Membrane contact sites regulate vacuolar fission via sphingolipid metabolism
Curation statements for this article:-
Curated by eLife

eLife assessment
This manuscript presents valuable findings that contribute to our understanding of how sphingolipids and membrane contact sites, formed by the tethering protein family tricalbins, are involved in regulating vacuolar morphology in S. cerevisiae. The evidence supporting the authors' claims is largely solid. While the reported correlation between sphingolipid levels and vacuole homeostasis is interesting and intriguing, more work is needed to thoroughly substantiate the proposed mechanism. This study will be of interest to cell biologists focusing on intracellular organization and lipid metabolism.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Membrane contact sites (MCSs) are junctures that perform important roles including coordinating lipid metabolism. Previous studies have indicated that vacuolar fission/fusion processes are coupled with modifications in the membrane lipid composition. However, it has been still unclear whether MCS-mediated lipid metabolism controls the vacuolar morphology. Here, we report that deletion of tricalbins (Tcb1, Tcb2, and Tcb3), tethering proteins at endoplasmic reticulum (ER)–plasma membrane (PM) and ER–Golgi contact sites, alters fusion/fission dynamics and causes vacuolar fragmentation in the yeast Saccharomyces cerevisiae . In addition, we show that the sphingolipid precursor phytosphingosine (PHS) accumulates in tricalbin-deleted cells, triggering the vacuolar division. Detachment of the nucleus–vacuole junction (NVJ), an important contact site between the vacuole and the perinuclear ER, restored vacuolar morphology in both cells subjected to high exogenous PHS and Tcb3-deleted cells, supporting that PHS transport across the NVJ induces vacuole division. Thus, our results suggest that vacuolar morphology is maintained by MCSs through the metabolism of sphingolipids.
Article activity feed
-

-
-
-

Author Response
The following is the authors’ response to the previous reviews.
Reviewer #1 (Public Review):
The manuscript investigates the role of membrane contact sites (MCSs) and sphingolipid metabolism in regulating vacuolar morphology in the yeast Saccharomyces cerevisiae. The authors show that tricalbin (1-3) deletion leads to vacuolar fragmentation and the accumulation of the sphingolipid phytosphingosine (PHS). They propose that PHS triggers vacuole division through MCSs and the nuclear-vacuolar junction (NVJ). The study presents some solid data and proposes potential mechanisms underlying vacuolar fragmentation driven by this pathway. Although the manuscript is clear in what the data indicates and what is more hypothetical, the story would benefit from providing more conclusive evidence to support these hypothesis. Overall, …
Author Response
The following is the authors’ response to the previous reviews.
Reviewer #1 (Public Review):
The manuscript investigates the role of membrane contact sites (MCSs) and sphingolipid metabolism in regulating vacuolar morphology in the yeast Saccharomyces cerevisiae. The authors show that tricalbin (1-3) deletion leads to vacuolar fragmentation and the accumulation of the sphingolipid phytosphingosine (PHS). They propose that PHS triggers vacuole division through MCSs and the nuclear-vacuolar junction (NVJ). The study presents some solid data and proposes potential mechanisms underlying vacuolar fragmentation driven by this pathway. Although the manuscript is clear in what the data indicates and what is more hypothetical, the story would benefit from providing more conclusive evidence to support these hypothesis. Overall, the study provides valuable insights into the connection between MCSs, lipid metabolism, and vacuole dynamics.
We thank the positive review from the Reviewer #1. We hope that our hypotheses are supported by the "Author Response to Recommendations" and by further research in the future.
Reviewer #2 (Public Review):
This manuscript explores the mechanism underlying the accumulation of phytosphingosine (PHS) and its role in initiating vacuole fission. The study posits the involvement of membrane contact sites (MCSs) in two key stages of this process. Firstly, MCSs tethered by tricalbin between the endoplasmic reticulum (ER) and the plasma membrane (PM) or Golgi regulate the intracellular levels of PHS. Secondly, the amassed PHS triggers vacuole fission, most likely through the nuclear-vacuolar junction (NVJ). The authors propose that MCSs play a regulatory role in vacuole morphology via sphingolipid metabolism. While some results in the manuscript are intriguing, certain broad conclusions occasionally surpass the available data. Despite the authors' efforts to enhance the manuscript, certain aspects remain unclear. It is still uncertain whether subtle changes in PHS levels could induce such effects on vacuolar fission. Additionally, it is regrettable that the lipid measurements are not comparable with previous studies by the authors. Future advancements in methods for determining intracellular lipid transport and levels are anticipated to shed light on the remaining uncertainties in this study.
We thank the careful comment from Reviewer #2. As Reviewer #2 pointed out, the mechanism of how slight changes in PHS levels can induce the vacuolar fission event is still uncovered in this manuscript. We sincerely consider that this issue has to be resolved in further study.
