Fungal–bacteria interactions provide shelter for bacteria in Caesarean section scar diverticulum
Curation statements for this article:-
Curated by eLife

eLife assessment
This important study reports the fungal composition and its interaction with bacteria in the Caesarean section scar diverticulum. The data are solid and supportive of the conclusion. This work will be of interest to researchers and clinicians who work on women's health.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Caesarean section scar diverticulum (CSD) is a significant cause of infertility among women who have previously had a Caesarean section, primarily due to persistent inflammatory exudation associated with this condition. Even though abnormal bacterial composition is identified as a critical factor leading to this chronic inflammation, clinical data suggest that a long-term cure is often unattainable with antibiotic treatment alone. In our study, we employed metagenomic analysis and mass spectrometry techniques to investigate the fungal composition in CSD and its interaction with bacteria. We discovered that local fungal abnormalities in CSD can disrupt the stability of the bacterial population and the entire microbial community by altering bacterial abundance via specific metabolites. For instance, Lachnellula suecica reduces the abundance of several Lactobacillus spp., such as Lactobacillus jensenii , by diminishing the production of metabolites like Goyaglycoside A and Janthitrem E . Concurrently, Clavispora lusitaniae and Ophiocordyceps australis can synergistically impact the abundance of Lactobacillus spp. by modulating metabolite abundance. Our findings underscore that abnormal fungal composition and activity are key drivers of local bacterial dysbiosis in CSD.
Article activity feed
-

-
-
-

Author Response
The following is the authors’ response to the previous reviews.
Thank you once again for your patience and guidance through this revision process. I would like to add an important aspect to our previous discussion regarding the identification and impact of potential contaminants in our study.
In recent years, advanced tools such as SCRuB (recently published in Nature Biotechnology, DOI:10.1038/s41587-023-01696-w) and the widely-used tool decontam have been developed to address the issue of contaminants in metagenomic studies. These tools primarily operate based on sequence similarity, identifying potential contaminants by marking and removing those found in only a minority of samples or those that display patterns indicative of laboratory contamination.
As the reviewer rightly pointed out, contaminants are often rare …
Author Response
The following is the authors’ response to the previous reviews.
Thank you once again for your patience and guidance through this revision process. I would like to add an important aspect to our previous discussion regarding the identification and impact of potential contaminants in our study.
In recent years, advanced tools such as SCRuB (recently published in Nature Biotechnology, DOI:10.1038/s41587-023-01696-w) and the widely-used tool decontam have been developed to address the issue of contaminants in metagenomic studies. These tools primarily operate based on sequence similarity, identifying potential contaminants by marking and removing those found in only a minority of samples or those that display patterns indicative of laboratory contamination.
As the reviewer rightly pointed out, contaminants are often rare species that appear in very few samples. Our study, focusing on high-abundance species in the vaginal microbiome, is less susceptible to the influences of such rare contaminants. This approach aligns with the methodology employed by leading research groups in the field, such as Professor Jacques Ravel's lab. Their decision not to use blank controls in several of their studies on the female reproductive tract microbiome likely stems from a similar understanding — that the impact of rare contaminants is minimal on the study's conclusions, especially when high-abundance species are the main focus.
We believe that the methodologies and tools currently available for contaminant identification and removal, while highly effective for their intended purpose, reinforce our decision to focus on high-abundance species. This focus minimizes the potential impact of rare contaminants on our study's conclusions. In light of this, our study's methodology remains robust and well-suited for achieving our research objectives.
In our revised manuscript, we will include a discussion of these points, further clarifying our approach and the rationale behind our methodological choices. We hope that this additional information will address the concerns raised and provide a clearer understanding of the context and reliability of our findings.
Thank you for considering these additional points. We look forward to your feedback on our revised manuscript.
-
eLife assessment
This important study reports the fungal composition and its interaction with bacteria in the Caesarean section scar diverticulum. The data are solid and supportive of the conclusion. This work will be of interest to researchers and clinicians who work on women's health.
-
Reviewer #2 (Public Review):
Summary:
Shotgun data have been analysed to obtain fungal and bacterial organisms abundance. Through their metabolic functions and through co-occurrence networks, a functional relationship between the two types of organisms can be inferred. By means of metabolomics, function-related metabolites are studied in order to deepen the fungus-bacteria synergy.
Strengths:
Data obtained in bacteria correlate with data from other authors.
The study of metabolic "interactions" between fungi and bacteria is quite new.
