Microphase separation produces interfacial environment within diblock biomolecular condensates
Curation statements for this article:-
Curated by eLife

eLife assessment
This important study investigates the structural organization of a series of diblock elastin-like polypeptide condensates. The methodology is highly compelling, as it combines multiscale simulations and fluorescence lifetime imaging microscopy experiments. The results increase our understanding of model biomolecular condensates.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The phase separation of intrinsically disordered proteins is emerging as an important mechanism for cellular organization. However, efforts to connect protein sequences to the physical properties of condensates, that is, the molecular grammar, are hampered by a lack of effective approaches for probing high-resolution structural details. Using a combination of multiscale simulations and fluorescence lifetime imaging microscopy experiments, we systematically explored a series of systems consisting of diblock elastin-like polypeptides (ELPs). The simulations succeeded in reproducing the variation of condensate stability upon amino acid substitution and revealed different microenvironments within a single condensate, which we verified with environmentally sensitive fluorophores. The interspersion of hydrophilic and hydrophobic residues and a lack of secondary structure formation result in an interfacial environment, which explains both the strong correlation between ELP condensate stability and interfacial hydrophobicity scales, as well as the prevalence of protein-water hydrogen bonds. Our study uncovers new mechanisms for condensate stability and organization that may be broadly applicable.
Article activity feed
-

-
-

Author response:
The following is the authors’ response to the previous reviews.
The authors have addressed my comments. As a final minor point, regarding comment 2, these condensates are likely viscoelastic rather than purely viscous. It is prudent to indicate that the data may refer to an apparent viscosity.
We added the following text to the manuscript to highlight the viscoelastic nature of ELP condensates, and the relationship of reported values with the steady state viscosity. “It is worth noting that the reported values, although related, may not quantitatively represent the steady-state viscosity. This discrepancy arises from the slow relaxation timescale inherent in ELP condensates with viscoelastic properties.”
-
eLife assessment
This important study investigates the structural organization of a series of diblock elastin-like polypeptide condensates. The methodology is highly compelling, as it combines multiscale simulations and fluorescence lifetime imaging microscopy experiments. The results increase our understanding of model biomolecular condensates.
-
Reviewer #1 (Public Review):
This is an interesting, informative, and well-designed study that combines theoretical and experimental methodologies to tackle the phenomenon of higher-resolution structures/substructures in model biomolecular condensates.
The authors have adequately addressed my previous concerns.
-
Reviewer #2 (Public Review):
Summary:
Latham A.P. et al. apply simulations and FLIM to analyse several di-block elastin-like polypetides and connect their sequence to the micro-structure of coacervates resulting from their phase-separation.
Strengths:
Understanding the molecular grammar of phase separating proteins and the connection with mesoscale properties of the coacervates is highly relevant. This work provides insights into micro-structures of coacervates resulting from di-block polypetides.
Weaknesses:
The results apply to a very specific architecture (di-block polypetides) with specific sequences.
-
-
Author response:
The following is the authors’ response to the original reviews.
In this letter, we respond to each of the reviewers’ comments. We support responses by referring to the revised manuscript and, where necessary, by including additional descriptions and analyses that we consider extrinsic to the manuscript itself. In this letter, all changes to the manuscript are shown in blue. As noted, the displayed figures have been added to the manuscript or the SI. We believe that we have successfully addressed all comments and that the quality of our paper has improved significantly.
Comment 1: In addition to the technical comments by the reviewers, I would encourage the authors to discuss the dependency of their observations, e.g. emergence of microphase separation, not only on the sequence of the polypeptides, but also on the …
Author response:
The following is the authors’ response to the original reviews.
In this letter, we respond to each of the reviewers’ comments. We support responses by referring to the revised manuscript and, where necessary, by including additional descriptions and analyses that we consider extrinsic to the manuscript itself. In this letter, all changes to the manuscript are shown in blue. As noted, the displayed figures have been added to the manuscript or the SI. We believe that we have successfully addressed all comments and that the quality of our paper has improved significantly.
Comment 1: In addition to the technical comments by the reviewers, I would encourage the authors to discuss the dependency of their observations, e.g. emergence of microphase separation, not only on the sequence of the polypeptides, but also on the solution conditions. Similarly, the distributions of ions in the condensate bulk, interphase, and diluted phase, and hence the interfacial free energy, are significantly affected both by the chemical composition of the condensate and the salt concentration itself, see: https://pubs.acs.org/doi/10.1021/acs.nanolett.1c03138
We thank the editor for this suggestion. Here, we have focused on the effect of sequence on condensate organization. We agree that how changes in solution condition affect condensate, including microphase separation of ELPs, is potentially interesting as well. We note this as a possible future direction at multiple places in the revised Conclusions and Discussion:
“The simulations successfully reproduced condensate stability variation upon amino acid substitution. While our study is performed at set salt concentration and temperature to isolate the contributions of amino acid hydrophobicity to condensate organization, future studies may consider implementing temperature [cite] or salt [cite] dependent models to explore how solution conditions affect the organization of ELP condensates.”
“Such a microenvironment arises from the collective behavior of many proteins, can deviate from that of individual chains, and is likely sensitive to the solution conditions,[cite] which are held constant in our study. Future work on systems with double amino acid substitutions or changes to salt concentration or temperature could elucidate the generality of the mean field interpretation and the additivity of individual contributions.”
Response to referee 1
Comment 0: This is an interesting, informative, and well-designed study that combines theoretical and experimental methodologies to tackle the phenomenon of higher-resolution structures/substructures in model biomolecular condensates. The results should be published. However, there is significant room for improvement in the presentation and interpretation of the results. As it stands, the precise definition of “frustration,” which is a main theme of this manuscript (as emphasized in the title), is not sufficiently well articulated. This situation should be rectified to avoid ””rustration” becoming a ”catch-all” term without a clear perimeter of applicability rather than a precise, informative description of the physical state of affairs. There are also a few other concerns, e.g., regarding interpretation of correlation of phase-separation critical temperature and transfer free energy of amino acid residues as well as the difference between critical temperature and onset temperature, and the way the simulated configurations are similar to that of gyroids.
We want to thank the reviewers for their insightful comments. We revised the manuscript extensively to improve its clarity and to address the reviewers’ concerns. In the following, we provide point-to-point responses to all the comments.
Comment 1: It is accurately pointed out on p.4 that elastin-like polypeptides (ELPs) undergo heat-induced phase separation and therefore exhibit lower critical solution temperatures (LCSTs). But it is not entirely clear how this feature is reproduced by the authors’ simulation. A relationship between simulated surface tension and “transition temperature” is provided in Fig.1C; but is the ”transition temperature” (authors cited ref.41 by Urry) the same as critical temperature? Apparently, Urry’s Tt is””critical onset temperature”, the temperature when phase separation happens at a given polymer concentration. This is different from the (global) critical temperature LCST - though the two may be correlated-or not-depending on the shape of the phase boundary. Moreover, is the MOFF coarse-grained forcefield (first step in the multi-scale simulation), by itself, capable of reproducing heat-induced phase separation in a way similar to the forcefield of Dignon et al., ACS Cent Sci 5, 821-230 (2019)? Or is this temperature-dependent effect appearing only subsequently, after the implementation of the MARTINI and/or all-atom steps? Clarification is needed. To afford a more informative context for the authors’ introductory discussion, the aforementioned Dignon et al. work and the review by Cinar et al. [Chem Eur J 25, 13049-13069 (2019)], both touching upon the physical underpinning of the LCST feature of elastin, should also be cited along with refs.41-43.