Reviewer #3 (Public Review):
In this manuscript, the authors investigated the effects of deletion of the ER-plasma membrane/Golgi tethering proteins tricalbins (Tcb1-3) on vacuolar morphology to demonstrate the role of membrane contact sites (MCSs) in regulating vacuolar morphology in Saccharomyces cerevisiae. Their data show that tricalbin deletion causes vacuolar fragmentation possibly in parallel with TORC1 pathway. In addition, their data reveal that levels of various lipids including ceramides, long-chain base (LCB)-1P, and phytosphingosine (PHS) are increased in tricalbin-deleted cells. The authors find that exogenously added PHS can induce vacuole fragmentation and by performing analyses of genes involved in sphingolipid metabolism, they conclude that vacuolar fragmentation in tricalbin-deleted cells is due to the accumulated PHS in these cells. Importantly, exogenous PHS- or tricalbin deletion-induced vacuole fragmentation was suppressed by loss of the nucleus vacuole junction (NVJ), suggesting the possibility that PHS transported from the ER to vacuoles via the NVJ triggers vacuole fission. Of note, the authors find that hyperosmotic shock increases intracellular PHS levels, suggesting a general role of PHS in vacuole fission in response to physiological vacuolar division-inducing stimuli. This work provides valuable insights into the relationship between MCS-mediated sphingolipid metabolism and vacuole morphology. The conclusions of this paper are mostly supported by their results, but inclusion of direct evidence indicating increased transport of PHS from the ER to vacuoles via NVJ in response to vacuolar division-inducing stimuli would have strengthened this study. There is another weakness in their claim that the transmembrane domain of Tcb3 contributes to the formation of the tricalbin complex which is sufficient for tethering ER to the plasma membrane and the Golgi complex. Their claim is based only on the structural simulation, but not on by biochemical experiments such as co-immunoprecipitation and pull-down.
We appreciate the careful feedback from Reviewer #3. We have responded in the "Recommendations to Authors" section and hope it can partially support the weakness in our claim regarding the physical interaction between Tcb1, 2, and 3.
Reviewer #1 (Recommendations For The Authors):
I would suggest that the authors include some of the data (e.g., Tcb interactions) that they refer to in the response to the reviewers. I think that this could enhance the message in this manuscript. Also, maybe it's a typo and you were referring to some other image panel, but in the rebuttal letter a "Fig. S3B" is mentioned, but I could not find it.
Following the suggestions of reviewers #1 and #3, we have added the data of co-immunoprecipitation which confirmed that Tcb3 binds to both Tcb1 and Tcb2 as Supplemental Figure 2. With this change, the person (Ms. Saku Sasaki) who performed this analysis was also added as a co-author.
Also, we appreciate the careful remark and apologize for the mistake. In the previous Author's response, we mentioned the vacuole observation using SD medium, but this data was Fig 5C, not Fig S3B.
Reviewer #3 (Recommendations For The Authors):
I would recommend that the authors include the IP data mentioned in their rebuttal letter to show the interactions among Tcb1-3. Also, the authors should quantify all lipid species in Fig 5B, as shown in Fig 3A.
Following the suggestions of reviewers #1 and #3, we have added the co-immunoprecipitation data (Fig S2). In a further study, we would like to test if the transmembrane domain of Tcb3 is sufficient for the interaction among Tcb1-3. Also, we quantified all lipid species and replaced the data in Fig 5B.
Minor points:
(1) The function of vps4 is not mentioned in the manuscript.
(2) The function of Sur2p is not mentioned in the manuscript. It should be clearly mentioned that DHS is converted to PHS by Sur2p.
(1) We have added text sections which mention that VPS4 is needed for normal ESCRT function, and its deletion is an example for inhibition of GFP-Cps1p transport into the vacuole.
(2) We have added the text in the manuscript that states Sur2p is the hydroxylase that catalysis the conversion of DHS to PHS.
-
eLife assessment
This manuscript presents valuable findings that contribute to our understanding of how sphingolipids and membrane contact sites, formed by the tethering protein family tricalbins, are involved in regulating vacuolar morphology in S. cerevisiae. The evidence supporting the authors' claims is largely solid. While the reported correlation between sphingolipid levels and vacuole homeostasis is interesting and intriguing, more work is needed to thoroughly substantiate the proposed mechanism. This study will be of interest to cell biologists focusing on intracellular organization and lipid metabolism.
-
Reviewer #1 (Public Review):
The manuscript investigates the role of membrane contact sites (MCSs) and sphingolipid metabolism in regulating vacuolar morphology in the yeast Saccharomyces cerevisiae. The authors show that tricalbin (1-3) deletion leads to vacuolar fragmentation and the accumulation of the sphingolipid phytosphingosine (PHS). They propose that PHS triggers vacuole division through MCSs and the nuclear-vacuolar junction (NVJ). The study presents some solid data and proposes potential mechanisms underlying vacuolar fragmentation driven by this pathway. Although the manuscript is clear in what the data indicates and what is more hypothetical, the story would benefit from providing more conclusive evidence to support these hypothesis. Overall, the study provides valuable insights into the connection between MCSs, lipid …
Reviewer #1 (Public Review):
The manuscript investigates the role of membrane contact sites (MCSs) and sphingolipid metabolism in regulating vacuolar morphology in the yeast Saccharomyces cerevisiae. The authors show that tricalbin (1-3) deletion leads to vacuolar fragmentation and the accumulation of the sphingolipid phytosphingosine (PHS). They propose that PHS triggers vacuole division through MCSs and the nuclear-vacuolar junction (NVJ). The study presents some solid data and proposes potential mechanisms underlying vacuolar fragmentation driven by this pathway. Although the manuscript is clear in what the data indicates and what is more hypothetical, the story would benefit from providing more conclusive evidence to support these hypothesis. Overall, the study provides valuable insights into the connection between MCSs, lipid metabolism, and vacuole dynamics.