The inclusion of metabolomics data to support the results is a great contribution.Weaknesses:
All my concerns have been clarified
-
-
Author Response
The following is the authors’ response to the original reviews.
necessary clarifications on some of the reviewers' suggestions.
Reviewer #1 (Public Review):
Weaknesses:
- This is a pilot study with only 24 cases and 24 controls. Because the human microbiota entails individual variability, this work should be confirmed with a higher sample size to achieve enough statistical power.
Thank you for your suggestion. Unlike the high sparsity of 16s rRNA, the data density of metagenomic data is higher. Based on the experience of previous research, the sample size used this time can basically meet the requirements. However, your suggestion is very valuable, increasing the sample size allows better in-depth analysis. Due to limitations of objective factors, it is difficult for us to continue to increase the sample size in this …
Author Response
The following is the authors’ response to the original reviews.
necessary clarifications on some of the reviewers' suggestions.
Reviewer #1 (Public Review):
Weaknesses:
- This is a pilot study with only 24 cases and 24 controls. Because the human microbiota entails individual variability, this work should be confirmed with a higher sample size to achieve enough statistical power.
Thank you for your suggestion. Unlike the high sparsity of 16s rRNA, the data density of metagenomic data is higher. Based on the experience of previous research, the sample size used this time can basically meet the requirements. However, your suggestion is very valuable, increasing the sample size allows better in-depth analysis. Due to limitations of objective factors, it is difficult for us to continue to increase the sample size in this study.
- The authors do not report here the use of blank controls. The use of this type of control is important to "subtract" the potential background from plasticware, buffer or reagents from the real signal. Lack of controls may lead to microbiome artefacts in the results. This can be seen in the results presented where the authors report some bacterial contaminants (Agrobacterium tumefaciensis, Aequorivita lutea, Chitinophagaceae, Marinobacter vinifirmus, etc) as part of the most common bacteria found in cervical samples.
Thank you for your suggestion. Applying blank controls in low biomass areas can effectively avoid contamination caused by the environment or kits. This opinion is consistent with that published by Raphael Eisenhofer et al. in Trends in Microbiology. When designing this study, we considered that this study described a biomass-rich site, and the abundance of dominant species was much higher than that of the possible 'kitome', so we did not set a blank control. On the other hand, our main discussion object in this study is high-abundance species, and the species filtering threshold for some analyzes was raised to 50%. Therefore, we believe that the absence of the blank control has little effect on the conclusions of this study. However, your opinion is spot on. Failure to set up a negative control will affect our future research on rare species. We will add a description in the Limitations section of the Discussion section.
- Samples used for this study were collected from the cervix. Why not collect samples from the uterine cavity and isthmocele fluid (for cases)? In their previous paper using samples from the same research protocol ((IRB no. 2019ZSLYEC-005S) they used endometrial tissue from the patients, so access to the uterine cavity was guaranteed.
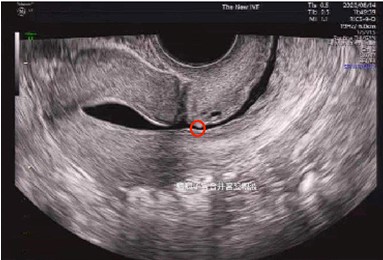
Thank you for your suggestion. In Author response image 1 we show the approximate location of our cervical swab sampling. There are two main reasons for choosing cervical swabs:
The adsorption of swabs allows us to obtain sufficient nucleic acid for high-depth sequencing, while the isthmocele fluid varies greatly among patients, which will introduce unnecessary batch effects.
Since the female reproductive tract is a continuous whole, our sampling location is close to the lesion in the cervix, which can be effectively studied. On the other hand, the microbial biomass of the endometrium is probably two orders of magnitude lower than that of the cervix, and it is difficult to avoid contamination of the lower genital tract when sampling.
Based on the above reasons, we selected cervical swabs for our microbial data.
Author response image 1.
- Through the use of shotgun genomics, results from all the genomes of the organisms present in the sample are obtained. However, the authors have only used the metagenomic data to infer the taxonomical annotation of fungi and bacteria.
Thank you for your suggestion. The advantage of metagenomics is that it can obtain all the nucleic acid information of the entire environment. However, in the study of the female reproductive tract, the database of viruses and archaea is still immature, in order to ensure the accuracy of the results, we did not conduct the study. Looking forward to the emergence of a mature database in the future.