We thank the reviewer for their comment. First, we apologize for the lack of clarity between the global lower critical solution temperature, Tc, and the transition temperature, Tt. We have modified the manuscript to be more explicit that the transition temperature we utilize is dependent on the solution conditions, instead of the global lower critical solution temperature.
Author response image 1.
Tt as a function of concentration for ELP[V5A2G3] constructs of different chain lengths. Logarithmic fits to the data for each construct using Eq. 1 are also shown. It is evident that the different curves converge to the critical temperature Tc at the critical concentration Cc. Figure reproduced from ref.[2] CC BY 4.0.
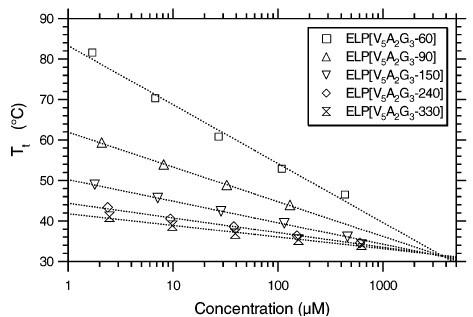
However, as shown by Chilkoti and coworkers [1, 2] and in Author response image 1, the critical temperature of ELPs Tc is indeed linearly related to Tt with the following relationship

The above equation highlights the dependence of Tt on the chain length (length) and polymer concentration (conc). The parameter Cc is the corresponding theoretical polypeptide concentration that would be required to achieve Tc, and k is the proportionality constant. Instead of making computationally expensive predictions of condensate critical temperatures, we focused on the surface tension, which can be more readily determined from single constant temperature simulations as detailed in the Methods section. This decision was made so to make it computationally feasible to systematically probe the properties of all 20 amino acids in diblock ELPs in our multiscale model. Furthermore, an expected relationship between the critical temperature and the surface tension can be inferred based on the Flory Huggins theory. In particular, relationships between the Flory Huggins parameter, χ, and interfacial tension (τ) have been investigated, and the relationship can be approximated as

where α is a positive constant, whose exact value depends on the proximity of χ to the critical value of χ necessary for phase separation (χC).[3, 4] As detailed in new Supplemental Theory of the Supporting Information, for systems undergoing LCST,
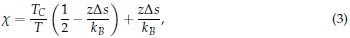
with
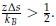 Therefore, we have
Therefore, we have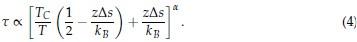
Several conclusions can be drawn from Eq. 4. First, for α = 1, τ is linearly proportional to Tc. Secondly, τ decreases at larger values for Tc since
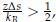 trend that is consistent with results presented in Figure 1 of the main text. Finally, as detailed in the Supplemental Theory, the inverse relationship between τ and Tc is only expected for systems exhibiting LCSTs. For systems with UCST, τ increases at larger Tc. Therefore, reproducing the correct trend supports the model’s ability to capture the temperature-dependent effect specific to the ELP system.
trend that is consistent with results presented in Figure 1 of the main text. Finally, as detailed in the Supplemental Theory, the inverse relationship between τ and Tc is only expected for systems exhibiting LCSTs. For systems with UCST, τ increases at larger Tc. Therefore, reproducing the correct trend supports the model’s ability to capture the temperature-dependent effect specific to the ELP system.We modified the text to define the physical meaning of Tt more explicitly. Furthermore, we added a new section in the Supporting Information titled Supplemental Theory to detail the relationship between Tt, Tc, the Flory-Huggins parameter χ, and the surface tension τ. The updated text now reads:
“Utilizing the simulated condensate conformations, we computed various quantities to benchmark against experimental measurements. While the critical temperature has been widely used as a measure for condensate stability, determining it computationally is expensive. As an alternative, we computed the surface tension, τ, using 100-µs-long MARTINI simulations performed with the NPNAT ensemble.[cite] As detailed in the Supplemental Theory in the Supporting information, an inverse relationship is expected between τ and the critical temperature, Tc, for systems exhibiting LCSTs. We further approximate Tc with the transition temperatures (Tt) of ELP sequences,[cite] which are the temperatures at which ELPs undergo an LCST transition at a specified solution condition. Tt was shown to be linearly proportional to TC[cite]. As expected, a negative correlation can be readily seen between computed surface tension and experimental Tt (Fig. 1C). This observed negative correlation between Tt and τ supports the simulation approach’s accuracy in reproducing the sequence-dependent changes in ELP phase behavior.”
The reviewer is correct that MOFF does not explicitly account for temperature-dependent effects in its interaction parameters. But as mentioned above and indicated by the reviewer, the following steps with explicit solvent simulations in the multiscale strategy succeed in capturing sequence-dependent differences in ELP systems, which are evident in both transition temperature and surface tension.
We cited the two references suggested by the reviewer in the introduction. We further added the following text in the discussion section to suggest explicitly exploring temperature-dependent effects as an interesting future direction.
“While our study is performed at set salt concentration and temperature to isolate the contributions of amino acid hydrophobicity to condensate organization, future studies may consider implementing temperature[cite] or salt[cite] dependent models to explore how solution conditions effect the organization of ELP condensates.”
Comment 2: “Frustration” and ”frustrated” are used prominently in the manuscript to characterize certain observed molecular configurations (11 times total, in both the title and in the abstract). Apparently, it is the most significant conceptual pronouncement of this work, hence its precise meaning is of central importance to the authors’ thesis. Whereas one should recognize that the theoretical and experimental observations are striking without invocation of the “frustration” terminology, usage of the term can be useful if it offers a unifying conceptual framework. However, as it stands, a clear definition of the term “frustration” is lacking, leaving readers to wonder what molecular configurations are considered “frustrated” and what are not (i.e., is the claim of observation of frustration falsifiable?). For instance, “frustrated microphase separation” appears in both the title and abstract. A logical question one may ask is: “Are all microphase separations frustrated”? If the answer is in the affirmative, does invocation of the term “frustration” add anything to our physical insight? If the answer is not in the affirmative, then how does one distinguish between microphase separations that are frustrated from those that are not frustrated? Presumably all simulated and experimental molecular configurations in the present study are those of lowest free energy for the given temperature. In other words, they are what they are. In the discussion about frustrated phase separation on p.13, for example, the authors appear to refer to the fact that chain connectivity is preventing hydrophobic residues to come together in a way to achieve the most favorable interactions as if there were no chain connectivity (one may imagine in that case all the hydrophobic residues will form a large cluster without microphase separation). Is this what the authors mean by “frustration”? If that’s true, isn’t that merely stating the obvious, at least for the observed microphase separation? In general, does “frustration” always mean deviation of actual, physical molecular configurations from certain imagined/hypothetical/reference molecular configurations, and therefore dependent upon the choice of the imagined reference configuration? If this is how the authors apply the term “frustration” in the present work, what is the zero-frustration reference state/configuration for microphase separation? And, similarly, what is the zero-frustration reference state/configuration when frustrated EPS-water interactions are discussed (p.14-p.15, Fig.5)? How do non-frustrated water-protein interactions look like? Is the classic clathrate-like organization of water hydrogen bonds around small nonpolar solute “frustrated”?