-
Reviewer #2 (Public Review):
This manuscript explores the mechanism underlying the accumulation of phytosphingosine (PHS) and its role in initiating vacuole fission. The study posits the involvement of membrane contact sites (MCSs) in two key stages of this process. Firstly, MCSs tethered by tricalbin between the endoplasmic reticulum (ER) and the plasma membrane (PM) or Golgi regulate the intracellular levels of PHS. Secondly, the amassed PHS triggers vacuole fission, most likely through the nuclear-vacuolar junction (NVJ). The authors propose that MCSs play a regulatory role in vacuole morphology via sphingolipid metabolism.
While some results in the manuscript are intriguing, certain broad conclusions occasionally surpass the available data. Despite the authors' efforts to enhance the manuscript, certain aspects remain unclear. It is …
Reviewer #2 (Public Review):
This manuscript explores the mechanism underlying the accumulation of phytosphingosine (PHS) and its role in initiating vacuole fission. The study posits the involvement of membrane contact sites (MCSs) in two key stages of this process. Firstly, MCSs tethered by tricalbin between the endoplasmic reticulum (ER) and the plasma membrane (PM) or Golgi regulate the intracellular levels of PHS. Secondly, the amassed PHS triggers vacuole fission, most likely through the nuclear-vacuolar junction (NVJ). The authors propose that MCSs play a regulatory role in vacuole morphology via sphingolipid metabolism.
While some results in the manuscript are intriguing, certain broad conclusions occasionally surpass the available data. Despite the authors' efforts to enhance the manuscript, certain aspects remain unclear. It is still uncertain whether subtle changes in PHS levels could induce such effects on vacuolar fission. Additionally, it is regrettable that the lipid measurements are not comparable with previous studies by the authors. Future advancements in methods for determining intracellular lipid transport and levels are anticipated to shed light on the remaining uncertainties in this study.
-
Reviewer #3 (Public Review):
In this manuscript, the authors investigated the effects of deletion of the ER-plasma membrane/Golgi tethering proteins tricalbins (Tcb1-3) on vacuolar morphology to demonstrate the role of membrane contact sites (MCSs) in regulating vacuolar morphology in Saccharomyces cerevisiae. Their data show that tricalbin deletion causes vacuolar fragmentation possibly in parallel with TORC1 pathway. In addition, their data reveal that levels of various lipids including ceramides, long-chain base (LCB)-1P, and phytosphingosine (PHS) are increased in tricalbin-deleted cells. The authors find that exogenously added PHS can induce vacuole fragmentation and by performing analyses of genes involved in sphingolipid metabolism, they conclude that vacuolar fragmentation in tricalbin-deleted cells is due to the accumulated PHS …
Reviewer #3 (Public Review):
In this manuscript, the authors investigated the effects of deletion of the ER-plasma membrane/Golgi tethering proteins tricalbins (Tcb1-3) on vacuolar morphology to demonstrate the role of membrane contact sites (MCSs) in regulating vacuolar morphology in Saccharomyces cerevisiae. Their data show that tricalbin deletion causes vacuolar fragmentation possibly in parallel with TORC1 pathway. In addition, their data reveal that levels of various lipids including ceramides, long-chain base (LCB)-1P, and phytosphingosine (PHS) are increased in tricalbin-deleted cells. The authors find that exogenously added PHS can induce vacuole fragmentation and by performing analyses of genes involved in sphingolipid metabolism, they conclude that vacuolar fragmentation in tricalbin-deleted cells is due to the accumulated PHS in these cells. Importantly, exogenous PHS- or tricalbin deletion-induced vacuole fragmentation was suppressed by loss of the nucleus vacuole junction (NVJ), suggesting the possibility that PHS transported from the ER to vacuoles via the NVJ triggers vacuole fission. Of note, the authors find that hyperosmotic shock increases intracellular PHS levels, suggesting a general role of PHS in vacuole fission in response to physiological vacuolar division-inducing stimuli.
This work provides valuable insights into the relationship between MCS-mediated sphingolipid metabolism and vacuole morphology. The conclusions of this paper are mostly supported by their results, but inclusion of direct evidence indicating increased transport of PHS from the ER to vacuoles via NVJ in response to vacuolar division-inducing stimuli would have strengthened this study.
There is another weakness in their claim that the transmembrane domain of Tcb3 contributes to the formation of the tricalbin complex which is sufficient for tethering ER to the plasma membrane and the Golgi complex. Their claim is based only on the structural simulation, but not on by biochemical experiments such as co-immunoprecipitation and pull-down.
-
-
Author Response
The following is the authors’ response to the original reviews.
Reviewer #1 (Public Review):
The manuscript investigates the role of membrane contact sites (MCSs) and sphingolipid metabolism in regulating vacuolar morphology in the yeast Saccharomyces cerevisiae. The authors show that tricalbin (1-3) deletion leads to vacuolar fragmentation and the accumulation of the sphingolipid phytosphingosine (PHS). They propose that PHS triggers vacuole division through MCSs and the nuclear-vacuolar junction (NVJ). The study presents some solid data and proposes potential mechanisms underlying vacuolar fragmentation driven by this pathway. However, there are some concerns regarding the strength and interpretation of their lipid data, and the robustness of some conclusions. The manuscript would benefit from addressing these …
Author Response
The following is the authors’ response to the original reviews.