Reviewer #1 (Recommendations For The Authors):
- It would be interesting to use another series of functional data coming from the metagenomic analyses (not only taxonomic) to expand and reinforce the results presented.
Thank you for your suggestion. We have dissected the functional data of microbiota in the article.
- The authors have previously published the 16S rRNA sequencing and transcriptomic analysis of the same set of patients. It would be nice to see the integration of all the datasets produced.
Thank you for your suggestion. There is no doubt that integrating all the data will have more dimensional results. In our previous study we focused on microbe-host interactions. However, there is an unanswered question: What are the characteristics of the regulatory network within microbiota? Therefore, we answered this question in this study, exploring the complex interaction processes within microbial communities. In addition to direct effects, interactions between microbiota may also occur through special metabolite experiments. Therefore, we introduced the analysis of the untargeted metabolome. However, 16s rRNA can only provide bacterial information, so we did not integrate the data. In addition, the transcriptome provides host information and is not the focus of this study. However, your suggestion is very valuable, and we will integrate all the data in the next study on the exploration of treatment methods.
Reviewer #2 (Public Review):
Weaknesses: Methodological descriptions are minimal.
Some example:
*The CON group (line 147) has not been defined. I supposed it is the control group.
- There are no statistics related to shotgun sequencing. How many reads have been sequenced? How many have been removed from the host? How many are left to study bacteria and fungi? Are these reads proportional among the 48 samples? If not, what method has been used to normalise the data?
- ggClusterNet has numerous algorithms to better display the modules of the microbiome network. Which one has been used?
Thank you for your suggestion. We have added details to the method.
Reviewer #2 (Recommendations For The Authors):
I think the author should take into account the points described in the "Weaknesses" section. The lack of detail extends to almost all the analyses that have been included in the manuscript. Although the results are sound, I think it is important to understand what has been analysed and how it has been analysed. It is important that all work is reproducible and this requires vital information.
For example, what parameters have been used for bowtie2? has a local analysis been used? or end-to-end ? Some parameters like --very-sensitive are important for this kind of analysis. You can also use specific programs like kneaddata.
The Raw data preprocessing section should be more detailed.
The same with the "Taxa and functional annotation" section, how have the data been normalised? has any Zero-Inflated Gamma probabilistic model algorithm been taken into account? How were the 0 (no species detected) in the shallow samples treated?
Which algorithms have been used for LEfSe ? Kluskal-Wallis->(Wilcoxon)->LDA ?
Which p-value has been used as cut-off ? this p-value has been corrected for multiple testing?
- Information on ggClusterNet should be included and explained.
The first section of the results and Table 1 should be in the Materials and Methods.
Thank you for your suggestion. We have added details to the method.
In the fungi section, it is mentioned that 431 species have been found. They should be included in a supplementary table.
How many bacteria were found? Please include them also in a supplementary table.
Thank you for your suggestion. We have added the corresponding table.
Reviewer #3 (Public Review):
Major
- Smoke or drink conditions, as well as diseases like hypertension and diabetes are important factors that could influence the metabolism of the host, thus the authors should add them in the exclusion criteria in the Methods.
Thanks to reviewer #3 for professional comments. We have made corresponding additions in the method section. We also followed this standard when recruiting subjects.
- The sample size of this study is not large enough to draw a convincing conclusion.
Thank you for your suggestion. Unlike the high sparsity of 16s rRNA, the data density of metagenomic data is higher. Based on the experience of previous research, the sample size used this time can basically meet the requirements. However, your suggestion is very valuable, increasing the sample size allows better in-depth analysis. Due to limitations of objective factors, it is difficult for us to continue to increase the sample size in this study.
Reviewer #3 (Recommendations For The Authors):
Please recruit more samples.
In addition, there are many formatting and grammatical mistakes in the manuscript.
Minor
- In Line 24-25 of the "Composition and characteristics of fungal communities", the format of "Goyaglycoside A and Janthitrem E." shouldn't be italic.
- In Line 126 of the "Metabolite detection using liquid chromatography (LC) and mass spectrometry (MS)", the "10 ul" should be changed to "Ten ul". Beginning with arabic numerals in a sentence should be avoided.
- In Line 170 of the "Composition and characteristics of bacterial communities", the "162 differential species" should be "One hundred and sixty-two differential species".
- In Line 187 of the "Composition and characteristics of fungal communities", the "42 differential" should be "Forty-two differential".
Thanks to reviewer #3 for professional comments. We have completely revised the language of the article.