We thank the reviewer for their insightful comment, and agree that the concept of “frustration” is both important to our conclusions and, upon review, is too vague in our previous draft of the manuscript.
For conceptual simplicity and to maximize transferability to real biological systems, we will focus our discussion of frustration on one specific type, which we term “chain frustration.” Chain frustration occurs in states where tertiary interactions between chemically distinct polymer blocks favor phase separation, while chain connectivity prevents macroscopic phase separation from occurring.[5] This frustration leads to microphase separation with microdomains of different monomers.
We agree with the reviewer that “all microphase separations” are frustrated, and have revised the title to
“Microphase Separation Produces Interfacial Environment within Diblock Biomolecular Condensates”
Furthermore, we also removed frustration from the abstract to read
“The interspersion of hydrophilic and hydrophobic residues and a lack of secondary structure formation result in an interfacial environment, which explains both the strong correlation between ELP condensate stability and interfacial hydrophobicity scales, as well as the prevalence of protein-water hydrogen bonds.”
We have limited our discussion of the frustration to the incomplete separation of hydrophobic and hydrophobic groups. As pointed out by the reviewer, in this case, frustration refers to the fact that chain connectivity is preventing hydrophobic residues from coming together in a way to achieve the most favorable interactions as if there were no chain connectivity. The reference would be a perfectly macroscopic phase separation that partitions hydrophobic from hydrophilic groups.
While the frustration from chain connectivity is well understood for block copolymers[5], its effect on producing the interfacial solvation environment, to the best of our knowledge, has not been emphasized before. We have revised the text at the point where we mention frustration to clearly define its meaning.
“Therefore, while microphase separation occurs in ELP condensates, frustration remains in the system. Hydrophilic residues cannot completely separate from hydrophobic ones due to constraints imposed by the acid sequence, creating unique microenvironments.”
When discussing the interactions between ELP and water, we used the hydrogen bond analysis to emphasize the interfacial environment. For example, the hydrophobic residues tend to “repel” water molecules, reducing the hydrogen bond density; on the other hand, hydrophilic residues and backbone retain water molecules. This difference resulted in the positive and negative correlation with Tt shown in Fig 5C. The behavior of water molecules is, therefore, inhomogeneous inside the condensate. We expect water molecules to become frustrated due to the simultaneous contact with both hydrophobic and hydrophilic chemical groups, and a perfect reference state would be the pure water environment. However, since this point is not central to our study, to avoid confusion, we have avoided mentioning frustration and revised the text to read amino acid sequence, creating unique microenvironments.”
“The water hydrogen bond density also highlights an interfacial environment of blended hydrophobic and hydrophilic regions.”
After revising the text, frustration only appears three times in the manuscript.
Comment 3: In the discussion about the correlation of various transfer free energy scales for amino acids and Urry’s critical onset temperature (ref.41) on p.11 and Fig.4, is there any theoretical relationship to be expected between the interactions among amino acids of ELPs and their critical onset temperatures? While a certain correlation may be intuitively expected if the free energy scale ”is working”, is there any theoretical insight into the mathematical form of this relationship? A clarifying discussion is needed because it bears logically on whether the observed correlation or lack thereof for different transfer energy scales is a good indication of the adequacy of the energy scales in describing the actual physical interactions at play. This question requires some prior knowledge of the expected mathematical relationship between interaction parameters and onset temperature.
We thank the reviewer for their comment. The exact relationship between the interactions between amino acids and their transition temperature can be understood in terms of the Flory-Huggins theory, which describes the thermodynamics of polymer mixtures using a lattice model. The chemical composition of the mixture is built into the polymer-solvent interaction parameter
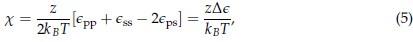
Where
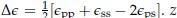 is the coordination number, T is the temperature, kB is the Boltzmann constant, and {ϵpp, ϵss, ϵps} are the strength of polymer-polymer, solventsolvent, and polymer-solvent interactions respectively.[6]
is the coordination number, T is the temperature, kB is the Boltzmann constant, and {ϵpp, ϵss, ϵps} are the strength of polymer-polymer, solventsolvent, and polymer-solvent interactions respectively.[6]From the original derivation of Flory-Huggins theory, it can be shown that phase separation occurs when χ is greater than its critical value, or
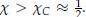 χC, we can derive the critical temperature as
χC, we can derive the critical temperature as
Δϵ can indeed be interpreted as the free energy cost of transferring a polymer bead from a solution phase to a polymer phase. It corresponds to the change of energy from a mixed state, with contacts between polymer and solvent (ϵps), to the demixed state with only polymer-polymer (ϵpp) and solvent-solvent (ϵss) contacts.
Therefore, the transfer free energy, and the interactions among amino acids of ELPs, are expected to correlate with the critical temperature. The above discussion has been incorporated into the new section Supplemental Theory in the Supporting Information. There, we also discuss the more general scenario where Δϵ is temperature dependent, which is essential for giving rise to LCST.
We have modified the main text in the discussions of Figure 4 to better explain these mathematical relationships and their necessary assumptions in order to help interpret our simulations. Here is an expert from where we discuss Figure 4:
“The strong dependence of molecular organization on amino acid hydrophobicity suggests that the solvation environment of individual residues might be a determining factor for condensate stability. Indeed, as shown in the Supplemental Theory of the Supporting Information, the critical temperature is closely related to the free energy cost of transferring polymer beads from a solution state to a polymer-only environment. This transfer free energy is often used to quantify the hydrophobicity of amino acids [cite]. To explore their relationship more quantitatively, we compared the transition temperature for ELP condensates measured by Urry [cite] to several hydrophobicity scales.”
Comment 4: To provide a more comprehensive context for the present study, it is useful to compare the microphase separation seen in the authors’ simulation with the micelle-like structures observed in recent simulated condensed/aggregated states of hydrophobic-polar (HP) model sequences in Statt et al., J Chem Phys 152, 075101 (2020) [see esp. Fig.6] and Wesse´n et al., J Phys Chem B 126, 9222-9245 (2022) [see, e.g., Fig.10].
We thank the reviewer for this suggestion. The results of Statt et al. and Wessen et al.´ indeed provide a nice comparison to our results. While we capture some of the same behavior they observe, the full array of chemical space in our model seems to give some additional morphologies as well.
First, as predicted by the self-consistent field theory, block copolymers are expected to form primarily lamellar like micelles that clearly seperate the dense and dilute phase when the volume fraction, f, is 0.5 (Response to Comment 5). This prediction is indeed consistent with results from simulations with the HP model, and is consistent with our simulations when the substituted amino acid, X, is sufficiently polar.
However, this observation is only one of several behaviors we observe. In particular, our simulations also produce gyroid-like structures, which are predicted to emerge at small volume differences, i.e. f ≈ 0.4 or f ≈ 0.6. These different configurations likely emerge due to the more realistic representation of amino acids in our model, which presents more frustration than the HP model. In particular, the backbone atoms are inherently hydrophilic and cannot separate from the hydrophobic side chains. Therefore, under microphase separation, it is inherently difficult to separate the different chemical groups to form lamellar or micelle-like structures. This produces a condensate interior with interfacial properties that may not be captured by the HP model.