Reviewer #1 (Public Review):
The manuscript investigates the role of membrane contact sites (MCSs) and sphingolipid metabolism in regulating vacuolar morphology in the yeast Saccharomyces cerevisiae. The authors show that tricalbin (1-3) deletion leads to vacuolar fragmentation and the accumulation of the sphingolipid phytosphingosine (PHS). They propose that PHS triggers vacuole division through MCSs and the nuclear-vacuolar junction (NVJ). The study presents some solid data and proposes potential mechanisms underlying vacuolar fragmentation driven by this pathway. However, there are some concerns regarding the strength and interpretation of their lipid data, and the robustness of some conclusions. The manuscript would benefit from addressing these concerns and providing more conclusive evidence to support the proposed conclusions. Overall, the study provides valuable insights into the connection between MCSs, lipid metabolism, and vacuole dynamics, but further clarification will be highly valuable to strengthen the conclusions.
We thank the thoughtful and positive feedback from Reviewer #1. Nevertheless, there are concerns raised regarding the strength and interpretation of the lipid data, as well as the robustness of specific conclusions. We acknowledge the importance of addressing the raised concerns and provide more conclusive evidence to support our proposed conclusions. We have responded in the "Recommendations to Authors" section and hope that our research has been further strengthened.
Reviewer #2 (Public Review):
This manuscript investigates the mechanism behind the accumulation of phytosphingosine (PHS) and its role in triggering vacuole fission. The study proposes that membrane contact sites (MCSs) are involved in two steps of this process. First, tricalbin-tethered MCSs between the endoplasmic reticulum (ER) and the plasma membrane (PM) or Golgi modulate the intracellular amount of PHS. Second, the accumulated PHS induces vacuole fission, most likely via the nuclear-vacuolar junction (NVJ). The authors suggest that MCSs regulate vacuole morphology through sphingolipid metabolism.
While some of the results in the manuscript are interesting the overall logic is hard to follow. In my assessment of the manuscript, my primary concern lies in its broad conclusions which, in my opinion, exceed the available data and raise doubts. Here are some instances where this comes into play for this manuscript:
We greatly appreciate the careful insights into our research from Reviewer #2. We have sincerely addressed the points one by one in the following.
Major points for revision
- The rationale to start investigating a vacuolar fission phenotype in the beginning is very weak. It is basically based on a negative genetic interaction with NVJ1. Based on this vacuolar fragmentation is quantified. The binning for the quantifications is already problematic as, in my experience, WT cells often harbor one to three vacuoles. How are quantifications looking when 1-3 vacuoles are counted as "normal" and more than 3 vacuoles as "fragmented"? The observed changes seem to be relatively small and the various combinations of TCB mutants do not yield a clear picture.
The number of vacuoles at a steady state could be influenced by various environmental factors, including the composition of the medium (manufacturer supplying the reagent and local water hardness) and the background of the strain. Possibly due to those causes, our observations differ from the experience of Reviewer #2. Indeed, we observed that WT cells always have one vacuole in YPD medium. Whereas in SD medium (Fig S3B only), WT cells have mainly one or two vacuoles per cell. In both cases, we observed that some of the mutants showed a different phenotype from the WT and that those differences are supported by student’s t-test and two-way ANOVA analysis.
- The analysis of the structural requirements of the Tcb3 protein is interesting but does not seem to add any additional value to this study. While it was used to quantify the mild vacuolar fragmentation phenotype it does not reoccur in any following analysis. Is the tcb3Δ sufficient to yield the lipid phenotype that is later proposed to cause the vacuolar fragmentation phenotype?
We do not know whether tcb3Δ alone is sufficient to increase PHS as we have not examined it. Nevertheless, as another approach, we analyzed the difference in IPC level between tcb1Δ2Δ3Δ triple deletion and tcb3Δsingle deletion in a sec18 mutant background and showed that the reduction of IPC synthesis is similar between tcb1Δ2Δ3Δand tcb3Δ alone (unpublished). This result suggests that out of all tricalbins (Tcb1, Tcb2 and Tcb3), Tcb3 plays a central role. In addition, the IPC synthesis reduction phenotype was small in tcb1Δ alone and tcb2Δ alone, but a strong phenotype appeared in the tcb1Δtcb2Δ combined deletion (as strong as in tcb3Δ alone). The relationship between Tcb1 Tcb2 and Tcb3 indicated by these results is also consistent with the results of the structural analysis in this study. We have shown that Tcb3 physically interacts with Tcb1 and Tcb2 by immunoprecipitation analysis (unpublished). In the future, we plan to investigate the relationship between Tcb proteins in more detail, along with the details of the interactions between Tcb1, Tcb2, and Tcb3.
- The quantified lipid data also has several problems. i) The quantified effects are very small. The relative change in lipid levels does not allow any conclusion regarding the phenotypes. What is the change in absolute PHS in the cell. This would be important to know for judging the proposed effects. ii) It seems as if the lipid data is contradictory to the previous study from the lab regarding the role of tricalbins in ceramide transfer. Previously it was shown that ceramides remain unchanged and IPC levels were reduced. This was the rationale for proposing the tricalbins as ceramide transfer proteins between the ER and the mid-Golgi. What could be an explanation for this discrepancy? Does the measurement of PHS after labelling the cells with DHS just reflect differences in the activity of the Sur2 hydroxylase or does it reflect different steady state levels.