-
eLife assessment
This study has uncovered some interesting findings about the fungal composition and its interaction with bacteria in Caesarean section scar diverticulum (CSD). While the study's findings are valuable and with translation possibilities, the strength of the conclusions obtained is incomplete due to the small sample size and methodological issues indicated by the reviewers such as the lack of controls and the location of samples analyzed.
-
Reviewer #1 (Public Review):
Summary:
Chen et al. describe the bacterial and fungal composition of cervical samples from women with/without Cesarean-section scar diverticulum (CSD) using whole metagenomic sequencing. Also, they report the metabolomic profile associated with CSD and built correlation networks at the taxonomical and taxonomic-metabolic levels to establish potential bacteria-fungi interactions. These interactions could be used, long-term, as therapeutic options to treat or prevent CSD.
After reviewing the manuscript, the authors have not integrated any of my previous recommendations into the new version of the work. Therefore, in my opinion, the limitations or weaknesses of the study remain the same.
I find it especially worrying that they do not consider the use of white controls necessary, arguing that "we considered …
Reviewer #1 (Public Review):
Summary:
Chen et al. describe the bacterial and fungal composition of cervical samples from women with/without Cesarean-section scar diverticulum (CSD) using whole metagenomic sequencing. Also, they report the metabolomic profile associated with CSD and built correlation networks at the taxonomical and taxonomic-metabolic levels to establish potential bacteria-fungi interactions. These interactions could be used, long-term, as therapeutic options to treat or prevent CSD.
After reviewing the manuscript, the authors have not integrated any of my previous recommendations into the new version of the work. Therefore, in my opinion, the limitations or weaknesses of the study remain the same.
I find it especially worrying that they do not consider the use of white controls necessary, arguing that "we considered that this study described a biomass-rich site, and the abundance of dominant species was much higher than that of the possible 'kitome', so we did not set a blank control" while describing among the most predominant species in the reproductive tract bacteria that do not colonize humans and that have been previously described as contaminants.
Lack of experimental controls can lead to artifactual results and compromise the evidence presented and the significance of the results.
-
Reviewer #2 (Public Review):
Summary:
Shotgun data have been analysed to obtain fungal and bacterial organisms abundance. Through their metabolic functions and through co-occurrence networks, a functional relationship between the two types of organisms can be inferred. By means of metabolomics, function-related metabolites are studied in order to deepen the fungus-bacteria synergy.
Strengths:
Data obtained in bacteria correlate with data from other authors.
The study of metabolic "interactions" between fungi and bacteria is quite new.
The inclusion of metabolomics data to support the results is a great contribution.Weaknesses:
Most of them have been solved in the revision, but for the future it will be nice to integrate this data with others from 16s.
-
-
eLife assessment
This study has uncovered some interesting findings about the fungal composition and its interaction with bacteria in Caesarean section scar diverticulum (CSD). While the study's findings are valuable and with translation possibilities, the strength of the conclusions obtained is incomplete due to the small sample size and methodological issues indicated by the reviewers such as the lack of controls and the location of samples analyzed.
-
Reviewer #1 (Public Review):
Summary:
Chen et al. describe the bacterial and fungal composition of cervical samples from women with/without Cesarean-section scar diverticulum (CSD) using whole metagenomic sequencing. Also, they report the metabolomic profile associated with CSD and built correlation networks at the taxonomical and taxonomic-metabolic levels to establish potential bacteria-fungi interactions. These interactions could be used, long-term, as therapeutic options to treat or prevent CSD.Strengths:
- The authors have used advanced techniques in shotgun sequencing which is a powerful tool able to characterize the microbiome at the species (or lower) level and metabolomics.
- These are novel results showing the interaction of bacteria and fungi and present a wider view of the role of the microbiome in female infertility.Weakne…
Reviewer #1 (Public Review):
Summary:
Chen et al. describe the bacterial and fungal composition of cervical samples from women with/without Cesarean-section scar diverticulum (CSD) using whole metagenomic sequencing. Also, they report the metabolomic profile associated with CSD and built correlation networks at the taxonomical and taxonomic-metabolic levels to establish potential bacteria-fungi interactions. These interactions could be used, long-term, as therapeutic options to treat or prevent CSD.Strengths:
- The authors have used advanced techniques in shotgun sequencing which is a powerful tool able to characterize the microbiome at the species (or lower) level and metabolomics.