We make note of the micelle-like topologies predicted by HP models in the revised text, citing both Statt et al. and Wessen et al.:´
“Surprisingly, microphase separation did not produce lamellar morphology as expected for block copolymers with equal volume fraction of the two blocks (Fig. S3 in the Supporting Information) [cite]. In particular, the condensates appear to form gyroid-like structures (Fig. S4 in the Supporting Information), in which the V and X blocks form two interpenetrating networks. This morphology also differs from micelle-like structures seen in simplified hydrophobicpolar (HP) polymers [cite]. It promotes interfacial contacts while maintaining substantial self-interactions as well. Weak interfacial tension between different ELP blocks has also been noted by Hassouneh et al.[cite]”
Comment 5: ”Gyroid-like morphology” is mentioned several times in the manuscript (p.4, p.8, p.17, Fig.S3). This is apparently an interesting observation, but a clear explanation is lacking. A more detailed and specific discussion, perhaps with additional graphical presentations, should be provided to demonstrate why the simulated condensed-phase ELP configurations are similar to the classical description of gyroid as in, e.g., Terrones & Mackay, Chem Phys Lett 207, 45-50 (1993) and Lambert et al., Phil Trans R Soc A 354, 2009-2023 (1996).
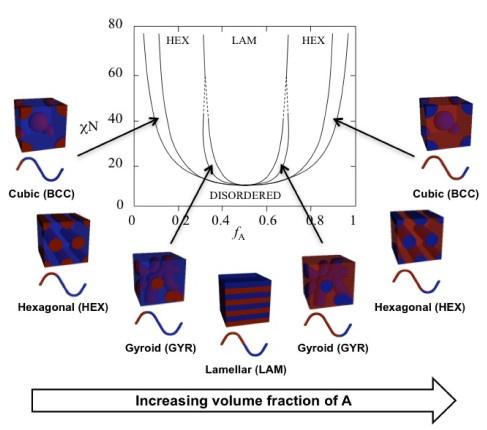
We thank the reviewer for their comment. Gyroids are canonical structures for diblock copolymers.[5, 7, 8, 9] Their stability is predicted using self-consistent field theory (SCFT), and occurs due to the balance of the volume fraction of polymer block A (fA), the length of the polymer (N), and the Flory-Huggins interaction parameter (χ).[8, 9] The prediction from SCFT suggests that gyroids occur at smaller values of χN and values fA near, but not equal to 0.5 (Author response image 2).[10] We hypothesize that these configurations emerge at equal molar fraction of V and X amino acids due to small differences in solvation volume between each half of the polymer chain.
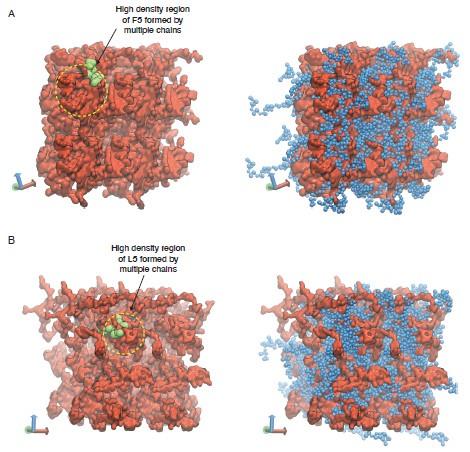
Our support for gyroid-like structures is mainly from observations of two interpenetrating networks formed by the two ELP blocks. We have revised Figure S4 to clearly highlight the two networks as shown in Author response image 3.
We have revised the main text to clearly define the gyroid-like structures as interpenetrating networks, and added the theoretical phase diagram of diblock copolymers predicted by SCFT as Figure S3 in the Supporting Information.
“In particular, the condensates appear to form gyroid-like structures (Fig. S4 in the Supporting Information), in which the V and X blocks form two interpenetrating networks. This morphology also differs from micelle-like structures seen in simplified hydrophobic-polar (HP) polymers [cite]. It promotes interfacial contacts while maintaining substantial self-interactions as well. Weak interfacial tension between different ELP blocks has also been noted by Hassouneh et al.[cite]”
We note, however, that proving that our observations are indeed gyroid structures requires more sophisticated mathematical analysis that is beyond the scope of the study. It is also possible that these structures are metastable in our simulations. We emphasize these caveats in the updated Discussion Section.
“Further studies on the thermodynamic stability of these morphologies and comparing them with predictions from the self-consistent field theory shall provide more insights into the driving forces for their emergence [cite].”
Author response image 2.
Theoretical phase diagram[8] and corresponding morphologies for diblock copolymers. The phases are labeled as: body centered cubic (BCC), hexagonal cylinders (HEX), gyroid (GYR), and lamellar (LAM). fA is the volume fraction of a single polymer block, denoted A, χ is the Flory-Huggins interaction parameter, and N is the total degree of polymerisation. Figure reproduced from ref.[10] CC BY 4.0.
Author response image 3.
Representative configurations of (A) V5F5 and (B) V5L5 condensates from MARTINI simulations. The valine substituted half of the chain is colored blue (V5) and the X substituted half of the chain is colored red (X5). To highlight the interpenetrating networks formed by the two halves, only the X substituted half of the chain is shown on the left. Simulation interfaces are once repeated periodically in the positive x and positive y dimensions for clarity. High density regions formed by the multiple X substituted half of the chains are highlighted in yellow circles, with one of the chain shown in green.
Response to referee 2
Comment 1: The experimental characterization relies on BODIPY and SBD reporting, respectively, on viscosity and polarity. The fluorescent signal of these dyes can possibly depend on many other factors, including quenching. Additional controls are required, or a more extensive discussion with additional references, and a mention to potential limitations of this approach.
We agree with the reviewer that the fluorescence lifetime signal will be affected by many factors. Compared with the fluorescence intensity, the fluorescence lifetime mainly depends on the dyes’ self properties and environmental factors. BODIPY and SBD have been used in biological systems to detect the microviscosity and micropolarity of condensates. Our group published the same SBD and BODIPY fluorophores in previous work to quantify the microenvironment of protein aggregation and condensations. The extended data (ChemBioChem 20:1078–1087. doi: 10.1002/cbic.201800782; Aggregate 4:e301. doi:10.1002/agt2.301; Nat Chem Biol 1–9. doi:10.1038/s41589-023-01477-1) shows evidences that the BODIPY is only sensitive to the viscosity while SBD is only sensitive to the polarity, but nonsensitive to other environmental factors. As for the quenched issue, the fluorophores with extended pi-rich structure display aggregation-caused quenching (ACQ) effect in high probe concentration, which will lower the fluorescence lifetime and intensity. We usually labeled the 20% molar ratio of the ELPs using NHS-ester fluorophores to get stock solutions. Due to the labeling efficiency, the exact labeling ratio is much lower than 20%. The labeled ELP stock solution will be further mixed with unlabeled ELP to get ELP solutions with low labeling fractions. We measured the ELPs labeled with a different fraction of dyes. The result shows that only BODIPY performs slight ACQ phenomena at a high
Author response image 4.
FLIM images of ELP condensates labeled with different fractions of dyes. A) FLIM images of V30A30 condensates with 5%, 2.5%, and 1% BODIPY labels. B) FLIM images of V30A30 condensates with 5%, 2.5%, and 1% fraction of SBD. Droplets were formed with a final concentration of 70 µM ELP labeled with different fractions of BODIPY or SBD in 2 M NaCl solution. Scale bar:5 µm.