i) As Reviewer #2 pointed out, it is a slight change, but we cannot say that it is not sufficient. We have shown that PHS increases in the range of 10~30% depending on the concentration of NaCl that induces vacuole division (This result is related to the answers to the following questions by Reviewer #3 and to the additional data in the new version). This observation supports the possibility that a small increase in PHS levels may have an effect on vacuole fragmentation. We did not analyze total PHS level by using methods such as liquid chromatography-mass spectrometry or ninhydrin staining of TLC-separated total lipids. The reason for this is that radiolabeling of sphingolipids using the precursor [3H]DHS provides higher sensitivity and makes it easier to detect differences. Moreover, using [3H]DHS labeling, we only measure PHS that is synthesized in the ER and that doesn’t originate from degradation of complex sphingolipids or dephosphorylation of PHS-1P in other organelles.
ii) In our previous study (Ikeda et al. iScience. 2020), we separated the lipid labeled with [3H]DHS into ceramides and acylceramides. There was no significant change in ceramide levels, but acylceramides increased in tcb1Δ2Δ3Δ. Since we did not separate these lipids in the present study, the data shows the total amount of both ceramide and acylceramide. We apologize that the term in Figure 3A was wrong. We have corrected it. Also, we have used [3H]DHS to detect IPC levels, which differs from the previous analysis used [3H]inositol. This means the lipid amounts detected are completely different. Since the amount of inositol incorporated into cells varies from cell to cell, the amount loaded on the TLC plate is adjusted so that the total amount (signal intensity) of radioactively labeled lipids is almost the same. In contrast, for DHS labeling, the amount of DHS attached to the cell membrane is almost the same between cells, so we load the total amount onto the TLC plate without adjustment. In addition, the reduction in IPC levels due to Tcb depletion that we previously reported was seen only in sec12 or sec18 mutation backgrounds, and no reduction in IPC levels was observed in the tcb1Δ2Δ3Δ by [3H]inositol labeling (Ikeda et al. iScience. 2020). Therefore, we cannot simply compare the current results with the previous report due to the difference in experimental methods.
The labeling time for [3H]DHS is 3 hours, and we are not measuring steady-state amounts, but rather analyzing metabolic reactions. Since [3H]DHS is converted to PHS by Sur2 hydroxylase in the cell, the possibility that differences in PHS amounts reflect differences in Sur2 hydroxylase activity cannot be ruled out. However, this possibility is highly unlikely since we have previously observed that the distribution of ceramide subclasses is hardly affected by tcb1Δtcb2Δtcb3Δ (Ikeda et al. iScience 2020). We have added to the discussion that the possibility of differences in Sur2 hydroxylase activity cannot be excluded.
- Determining the vacuole fragmentation phenotype of a lag1Δlac1Δ double mutant does not allow the conclusion that elevated PHS levels are responsible for the observed phenotype. This just shows that lag1Δlac1Δ cells have fragmented vacuoles. Can the observed phenotype be rescued by treating the cells with myriocin? What is the growth rate of a LAG1 LAC1 double deletion as this strain has been previously reported to be very sick. Similarly, what is the growth phenotype of the various LCB3 LCB4 and LCB5 deletions and its combinations.
As Reviewer #2 pointed out, the vacuolar fragmentation in lag1Δlac1Δ itself does not attribute to the conclusion that increased PHS levels are the cause. Since this mutant strain has decreased level of ceramide and its subsequent product IPC/MIPC in addition to the increased level of the ceramide precursors LCB or LCB-1P, we have changed the manuscript as follows. As noted in the following comment by reviewer #2, myriocin treatment has been reported to induce vacuolar fragmentation, so we do not believe that experiments on recovery by myriocin treatment will lead to the expected results.
・ Previous Version: We first tested whether increased levels of PHS cause vacuolar fragmentation. Loss of ceramide synthases could cause an increase in PHS levels. Our analysis showed that vacuoles are fragmented in lag1Δlac1Δ cells, which lack both enzymes for LCBs (DHS and PHS) conversion into ceramides (Fig 3B). This suggests that ceramide precursors, LCBs or LCB-1P, can induce vacuolar fragmentation.
・Current Version: We first evaluated whether the increases in certain lipids are the cause of vacuolar fragmentation in tcb1Δ2Δ3Δ. Our analysis showed that vacuoles are fragmented in lag1Δlac1Δ cells, which lack both enzymes for LCBs (DHS and PHS) conversion into ceramides (Fig 3B). This suggests that the increases in ceramide and subsequent products IPC/MIPC are not the cause of vacuolar fragmentation, but rather its precursors LCBs or LCB-1P.
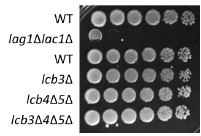
As reviewer #2 pointed out, the lag1Δlac1Δ double mutant is very slow growing as shown below (Author response image 1). We also examined the growth phenotype of LCB3, LCB4, and LCB5 deletion strains, and found that the growth of these strains was the same as the wild strains, with no significant differences in growth (Author response image 1).
Author response image 1.
Cells (FKY5687, FKY5688, FKY36, FKY37, FKY33, FKY38) were adjusted to OD 600 = 1.0 and fivefold serial dilutions were then spotted on YPD plates, then incubated at 25℃ for 3 days.
- The model in Figure 3 E proposes that treatment with PHS accumulates PHS in the endoplasmic reticulum. How do the authors know where exogenously added PHS ends up in the cell? It would also be important to determine the steady state levels of sphingolipids after treatment with PHS. Or in other words, how much PHS is taken up by the cells when 40 µM PHS is added?
