure. Needs to precise 'silkworm larval development'.
Response: Thank you very much for your careful work. We have modified this figure title.
- Figure 3. In the title, is the word 'pattern' really necessary? In the legend, please indicate the meaning of the acronyms AMSG and PSG.
Response: Thank you very much for your careful work. We have modified this figure legend.
- Figure S7A. Typo 'Znic finger 1', 'Znic finger 2', 'Znic finger 3',
Response: Thank you very much for your careful work. We have fixed these typos. .
-
eLife assessment
This important study identifies the gene mamo as a new regulator of pigmentation in the silkworm Bombyx mori, a function that was previously unsuspected based on extensive work on Drosophila where the mamo gene is involved in gamete production. The evidence supporting the role of Bm-nano in pigmentation is convincing, including high-resolution linkage mapping of two mutant strains, expression profiling, and reproduction of the mutant phenotypes with state-of-the-art RNAi and CRISPR knock-out assays. While the discussion about genetic changes being guided or accelerated by the environment is extremely speculative and has little relevance for the findings presented, the work will be of interest to evolutionary biologists and geneticists studying color patterns and evolution of gene networks.
-
Reviewer #1 (Public Review):
Summary: This papers performs fine-mapping of the silkworm mutants bd and its fertile allelic version, bdf, narrowing down the causal intervals to a small interval of a handful of genes. In this region, the gene orthologous to mamo is impaired by a large indel, and its function is later confirmed using expression profiling, RNAi, and CRISPR KO. All these experiments are convincingly showing that mamo is necessary for the suppression of melanic pigmentation in the silkworm larval integument.
The authors also use in silico and in vitro assays to probe the potential effector genes that mamo may regulate.
Strengths: The genotype-to-phenotype workflow, combining forward (mapping) and reverse genetics (RNAi and CRISPR loss-of-function assays) linking mamo to pigmentation are extremely convincing.
This revision is …
Reviewer #1 (Public Review):
Summary: This papers performs fine-mapping of the silkworm mutants bd and its fertile allelic version, bdf, narrowing down the causal intervals to a small interval of a handful of genes. In this region, the gene orthologous to mamo is impaired by a large indel, and its function is later confirmed using expression profiling, RNAi, and CRISPR KO. All these experiments are convincingly showing that mamo is necessary for the suppression of melanic pigmentation in the silkworm larval integument.
The authors also use in silico and in vitro assays to probe the potential effector genes that mamo may regulate.
Strengths: The genotype-to-phenotype workflow, combining forward (mapping) and reverse genetics (RNAi and CRISPR loss-of-function assays) linking mamo to pigmentation are extremely convincing.
This revision is a much improved manuscript and I command the authors for many of their edits.
I find the last part of the discussion, starting at "It is generally believed that changes in gene expression patterns are the result of the evolution of CREs", to be confusing.
In this section, I believe the authors sequentially:
- emphasize the role of CRE in morphological evolution (I agree)
- emphasize that TF, and in particular their own CRE, are themselves important mutational targets of evolution (I agree, but the phrasing need to insist the authors are here talking about the CRE found at the TF locus, not the CRE bound by the TF).
- use the stickleback Pel enhancer as an example, which I think is a good case study, but the authors also then make an argument about DNA fragility sites, which is hard to connect with the present study.
- then continue on "DNA fragility" using the peppered moth and butterfly cortex locus. There is no evidence of DNA fragility at these loci, so the connection does not work. "The cortex gene locus is frequently mutated in Lepidoptera", the authors say. But a more accurate picture would be that the cortex locus is repeatedly involved in the generation of color pattern variants. Unlike for Pel fragile enhancer, we don't know if the causal mutations at this locus are repeatedly the same, and the haplotypes that have been described could be collateral rather than causal. Overall, it is important to clarify the idea that mutation bias is a possible factor explaining "genetic hotspots of evolution" (or genetic parallelism sensu 10.1038/nrg3483), but it is also possible that many genetic hotspots are repeated mutational targets because of their "optimal pleiotropy" (e.g. hub position in GRNs, such as mamo might be), or because of particularly modular CRE region that allow fine-tuning. Thus, I find the "fragility" argument misleading here. In fact the finding that "bd" and "bdf" alleles are different in nature is against the idea of a fragility bias (unless the authors can show increased mutation rates at this locus in a wild silkmoth species?). These alleles are also artificially-selected ie. they increased in frequency by breeding rather than natural selection in the wild, so while interesting for our understand of the genotype-phenotype map, they are not necessarily representative of the mutations that may underlie evolution in the wild.