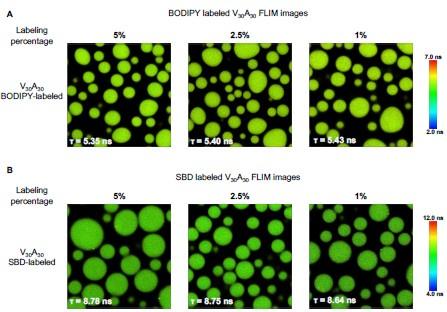
To mostly avoid the potential ACQ effect and achieve enough fluorescence signals, we finally use the ELP labeled with a lower fraction of dyes, 1% of BODIPY and 2.5 % of SBD, to perform the FLIM experiments. The data in Figure 3 will be corrected with the following data.
Author response image 5.
Structures of NHS-BODIPY and NHS-SBD, and representative FLIM images of V30A30, A30V30, V30G30 and G30V30 labeled with respective fluorophores. The fluorescence lifetime of each image is the average acquired from three independent experiments. Scale bar: 5 µm.
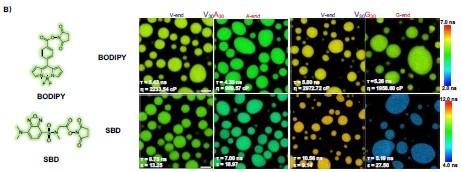
We revised the text in the section Microphase separation of ELP condensates as follows “To experimentally test the microphase separation behavior uncovered in simulations, we studied the micro-physicochemical properties of the V-end and X-end of the peptides. We constructed diblock peptides with the combination of 30 pentameric repeats of V block and X (A or G) block, namely V30A30 and V30G30 (Experimental Sequences Section in the Supporting Information). The amino-termini of V30A30 and V30G30 sequences were subsequently labeled with environmentally sensitive BODIPY or SBD fluorophores [cite], whose lifetime could be measured to quantify the viscosity or polarity of the V-end (Fig. 3A, left panel) [cite]. These probes have been reported to be only sensitive to single physicochemical properties.[cite] To avoid artifacts induced by fluorophore labeling, we usually used ELPs labeled with a low fraction of dyes. We also constructed A30V30 and G30V30 diblock peptides, wherein the viscosity or polarity of the A-end or the G-end could be measured by fluorophores that are attached at the amino-terminus (Fig. 3A, right panel). Using FLIM, we found that the lifetime of BODIPY for the V-end (5.43 ns) was longer than that for the A-end (4.35 ns), suggesting that the V-end indeed has a higher microviscosity than the A-end (ηV= 2233.54 cp vs ηA= 969.57 cp). Accordingly, the lifetime of SBD was longer for the V-end (8.75 ns) than the A-end (7.00 ns), indicating that the micropolarity of the V-end was lower than the A-end (ϵV= 13.25 vs ϵA = 18.97). These observations could be largely attributed to the greater extent of dehydration at the V-end due to its higher local peptide density. We further showed that the observed differences are not results of possible artifacts arising from any subtle distinctions between the two sequences V30A30 and A30V30 (Experimental Characterization of ELP Condensates Section in the Supporting Information, Fig. S8-S9 in the Supporting Information). Similar results were observed using the V-G sequences. FLIM experiments revealed that the V-end was more viscous than the G-end (ηV= 2972.72 cp vs ηG= 1958.60 cp) and the V-end was less polar than the G-end (ϵV= 9.14 vs ϵG = 27.50). These experimental observations provided the first line of evidence to support the microphase separation, as suggested by the simulation results.”
We revised the text in the section Experimental methods as follows
“The proteins of interest were labeled with NHS ester fluorophore. We used ELPs with 1% BODIPY labels or 2.5% SBD labels to form condensates, which avoid the artifacts induced by fluorophores. Droplets were formed with the final concentration of 70 µM ELP in 2 M NaCl for V-A and 1.5 M NH4SO4 for V-G diblock, respectively. A drop of droplets containing solution was placed on a 0.17 mm coverslip with a 500 µm spacer. Images were acquired by Leica Falcon Fluorescence Microscope equipped with Wil pulse laser and 63X/0.12 oil-immersion objective. The BODIPY was excited at 488 nm and the SBD was excited at 448 nm. The fluorescence lifetime fitting and image analysis were performed in LAS X and Image J.”
We also used a lower concentration of free dyes to remeasure the properties of the ELP condensates. The Figure S9 data are corrected as follows. The slight differences between the results are caused by experimental errors, which don’t affect the conclusion.
Author response image 6.
FLIM image of unlabeled ELP condensates. A) Chemical structure of free fluorophore, which can measure the physicochemical properties of condensates without labeling. B) Representative FLIM images of V30A30 and A30V30. The mix is the mixture of V30A30 (35 µM) and A30V30 (35 µM). Droplets were formed with a final concentration of 70 µM ELP in 2 M NaCl solution with 1 µM fluorophore. C) Representative FLIM images of V30G30 and G30V30. Droplets were formed with a final concentration of 70 µM ELP in 1.5 M (NH4)2SO4 solution with 1 µM fluorophore. The mix is the mixture of V30G30(35 µM) and G30V30 (35 µM). Scale bar, 5 µm. The fluorescence lifetime of each image is the average from three independent measurements.
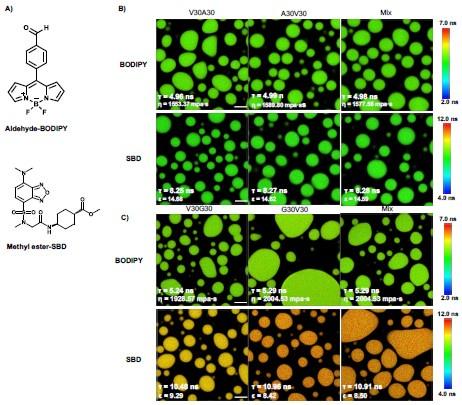
We also revised the Sequence dependence of micro-viscosity and polarity section of the Supporting Information as follows
“Since we used V30X30 and X30V30 to quantify the V- and X-end of the V-X blocks, it is possible that the observed differences arose from the innate property of the V30X30 and X30V30 sequences. To rule out this artifact, we formed the ELP condensates with sequences of V30X30, X30V30, or the V30X30 and X30V30 mixture. The condensates were subsequently treated with the aldehydeBODIPY and methyl-ester SBD fluorophores without the NHS ester reactive warhead (Fig. S9A in the Supporting Information). After brief incubation, aldehyde-BODIPY and methyl-ester SBD fluorophores were recruited into and homogeneously distributed in the ELP condensates. The fluorescence lifetime of aldehyde-BODIPY was the same for V30A30 (4.96 ns), A30V30 (4.99 ns), and their mixture (4.98 ns) (Fig. S9B in the Supporting Information, upper panel). Interestingly, this value is around the average (4.89 ns) of the A-end (4.35 ns) and the V-end (5.43 ns) labeled NHS-BODIPY. For the SBD measurement, methyl-ester SBD resulted in almost identical lifetime values of V30A30 (8.25 ns), A30V30 (8.27 ns), and their mixture (8.28 ns) (Fig. S9B in the Supporting Information, lower panel), again around the average values (7.88 ns) of the A-end (7.00 ns) and the V-end (8.75 ns) labeled NHS-SBD. In addition to the V-A blocks, similar observations were made for the V-G blocks as V30G30 and G30V30 sequences (Fig. S9C in the Supporting Information). The slight difference between the results is attributed to the experiment errors. Because the fluorophores did not covalently label the amino-terminus of the ELP peptides, their lifetime reports closer to the averaged property of the condensates instead of the microscopic property of the V-end or the X-end when the number of molecules is sufficient and the molecular distribution has no preference.