- These are novel results showing the interaction of bacteria and fungi and present a wider view of the role of the microbiome in female infertility.Weaknesses:
- This is a pilot study with only 24 cases and 24 controls. Because the human microbiota entails individual variability, this work should be confirmed with a higher sample size to achieve enough statistical power.
- The authors do not report here the use of blank controls. The use of this type of control is important to "subtract" the potential background from plasticware, buffer or reagents from the real signal. Lack of controls may lead to microbiome artefacts in the results. This can be seen in the results presented where the authors report some bacterial contaminants (Agrobacterium tumefaciensis, Aequorivita lutea, Chitinophagaceae, Marinobacter vinifirmus, etc) as part of the most common bacteria found in cervical samples.
- Samples used for this study were collected from the cervix. Why not collect samples from the uterine cavity and isthmocele fluid (for cases)? In their previous paper using samples from the same research protocol ((IRB no. 2019ZSLYEC-005S) they used endometrial tissue from the patients, so access to the uterine cavity was guaranteed.
- Through the use of shotgun genomics, results from all the genomes of the organisms present in the sample are obtained. However, the authors have only used the metagenomic data to infer the taxonomical annotation of fungi and bacteria. -
Reviewer #2 (Public Review):
Summary: Shotgun data have been analysed to obtain fungal and bacterial organisms' abundance. Through their metabolic functions and through co-occurrence networks, a functional relationship between the two types of organisms can be inferred. By means of metabolomics, function-related metabolites are studied in order to deepen the fungus-bacteria synergy.
Strengths:
Data obtained from bacteria correlate with data from other authors.
The study of metabolic "interactions" between fungi and bacteria is quite new.
The inclusion of metabolomics data to support the results is a great contribution.Weaknesses: Methodological descriptions are minimal.
Some example:
*The CON group (line 147) has not been defined. I supposed it is the control group.
* There are no statistics related to shotgun sequencing. How many …Reviewer #2 (Public Review):
Summary: Shotgun data have been analysed to obtain fungal and bacterial organisms' abundance. Through their metabolic functions and through co-occurrence networks, a functional relationship between the two types of organisms can be inferred. By means of metabolomics, function-related metabolites are studied in order to deepen the fungus-bacteria synergy.
Strengths:
Data obtained from bacteria correlate with data from other authors.
The study of metabolic "interactions" between fungi and bacteria is quite new.
The inclusion of metabolomics data to support the results is a great contribution.Weaknesses: Methodological descriptions are minimal.
Some example:
*The CON group (line 147) has not been defined. I supposed it is the control group.
* There are no statistics related to shotgun sequencing. How many reads have been sequenced? How many have been removed from the host? How many are left to study bacteria and fungi? Are these reads proportional among the 48 samples? If not, what method has been used to normalise the data?
* ggClusterNet has numerous algorithms to better display the modules of the microbiome network. Which one has been used? -
Reviewer #3 (Public Review):
In the present study, Chen et al. revealed the fungal composition and explored its interaction with bacteria in Caesarean section scar diverticulum (CSD) patients. Performing metagenomic and mass spectrometry analysis, they found specific fungi could alter bacterial abundance through regulating the production of several metabolites such as Goyaglycoside A and Janthitrem E, which results in disruption of bacterial composition stability. Their study drew a conclusion that abnormal fungal composition and activity are essential drivers for bacterial dysbiosis in CSD patients. However, the results are not substantial enough and there are many format errors throughout the manuscript. In addition, I have some concerns or suggestions that may help to improve this work.
Major
1. Smoke or drink conditions, as well as …Reviewer #3 (Public Review):
In the present study, Chen et al. revealed the fungal composition and explored its interaction with bacteria in Caesarean section scar diverticulum (CSD) patients. Performing metagenomic and mass spectrometry analysis, they found specific fungi could alter bacterial abundance through regulating the production of several metabolites such as Goyaglycoside A and Janthitrem E, which results in disruption of bacterial composition stability. Their study drew a conclusion that abnormal fungal composition and activity are essential drivers for bacterial dysbiosis in CSD patients. However, the results are not substantial enough and there are many format errors throughout the manuscript. In addition, I have some concerns or suggestions that may help to improve this work.
Major
1. Smoke or drink conditions, as well as diseases like hypertension and diabetes are important factors that could influence the metabolism of the host, thus the authors should add them in the exclusion criteria in the Methods.
2. The sample size of this study is not large enough to draw a convincing conclusion. -