It has been found that the addition of PHS well suppresses the Gas1 trafficking (Gaigg et al. J Biol Chem. 2006) and endocytosis phenotypes in lcb-100 mutants (Zanolari et al. EMBO J. 2000). Their suppression depends on Lcb3 localized to the ER. Thus, we know that PHS added from outside the cell reaches the ER and is functional.
We also agree that it is important to measure the amount of PHS taken up into the cells. However, this is extremely difficult to do for the following reasons. The majority of PHS added to the medium remains attached to the surface layer of the cells. If we measure the lipids in the cells by MS, we would detect both lipids present on the outside and inside of the plasma membrane. This means we need to separate the outside from the inside of the cell's membrane to determine the exact amount of LCB that has taken up by the cells. Regretfully, this separation is currently technically difficult.
- Previous studies have observed that myriocin treatment itself results in vacuolar fragmentation (e.g. Hepowit et al. biorXivs 2022, Fröhlich et al. eLife 2015). Why does both, depletion and accumulation of PHS lead to vacuolar fragmentation?
It’s exactly as Reviewer #2 said. Consistent with previous results with myriocin treatment, we also observed vacuolar fragmentation in the lcb1-100 mutant strain. Then we have added these papers to the references for further discussion. Our discussion is as follows.
"Previous studies have observed that myriocin treatment results in vacuolar fragmentation (Hepowit et al. bioRxiv 2022; Now published in J Cell Sci. 2023, Fröhlich et al. eLife 2015). Myriocin treatment itself causes not only the depletion of PHS but also of complex sphingolipids such as IPC. This suggests that normal sphingolipid metabolism is important for vacuolar morphology. The reason for this is unclear, but perhaps there is some mechanism by which sphingolipid depletion affects, for example, the recruitment of proteins required for vacuolar membrane fusion. In contrast, our new findings show that both PHS increase and depletion cause vacuole fragmentation. Taken together, there may be multiple mechanisms controlling vacuole morphology and lipid homeostasis by responding to both increasing and decreasing level of PHS."
- The experiments regarding the NVJ genes are not conclusive. While the authors mention that a NVJ1/2/3 MDM1 mutant was shown to result in a complete loss of the NVJ the observed effects cannot be simply correlated. It is also not clear why PHS would be transported towards the vacuole. In the cited study (Girik et al.) the authors show PHS transport from the vacuole towards the ER. Here the authors claim that PHS is transported via the NVJ towards the vacuole. Also, the origin of the rationale of this study is the negative genetic interaction of tcb1/2/3Δ with nvj1Δ. This interaction appears to result in a strong growth defect according to the Developmental Cell paper. What are the phenotypes of the mutants used here? Does the additional deletion of NVJ genes or MDM1 results in stronger growth phenotypes?
We seriously appreciate the concerns in our research. As reviewer #2 pointed out, we have not shown evidence in this study to support that PHS is transported directly from the ER to the vacuole, so it is unclear whether PHS is transported to the vacuole and its physiological relevance. Girik et al. showed that the NVJ resident protein Mdm1 is important for PHS transport between vacuole and ER. Given the applied experimental method that tracks PHS released in the vacuole, indeed only transport of PHS from the vacuole to the ER was verified. However, assuming that Mdm1 transports PHS along its concentration gradient we consider that under normal conditions, PHS is transported from the ER (as the organelle of PHS synthesis) to the vacuole. We clarified this interpretation by adding the following sentences to the manuscript at line 313:
“The study applied an experimental method that tracks LCBs released in the vacuole and showed that Mdm1p is necessary for LCBs leakage into the ER. However, assuming that Mdm1p transports LCBs along its concentration gradient we consider that under normal conditions, LCBs is transported from the ER (as the organelle of PHS synthesis) to the vacuole.”
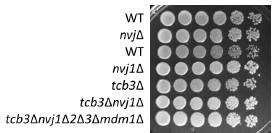
The negative genetic interaction between tcb1/2/3Δ and nvj1Δ is consistent with this model, but under our culture conditions we did not observe a negative interaction between the genes encoding the TCB3 and NVJ junction proteins (Author response image 2). We do not know if this is due to strain background, culture conditions, or whether the deletions of TCB1 and TCB2 are also required for the negative interaction. We would like to analyze details in the future.
Author response image 2.
Cells (FKY 3868, FKY5560, FKY6187, FKY6189, FKY6190, FKY6188, FKY6409) were adjusted to OD 600 = 1.0 and fivefold serial dilutions were then spotted on YPD plates, then incubated at 25℃ for 3 days.
Our results in this study show that deletion of the NVJ component gene partially suppresses vacuolar fission upon the addition of PHS. To clarify these facts, we have changed the sentences in Results and Discussion of our manuscript as follows. We hope that this change will avoid over-interpretation.
・ Previous: To test the role of NVJ-mediated “transport” for PHS-induced vacuolar fragmentation,
・Current: To test the role of NVJ-mediated “membrane contact” for PHS-induced vacuolar fragmentation,
・Previous: Taken together, we conclude from these findings that accumulated PHS in tricalbin deleted cells triggers vacuole fission via “non-vesicular transport of PHS” at the NVJ.
・Current: Taken together, we conclude from these findings that accumulated PHS in tricalbin deleted cells triggers vacuole fission via “contact between ER and vacuole” at the NVJ.
・Previous: Because both PHS- and tricalbin deletion-induced vacuolar fragmentations were partially suppressed by the lack of NVJ (Fig 4B, 4C), it is suggested that transport of PHS into vacuoles via the NVJ is involved in triggering vacuolar fragmentation.