Our results reveal that the V30X30 and X30V30 condensates exhibited similar macroscopic viscosity or polarity, suggesting that the previously observed different viscosity or polarity of V30X30 and X30V30 could be attributed to the microscopic property of the V-end or X-end.”
The FLIM technique combined with environment-sensitive fluorophores is a powerful tool for us to investigate the physicochemical properties of the microenvironment within the condensates. However, there are some limitations to this method. As the fluorophore is labeled in the protein, we can only detect the microenvironment surrounding the surface of the probe(the distance may be angstrom level). The fluorescence signal values we got are the statistical average of the fluorescence signals from the complex microenvironments. The signal from the probes is determined by the sampling position, orientation, and number of fluorescent probes. So the quantified values can be compared relatively, but these values can not accurately describe the physical or chemical states in different systems. In addition, the resolution in FLIM experiments is not enough to directly distinguish the microstructure in condensates.
Comment 2: It is unclear if, after the application of stretching, the micro-structure will eventually return to the original configuration or not. Overall, the point of this experiment remains somewhat unclear.
We thank the reviewer for this comment. The ELP condensates are actually viscous fluids and they could coalesce into larger droplets within seconds. Due to the high viscosity, ELP condensates show slow fluorescence recovery after photobleaching. As stretching the condensates, the micro-structure of condensates changes to show a response to the outer force. The fluorophores may be pulled out from the microenvironment. For such a dynamic system, we speculate that the microstructure will return to the original after the condensation system equilibrium, which may be a long process. However, it is hard to characterize whether these microstructures have completely returned to their original positions. The purpose of this experiment is to show the microenvironment properties of each terminal in another aspect. The experiment also shows evidence that the microenvironment around the V terminus is more dense than the A terminus.
Comment 3: The title is too generic and does not reflect the content of the work. There is no analysis of biological condensates. The results are specific to di-block polypetides with specific sequences. This should be clearly specified in text and title.
We have revised the title to ”Microphase Separation Produces Interfacial Environment within Diblock Biomolecular Condensates”
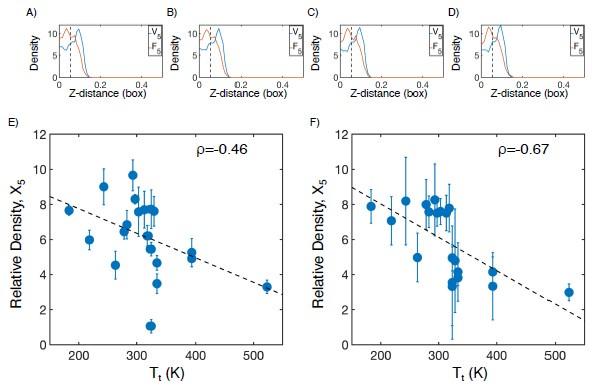
Comment 4: MD is out of the expertise of this reviewer. However, when looking at the density profiles (Figure S2), the simulation does not seem to be fully converged. The densities fluctuate inconsistently along the Z direction. The authors should comment on assessing simulation convergence. In many cases, the section used for the density values in the plot (i.e., below 0.06 box lengths away from the condensate center) does not seem representative of the dense phase. It should be justified, why these simulations can still be used for density/hydrogen bonding analysis.
We thank the reviewer for their comment, and agree that convergence of MD simulations is simultaneously important and difficult to control for. To demonstrate the convergence of our simulations, we have taken an example system (V5F5) and reproduced the density profile in 4 unique time windows of 50 ns each (Author response image 7A-D). We find that all distributions are nearly identical, indicating that further extending these simulations is unlikely to change our findings.
While we agree that the choice of 0.06 box lengths is arbitrary, it was chosen as an approximation for the interior of the condensate, where the more hydrophobic half of the protein chain tends to be at higher concentration. However, this choice is not important to our overall conclusion. Halving (Author response image 7E) or doubling (Author response image 7F) the cutoff maintains the inverse correlation between the protein density of the X5 half of the condensate and experimental transition temperature.
Finally, in our multiscale simulation approach, the all-atom portion of the simulation is mostly used to examine water structure and protein solvation. We can see that dividing the simulation into four independent time estimates does not substantially change these properties, resulting in low standard deviations in Figure 5 and Figure 6. Similarly, our previous work on the dielectric of ELP condensates has shown that choosing different starting structures from MARTINI simulations is unlikely to effect the estimate of similar quantities.[11]
Author response image 7.
Checking convergence of all-atom simulations of ELP condensates. (A-D) The relative mass density along the Z-distance from the condensate center is shown for the V-substituted and X-substituted halves of V5F5 in four independent time windows of 50 ns each. The Z−axis is defined as the direction perpendicular to the condensate-water interface. The dashed line represents a Z-distance of 0.06 box lengths away from the condensate center, which was the original cutoff for correlation analysis. E-F) Correlation between the mass fraction of the X5 half of the condensate and transition temperature (Tt) from Urry.[12] The condensate is defined as having a Z-distance of 0.03 box lengths (E) or 0.12 box lengths (F) away from the condensate center. ρ is the Pearson correlation coefficient between the two data sets, and the dashed diagonal line is the best fit line. Error bars represent standard deviations of the mean taken over box length intervals of 0.01.
References
(1) McDaniel JR, Radford DC, Chilkoti A (2013) A unified model for de novo design of elastin-like polypeptides with tunable inverse transition temperatures. Biomacromolecules 14:2866–2872.
](2) Meyer DE, Chilkoti A (2004) Quantification of the effects of chain length and concentration on the thermal behavior of elastin-like polypeptides. Biomacromolecules 5:846–851.
(3) Helfand E, Tagami Y (1972) Theory of the interface between immiscible polymers. J. Chem. Phys. 56:3592.
(4) Roe RJ (1975) Theory of the interface between polymers or polymer solutions. I. Two components system. J. Chem. Phys. 62:490–499.
(5) Shi AC (2021) Frustration in block copolymer assemblies. J. Phys. Condens. Matter 33.
(6) Flory PJ (1942) Thermodynamics of high polymer solutions. J. Chem. Phys. 10:51.
(7) Grason GM (2006) The packing of soft materials: Molecular asymmetry, geometric frustration and optimal lattices in block copolymer melts. Phys. Rep. 433:1–64.
(8) Matsen MW, Bates FS (1996) Unifying weak- and strong-segregation block copolymer theories. Macromolecules 29:1091–1098.
(9) Matsen MW, Schick M (1994) Stable and unstable phases of a diblock copolymer melt. Phys. Rev. Lett. 72:2660–2663.
(10) Swann JM, Topham PD (2010) Design and application of nanoscale actuators using block-copolymers. Polymers 2:454–469.