- Curiously, the last paragraph ("Some research suggests that common fragile sites...") elaborate on the idea that some sites of the genome are prone to mutation. The connection with mamo and the current article are extremely thin. There is here an attempt to connect meiotic and mitotic breaks to Bm-mamo, but this is confusing : it seems to propose Bm-mamo as a recruiter of epigenetic modulators that may drive higher mutation rates elsewhere. Not only I am not convinced by this argument without actual data, but this would not explain how the mutations at the Bm-mamo itself evolved.On a more positive note, I find it fascinating that the authors identified a TF that clearly articulates or orchestrate larval pattern development, and that when it is deleted, can generate healthy individuals. In other words, while it is a TF with many targets, it is not too pleiotropic. This idea, that the genetically causal modulators of developmental evolution are regulatory genes, has been described elsewhere (e.g. Fig 4c in 10.1038/s41576-020-0234-z, and associated refs). To me, the beautiful findings about Bm-mamo make sense in the general, existing framework that developmental processes and regulatory networks "shape" the evolutionary potential and trajectories of organisms. There is a degree of "programmability" in the genomes, because some loci are particularly prone to modulate a given type of trait. Here, Bm-mamo, as a potentially regulator of both CPs and melanin pathway genes, appear to be a potent modulator of epithelial traits. Claiming that there are inherent mutational biases behind this is unwarranted.
-
-
eLife assessment
This important study identifies the gene Bm-mamo as a new regulator of pigmentation in the silkworm Bombyx mori, a function that was previously unsuspected because the extensively studied Drosophila mamo gene is involved in gamete production. The evidence supporting the role of Bm-nano in pigmentation is convincing, including high-resolution linkage mapping of two mutant strains, expression profiling, and reproduction of the mutant phenotypes with state-of-the-art RNAi and CRISPR knock-out assays. The part of the work that concerns potential effector genes that Bm-mamo may regulate relies on in vitro and in silico analysis and can be used as a starting point for future in vivo studies. The work will be of interest to evolutionary biologists and geneticists interested in color patterns and in evolution of gene networks …
eLife assessment
This important study identifies the gene Bm-mamo as a new regulator of pigmentation in the silkworm Bombyx mori, a function that was previously unsuspected because the extensively studied Drosophila mamo gene is involved in gamete production. The evidence supporting the role of Bm-nano in pigmentation is convincing, including high-resolution linkage mapping of two mutant strains, expression profiling, and reproduction of the mutant phenotypes with state-of-the-art RNAi and CRISPR knock-out assays. The part of the work that concerns potential effector genes that Bm-mamo may regulate relies on in vitro and in silico analysis and can be used as a starting point for future in vivo studies. The work will be of interest to evolutionary biologists and geneticists interested in color patterns and in evolution of gene networks in general.
-
Reviewer #1 (Public Review):
Summary: This paper performs fine-mapping of the silkworm mutants bd and its fertile allelic version, bdf, narrowing down the causal intervals to a small interval of a handful of genes. In this region, the gene orthologous to mamo is impaired by a large indel, and its function is later confirmed using expression profiling, RNAi, and CRISPR KO. All these experiments are convincingly showing that mamo is necessary for the suppression of melanic pigmentation in the silkworm larval integument.
The authors also use in silico and in vitro assays to probe the potential effector genes that mamo may regulate.
Strengths: The genotype-to-phenotype workflow, combining forward (mapping) and reverse genetics (RNAi and CRISPR loss-of-function assays) linking mamo to pigmentation are extremely convincing.
Weaknesses:
The …
Reviewer #1 (Public Review):
Summary: This paper performs fine-mapping of the silkworm mutants bd and its fertile allelic version, bdf, narrowing down the causal intervals to a small interval of a handful of genes. In this region, the gene orthologous to mamo is impaired by a large indel, and its function is later confirmed using expression profiling, RNAi, and CRISPR KO. All these experiments are convincingly showing that mamo is necessary for the suppression of melanic pigmentation in the silkworm larval integument.
The authors also use in silico and in vitro assays to probe the potential effector genes that mamo may regulate.
Strengths: The genotype-to-phenotype workflow, combining forward (mapping) and reverse genetics (RNAi and CRISPR loss-of-function assays) linking mamo to pigmentation are extremely convincing.
Weaknesses:
The last section of the results, entitled "Downstream target gene analysis" is primarily based on in silico genome-wide binding motif predictions.