・Current: Based on the fact that both PHS- and tricalbin deletion-induced vacuolar fragmentations were partially suppressed by the lack of NVJ (Fig 4B, 4C), it is possible that the trigger for vacuolar fragmentation is NVJ-mediated transport of PHS into the vacuole.
- As a consequence of the above points, several results are over-interpreted in the discussion. Most important, it is not clear that indeed the accumulation of PHS causes the observed phenotypes.
We thank the suggestion by Reviewer #2. In particular, the concern that PHS accumulation really causes vacuolar fragmentation could only be verified by an in vitro assay system. This is an important issue to be resolved in the future.
Reviewer #3 (Public Review):
In this manuscript, the authors investigated the effects of deletion of the ER-plasma membrane/Golgi tethering proteins tricalbins (Tcb1-3) on vacuolar morphology to demonstrate the role of membrane contact sites (MCSs) in regulating vacuolar morphology in Saccharomyces cerevisiae. Their data show that tricalbin deletion causes vacuolar fragmentation possibly in parallel with TORC1 pathway. In addition, their data reveal that levels of various lipids including ceramides, long-chain base (LCB)-1P and phytosphingosine (PHS) are increased in tricalbin-deleted cells. The authors find that exogenously added PHS can induce vacuole fragmentation and by performing analyses of genes involved in sphingolipid metabolism, they conclude that vacuolar fragmentation in tricalbin-deleted cells is due to the accumulated PHS in these cells. Importantly, exogenous PHS- or tricalbin deletion-induced vacuole fragmentation was suppressed by loss of the nucleus vacuole junction (NVJ), suggesting the possibility that PHS transported from the ER to vacuoles via the NVJ triggers vacuole fission.
This work provides valuable insights into the relationship between MCS-mediated sphingolipid metabolism and vacuole morphology. The conclusions of this paper are mostly supported by their results, but there is concern about physiological roles of tricalbins and PHS in regulating vacuole morphology under known vacuole fission-inducing conditions. That is, in this paper it is not addressed whether the functions of tricalbins and PHS levels are controlled in response to osmotic shock, nutrient status, or ER stress.
We appreciate the comment, and we consider it an important point. To answer this, we have performed additional experiments. Please refer to the following section, "Recommendations For The Authors" for more details. These results and discussions also have been added to the revised Manuscript. We believe this upgrade makes our findings more comprehensive.
There is another weakness in their claim that the transmembrane domain of Tcb3 contributes to the formation of the tricalbin complex which is sufficient for tethering ER to the plasma membrane and the Golgi complex. Their claim is based only on the structural simulation, but not on biochemical experiments such as co-immunoprecipitation and pull-down.
We appreciate your valuable suggestion and would like to attempt to improve upon it in the future.
Author response to Recommendations:
The following is the authors' response to the Recommendations For The Authors. We have now incorporated the changes recommended by Reviewers to improve the interpretations and clarity of the manuscript.
Reviewer #1 (Recommendations For The Authors):
I would recommend the authors provide additional experimental data to fully support their claims or revise the writing of their manuscript to be more precise in their conclusions. In particular, I have suggestions/questions:
Fig. 1A: display the results as in 1B (that is, different colors for different number of vacuoles, and the x axes showing the different conditions, in this case WT vs tcb1∆2∆3∆.
In response to the suggestion of Reviewer #1, we have changed the display of results.
Fig. S1B: the FM4-64 pattern looks different in the KO strain as compared to those shown in Fig. 1A. Is there a reason for that? Also, no positive control of cps1p not in the vacuole lumen is shown.
Our apologies, this was probably due to the poor resolution of the images. We have made other observations and changed the Figure along with the positive control.
Line 172: the last condition in Fig. 2B (vi), should be compared to the tcb1∆tcb2∆ condition (shown in fig 1).
In response to the suggestion of Reviewer #1, we have changed the manuscript as follows: We found that cells expressing Tcb3(TM)-GBP and lacking Tcb1p and Tcb2p (Fig 2B (vi)) are even more fragmented than tcb1Δ2Δ in Fig 1B and are fragmented to a similar degree as tcb3Δ (Fig 1B and Fig 2B (ii)).
Fig 2E: the model shown here can be tested, is there binding (similar to kin recognition mechanism of some Golgi proteins) between the different Tcb TMDs?
As Reviewer #1 mentioned, we have confirmed by co-immunoprecipitation that Tcb3 binds to both Tcb1 and Tcb2 (unpublished). Furthermore, we will test if the binding can be observed with TMD alone in the future.
Fig 3A: you measured an increase in PHS that is metabolized from DHS (which is what you label). Are there other routes to produce PHS independently of DHS? I mean, how is the increase reporting on the total levels of this lipid?
PHS synthesized by Sur2 is converted to PHS-1P and phytoceramide. Conversely, PHS is reproduced by degradation of PHS1-P via Lcb3, Ysr3, and by degradation of phytoceramides via Ypc1 (Vilaça, Rita et al. Biochim Biophys Acta Mol Basis Dis. 2017. Fig1). Our analysis shows that these degradation substrates are not decreasing but rather accumulating in tcb1Δ2Δ3Δ strain, suggesting that the degradation system is not promoting PHS level. Therefore, the increase in detected PHS is most likely due to congestion/jams in metabolic processes downstream of PHS. Possible causes of the lipid metabolism disruption in Tcbdeletion cells have been discussed in the Discussion. To put it simply, (1) The reduced activity of a PtdIns4P phosphatase Sac1, due to MCS deficiency between ER and PM. (2) The impaired ceramide nonvesicular transport from the ER to the Golgi. (3) The low efficiency of PHS export by Rsb1, due to insufficient PHS diffusion between the ER and the PM.