(11) Ye S et al. (2023) Micropolarity governs the structural organization of biomolecular condensates. Nat. Chem. Biol. pp 1–9.
(12) Urry DW (1997) Physical chemistry of biological free energy transduction as demonstrated by elastic protein-based polymers. J. Phys. Chem. B 101:11007–11028.
-
eLife assessment
This important study investigates the structural organization of a series of diblock elastin-like polypeptide condensates. The methodology is highly compelling, as it combines multiscale simulations and fluorescence lifetime imaging microscopy experiments. The results increase our understanding of model biomolecular condensates.
-
Reviewer #1 (Public Review):
This is an interesting, informative, and well-designed study that combines theoretical and experimental methodologies to tackle the phenomenon of higher-resolution structures/substructures in model biomolecular condensates. However, there is significant room for improvement in the presentation and interpretation of the results. As it stands, the precise definition of "frustration," which is a main theme of this manuscript (as emphasized in the title), is not sufficiently well articulated. This situation should be rectified to avoid "frustration" becoming a "catch-all" term without a clear perimeter of applicability rather than a precise, informative description of the physical state of affairs. There are also a few other concerns, e.g., regarding interpretation of correlation of phase-separation critical …
Reviewer #1 (Public Review):
This is an interesting, informative, and well-designed study that combines theoretical and experimental methodologies to tackle the phenomenon of higher-resolution structures/substructures in model biomolecular condensates. However, there is significant room for improvement in the presentation and interpretation of the results. As it stands, the precise definition of "frustration," which is a main theme of this manuscript (as emphasized in the title), is not sufficiently well articulated. This situation should be rectified to avoid "frustration" becoming a "catch-all" term without a clear perimeter of applicability rather than a precise, informative description of the physical state of affairs. There are also a few other concerns, e.g., regarding interpretation of correlation of phase-separation critical temperature and transfer free energy of amino acid residues as well as the difference between critical temperature and onset temperature, and the way the simulated configurations are similar to that of gyroids. Accordingly, the manuscript should be revised to address the following:
(1) It is accurately pointed out on p.4 that elastin-like polypeptides (ELPs) undergo heat-induced phase separation and therefore exhibit lower critical solution temperatures (LCSTs). But it is not entirely clear how this feature is reproduced by the authors' simulation. A relationship between simulated surface tension and "transition temperature" is provided in Fig.1C; but is the "transition temperature" (authors cited ref.41 by Urry) the same as critical temperature? Apparently, Urry's Tt is "critical onset temperature", the temperature when phase separation happens at a given polymer concentration. This is different from the (global) critical temperature LCST - though the two may be correlated-or not-depending on the shape of the phase boundary. Moreover, is the MOFF coarse-grained forcefield (first step in the multi-scale simulation), by itself, capable of reproducing heat-induced phase separation in a way similar to the forcefield of Dignon et al., ACS Cent Sci 5, 821-230 (2019)? Or, is this temperature-dependent effect appearing only subsequently, after the implementation of the MARTINI and/or all-atom steps? Clarification is needed. To afford a more informative context for the authors' introductory discussion, the aforementioned Dignon et al. work and the review by Cinar et al. [Chem Eur J 25, 13049-13069 (2019)], both touching upon the physical underpinning of the LCST feature of elastin, should also be cited along with refs.41-43.
(2) "Frustration" and "frustrated" are used prominently in the manuscript to characterize certain observed molecular configurations (11 times total, in both the title and in the abstract). Apparently, it is the most significant conceptual pronouncement of this work, hence its precise meaning is of central importance to the authors' thesis. Whereas one should recognize that the theoretical and experimental observations are striking without invocation of the "frustration" terminology, usage of the term can be useful if it offers a unifying conceptual framework. However, as it stands, a clear definition of the term "frustration" is lacking, leaving readers to wonder what molecular configurations are considered "frustrated" and what are not (i.e.,is the claim of observation of frustration falsifiable?). For instance, "frustrated microphase separation" appears in both the title and abstract. A logical question one may ask is: "Are all microphase separations frustrated"? If the answer is in the affirmative, does invocation of the term "frustration" add anything to our physical insight? If the answer is not in the affirmative, then how does one distinguish between microphase separations that are frustrated from those that are not frustrated? Presumably all simulated and experimental molecular configurations in the present study are those of lowest free energy for the given temperature. In other words, they are what they are. In the discussion about frustrated phase separation on p.13, for example, the authors appear to refer to the fact that chain connectivity is preventing hydrophobic residues to come together in a way to achieve the most favorable interactions as if there were no chain connectivity (one may imagine in that case all the hydrophobic residues will form a large cluster without microphase separation). Is this what the authors mean by "frustration"? If that's true, isn't that merely stating the obvious, at least for the observed microphase separation? In general, does "frustration" always mean deviation of actual, physical molecular configurations from certain imagined/hypothetical/reference molecular configurations, and therefore dependent upon the choice of the imagined reference configuration? If this is how the authors apply the term "frustration" in the present work, what is the zero-frustration reference state/configuration for microphase separation? And, similarly, what is the zero-frustration reference state/configuration when frustrated EPS-water interactions are discussed (~p.14-p.15, Fig.5)? How do non-frustrated water-protein interactions look like? Is the classic clathrate-like organization of water hydrogen bonds around small nonpolar solute "frustrated"?
(3) In the discussion about the correlation of various transfer free energy scales for amino acids and Urry's critical onset temperature (ref.41) on p.11 and Fig.4, is there any theoretical relationship to be expected between the interactions among amino acids of ELPs and their critical onset temperatures? While a certain correlation may be intuitively expected if the free energy scale "is working", is there any theoretical insight into the mathematical form of this relationship? A clarifying discussion is needed because it bears logically on whether the observed correlation or lack thereof for different transfer energy scales is a good indication of the adequacy of the energy scales in describing the actual physical interactions at play. This question requires some prior knowledge of the expected mathematical relationship between interaction parameters and onset temperature.
(4) To provide a more comprehensive context for the present study, it is useful to compare the microphase separation seen in the authors' simulation with the micelle-like structures observed in recent simulated condensed/aggregated states of hydrophobic-polar (HP) model sequences in Statt et al., J Chem Phys 152, 075101 (2020) [see esp. Fig.6] and Wessén et al., J Phys Chem B 126, 9222-9245 (2022) [see, e.g., Fig.10].
(5) "Gyroid-like morphology" is mentioned several times in the manuscript (p.4, p.8, p.17, Fig.S3). This is apparently an interesting observation but a clear explanation is lacking. A more detailed and specific discussion, perhaps with additional graphical presentations, should be provided to demonstrate why the simulated condensed-phase ELP configurations are similar to the classical description of gyroid as in, e.g., Terrones & Mackay, Chem Phys Lett 207, 45-50 (1993) and Lambert et al., Phil Trans R Soc A 354, 2009-2023 (1996).
Comments on the revised manuscript:
The authors have adequately addressed my previous concerns.
-
Reviewer #2 (Public Review):
Summary:
Latham A.P. et al. apply simulations and FLIM to analyse several di-block elastin-like polypetides and connect their sequence to the micro-structure of coacervates resulting from their phase-separation.
Strengths:
Understanding the molecular grammar of phase separating proteins and the connection with mesoscale properties of the coacervates is highly relevant. This work provides insights into micro-structures of coacervates resulting from di-block polypetides.