While the authors identify a potential binding site using EMSA, it is unclear how much this general approach over-predicted potential targets. While I think this work is interesting, its potential caveats are not mentioned. In fact the Discussion section seems to trust the high number of target genes as a reliable result. Specifically, the authors correctly say: "even if there are some transcription factor-binding sites in a gene, the gene is not necessarily regulated by these factors in a specific tissue and period", but then propose a biological explanation that not all binding sites are relevant to expression control. This makes a radical short-cut that predicted binding sites are actual in vivo binding sites. This may not be true, as I'd expect that only a subset of binding motifs predicted by Positional Weight Matrices (PWM) are real in vivo binding sites with a ChIP-seq or Cut-and-Run signal. This is particularly problematic for PWM that feature only 5-nt signature motifs, as inferred here for mamo-S and mamo-L, simply because we can expect many predicted sites by chance.The last part of the current discussion ("Notably, the industrial melanism event, in a short period of several decades ... a more advanced self-regulation program") is flawed with important logical shortcuts that assign "agency" to the evolutionary process. For instance, this section conveys the idea that phenotypically relevant mutations may not be random. I believe some of this is due to translation issues in English, as I understand that the authors want to express the idea that some parts of the genome are paths of least resistance for evolutionary change (e.g. the regulatory regions of developmental regulators are likely to articulate morphological change). But the language and tone is made worst by the mention that in another system, a mechanism involving photoreception drives adaptive plasticity, making it sound like the authors want to make a Lamarckian argument here (inheritance of acquired characteristics), or a point about orthogenesis (e.g. the idea that the environment may guide non-random mutations).
Because this last part of the current discussion suffers from confused statements on modes and tempo of regulatory evolution and is rather out of topic, I would suggest removing it.
In any case, it is important to highlight here that while this manuscript is an excellent genotype-to-phenotype study, it has very few comparative insights on the evolutionary process. The finding that mamo is a pattern or pigment regulatory factor is interesting and will deserve many more studies to decipher the full evolutionary study behind this Gene Regulatory Network.
Minor Comment :
The gene models presented in Figure 1 are obsolete, as there are more recent annotations of the Bm-mamo gene that feature more complete intron-exon structures, including for the neighboring genes in the bd/bdf intervals. It remains true that the mamo locus encodes two protein isoforms.
An example of the Bm-mamo locus annotation, can be found at : https://www.ncbi.nlm.nih.gov/gene/101738295
RNAseq expression tracks (including from larval epidermis) can be displayed in the embedded genome browser from the link above using the "Configure Tracks" tool.Based on these more recent annotations, I would say that most of the work on the two isoforms remains valid, but FigS2, and particularly Fig.S2C, need to be revised.
-
Reviewer #2 (Public Review):
Summary:
The authors tried to identify new genes involved in melanin metabolism and its spatial distribution in the silkworm Bombyx mori. They identified the gene Bm-mamo as playing a role in caterpillar pigmentation. By functional genetic and in silico approaches, they identified putative target genes of the Bm-mamo protein. They showed that numerous cuticular proteins are regulated by Bm-mamo during larval development.Strengths:
-preliminary data about the role of cuticular proteins to pattern the localization of pigments
- timely question
- challenging question because it requires the development of future genetic and cell biology tools at the nanoscaleWeaknesses:
- statistical sampling limited
- the discussion would gain in being shorter and refocused on a few points, especially the link between …Reviewer #2 (Public Review):
Summary:
The authors tried to identify new genes involved in melanin metabolism and its spatial distribution in the silkworm Bombyx mori. They identified the gene Bm-mamo as playing a role in caterpillar pigmentation. By functional genetic and in silico approaches, they identified putative target genes of the Bm-mamo protein. They showed that numerous cuticular proteins are regulated by Bm-mamo during larval development.Strengths:
-preliminary data about the role of cuticular proteins to pattern the localization of pigments
- timely question
- challenging question because it requires the development of future genetic and cell biology tools at the nanoscaleWeaknesses:
- statistical sampling limited
- the discussion would gain in being shorter and refocused on a few points, especially the link between cuticular proteins and pigmentation. The article would be better if the last evolutionary-themed section of the discussion is removed.A recent paper has been published on the same gene in Bombyx mori (https://www.sciencedirect.com/science/article/abs/pii/S0965174823000760) in August 2023. The authors must discuss and refer to this published paper through the present manuscript.
-