Weaknesses:
The results apply to a very specific architecture (di-block polypetides) with specific sequences.
-
-
eLife assessment
This important study investigates the structural organization of a series of diblock elastin-like polypeptide condensates. The methodology is highly compelling, as it combines multiscale simulations and fluorescence lifetime imaging microscopy experiments. The results increase our understanding of model biomolecular condensates. The manuscript would benefit from more details for the concepts and terminology introduced, as well as the analysis of the simulations.
-
Reviewer #1 (Public Review):
This is an interesting, informative, and well-designed study that combines theoretical and experimental methodologies to tackle the phenomenon of higher-resolution structures/substructures in model biomolecular condensates. The results should be published. However, there is significant room for improvement in the presentation and interpretation of the results. As it stands, the precise definition of "frustration," which is a main theme of this manuscript (as emphasized in the title), is not sufficiently well articulated. This situation should be rectified to avoid "frustration" becoming a "catch-all" term without a clear perimeter of applicability rather than a precise, informative description of the physical state of affairs. There are also a few other concerns, e.g., regarding interpretation of correlation …
Reviewer #1 (Public Review):
This is an interesting, informative, and well-designed study that combines theoretical and experimental methodologies to tackle the phenomenon of higher-resolution structures/substructures in model biomolecular condensates. The results should be published. However, there is significant room for improvement in the presentation and interpretation of the results. As it stands, the precise definition of "frustration," which is a main theme of this manuscript (as emphasized in the title), is not sufficiently well articulated. This situation should be rectified to avoid "frustration" becoming a "catch-all" term without a clear perimeter of applicability rather than a precise, informative description of the physical state of affairs. There are also a few other concerns, e.g., regarding interpretation of correlation of phase-separation critical temperature and transfer free energy of amino acid residues as well as the difference between critical temperature and onset temperature, and the way the simulated configurations are similar to that of gyroids. Accordingly, the manuscript should be revised to address the following:
1. It is accurately pointed out on p.4 that elastin-like polypeptides (ELPs) undergo heat-induced phase separation and therefore exhibit lower critical solution temperatures (LCSTs). But it is not entirely clear how this feature is reproduced by the authors' simulation. A relationship between simulated surface tension and "transition temperature" is provided in Fig.1C; but is the "transition temperature" (authors cited ref.41 by Urry) the same as critical temperature? Apparently, Urry's Tt is "critical onset temperature", the temperature when phase separation happens at a given polymer concentration. This is different from the (global) critical temperature LCST - though the two may be correlated-or not-depending on the shape of the phase boundary. Moreover, is the MOFF coarse-grained forcefield (first step in the multi-scale simulation), by itself, capable of reproducing heat-induced phase separation in a way similar to the forcefield of Dignon et al., ACS Cent Sci 5, 821-230 (2019)? Or is this temperature-dependent effect appearing only subsequently, after the implementation of the MARTINI and/or all-atom steps? Clarification is needed. To afford a more informative context for the authors' introductory discussion, the aforementioned Dignon et al. work and the review by Cinar et al. [Chem Eur J 25, 13049-13069 (2019)], both touching upon the physical underpinning of the LCST feature of elastin, should also be cited along with refs.41-43.
2. "Frustration" and "frustrated" are used prominently in the manuscript to characterize certain observed molecular configurations (11 times total, in both the title and in the abstract). Apparently, it is the most significant conceptual pronouncement of this work, hence its precise meaning is of central importance to the authors' thesis. Whereas one should recognize that the theoretical and experimental observations are striking without invocation of the "frustration" terminology, usage of the term can be useful if it offers a unifying conceptual framework. However, as it stands, a clear definition of the term "frustration" is lacking, leaving readers to wonder what molecular configurations are considered "frustrated" and what are not (i.e., is the claim of observation of frustration falsifiable?). For instance, "frustrated microphase separation" appears in both the title and abstract. A logical question one may ask is: "Are all microphase separations frustrated"? If the answer is in the affirmative, does invocation of the term "frustration" add anything to our physical insight? If the answer is not in the affirmative, then how does one distinguish between microphase separations that are frustrated from those that are not frustrated? Presumably all simulated and experimental molecular configurations in the present study are those of lowest free energy for the given temperature. In other words, they are what they are. In the discussion about frustrated phase separation on p.13, for example, the authors appear to refer to the fact that chain connectivity is preventing hydrophobic residues to come together in a way to achieve the most favorable interactions as if there were no chain connectivity (one may imagine in that case all the hydrophobic residues will form a large cluster without microphase separation). Is this what the authors mean by "frustration"? If that's true, isn't that merely stating the obvious, at least for the observed microphase separation? In general, does "frustration" always mean deviation of actual, physical molecular configurations from certain imagined/hypothetical/reference molecular configurations, and therefore dependent upon the choice of the imagined reference configuration? If this is how the authors apply the term "frustration" in the present work, what is the zero-frustration reference state/configuration for microphase separation? And, similarly, what is the zero-frustration reference state/configuration when frustrated EPS-water interactions are discussed (~p.14-p.15, Fig.5)? How do non-frustrated water-protein interactions look like? Is the classic clathrate-like organization of water hydrogen bonds around small nonpolar solute "frustrated"?
3. In the discussion about the correlation of various transfer free energy scales for amino acids and Urry's critical onset temperature (ref.41) on p.11 and Fig.4, is there any theoretical relationship to be expected between the interactions among amino acids of ELPs and their critical onset temperatures? While a certain correlation may be intuitively expected if the free energy scale "is working", is there any theoretical insight into the mathematical form of this relationship? A clarifying discussion is needed because it bears logically on whether the observed correlation or lack thereof for different transfer energy scales is a good indication of the adequacy of the energy scales in describing the actual physical interactions at play. This question requires some prior knowledge of the expected mathematical relationship between interaction parameters and onset temperature.
4. To provide a more comprehensive context for the present study, it is useful to compare the microphase separation seen in the authors' simulation with the micelle-like structures observed in recent simulated condensed/aggregated states of hydrophobic-polar (HP) model sequences in Statt et al., J Chem Phys 152, 075101 (2020) [see esp. Fig.6] and Wessén et al., J Phys Chem B 126, 9222-9245 (2022) [see, e.g., Fig.10].
5. "Gyroid-like morphology" is mentioned several times in the manuscript (p.4, p.8, p.17, Fig.S3). This is apparently an interesting observation, but a clear explanation is lacking. A more detailed and specific discussion, perhaps with additional graphical presentations, should be provided to demonstrate why the simulated condensed-phase ELP configurations are similar to the classical description of gyroid as in, e.g., Terrones & Mackay, Chem Phys Lett 207, 45-50 (1993) and Lambert et al., Phil Trans R Soc A 354, 2009-2023 (1996).
-
Reviewer #2 (Public Review):
Summary:
Latham A.P. et al. apply simulations and FLIM to analyse several di-block elastin-like polypetides and connect their sequence to the micro-structure of coacervates resulting from their phase-separation.Strengths:
Understanding the molecular grammar of phase separating proteins and the connection with mesoscale properties of the coacervates is highly relevant. This work provides insights into micro-structures of coacervates resulting from di-block polypetides.Weaknesses:
The results apply to a very specific architecture (di-block polypetides) with specific sequences. -