Line 248: did the authors test if the NVJ MCS is unperturbed in the triple Tcb KO?
This is an exciting question. We are very interested in considering whether Tcb deficiency affects NVJ formation in terms of lipid transport. We would like to conduct further analysis in this regard in our future studies.
Reviewer #2 (Recommendations For The Authors):
I would suggest carefully evaluating the findings in this manuscript. Right now the connection between elevated PHS levels and vacuolar fragmentation are not really supported by the data. One of the major issues in the field of yeast sphingolipid biology is that quantification of the lipid levels is difficult and labor- and cost-intensive. But I think that it is very important to directly connect phenotypes with the lipid levels.
Minor points:
- In figure 1 c and d WT controls of the different treatments are lacking.
As reviewer #2 had pointed out, we have added data for the WT controls.
- The tcb1Δmutant appears to be sensitive in pH 5.0 media while the triple tricalbins mutant grows fine. Is that a known phenotype?
We have performed this assay on SD plates. Then, to check whether this phenotype of tcb1Δ was specific or general, we re-analyzed the same strain in YPD medium. In YPD medium, tcb1Δ strain grew normally, while the control, vma3Δ, was still pH sensitive. Therefore, the growth of this tcb1Δ strain is dependent on the nutrient conditions of the medium but does not appear to be pH sensitive. This new data was inserted as part of Supplementary Figure 1.
- Line 305. The is an "of" in the sentence that needs to be deleted.
As pointed out by Reviewer #2, we have corrected the sentence.
Reviewer #3 (Recommendations For The Authors):
In supplementary Fig 2, the authors show the involvement of the NVJ in hyperosmotic shockinduced vacuole fission, but the involvement of tricalbins and PHS in this process is not tested. Does osmotic shock affect the level or distribution of tricalbins and PHS? They will be able to test whether overexpression of tricalbins inhibits hyperosmotic shock-induced vacuole fission or not. Also, they will be able to perform the similar experiments upon ER stressinduced vacuole fission.
We appreciate Reviewer#3 for suggesting that it is important to test the involvement of PHS in hyperosmotic shock- or ER stress-induced vacuole fission. We have shown in a previous report that treatment with tunicamycin, which is ER stress inducer, increased the PHS level by about 20% (Yabuki et al. Genetics. 2019. Fig4). In addition, we tested the effect of hyperosmolarity on PHS levels for this time. Analysis of PHS under hyperosmotic shock conditions (0.2 M NaCl), in which vacuolar fragments were observed, showed an increase in PHS of about 10%. Furthermore, when the NaCl concentration was increased to 0.8 M, PHS levels increased up to 30%. In other words, we have shown that PHS increases in the range of tens of percent depending on the concentration of NaCl that induces vacuole division. This observation supports the possibility that a small increase in PHS levels may have an effect on vacuole fragmentation. Moreover, NaCl-induced vacuolar fragmentation, like that caused by PHS treatment, was also suppressed by PHS export from the cell by Rsb1 overexpression.
These new data are now inserted, commented and discussed in the manuscript as Figure 5. We hope that these results will provide further insight into the more general aspects of PHS involvement in the vacuole fission process.
Minor points:
- It is unclear for me whether endogenous Tcb3 is deleted in cells expressing Tcb3-GBP (FKY3903-3905 and FKY4754). They should clearly mention that these cells do not express endogenous Tcb3 in the manuscript.
We apologize that our description was not clear. In this strain, endogenous TCB3 gene is tagged with GBP and the original Tcb3 has been replaced by the tagged version. We have changed the description in our manuscript.
- The strength of the effect of PHS on vacuole morphology looks different in respective WT cells in Fig 3C, 4B, and S2B. Is this due to the different yeast strains they used?
Yes, we used BY4742 background for the strain in Figure 3C, SEY6210 background in Figure 4B, and HR background in Figure S2B. As a matter of fact, we observed that the strength of the PHS effect varies depending on their background. Strain numbers are now given in the legend so that the cells used for each data can be referenced in the strain list.
- p.3, line 44: the "SNARE" complex (instead of "protease")?
We thank for the remarks on the incorrect wording. We have corrected this sentence.
-
eLife assessment
This manuscript presents valuable findings that contribute to our understanding of how sphingolipids and membrane contact sites, formed by the tethering protein family tricalbins, are involved in regulating vacuolar morphology in S. cerevisiae. The evidence supporting the authors' claims is largely solid: while the reported correlation between sphingolipid levels and vacuole homeostasis is intriguing, the data do not completely substantiate the proposed mechanism. This study will be of interest to cell biologists focusing on intracellular organization and lipid metabolism.
-
Reviewer #1 (Public Review):
The manuscript investigates the role of membrane contact sites (MCSs) and sphingolipid metabolism in regulating vacuolar morphology in the yeast Saccharomyces cerevisiae. The authors show that tricalbin (1-3) deletion leads to vacuolar fragmentation and the accumulation of the sphingolipid phytosphingosine (PHS). They propose that PHS triggers vacuole division through MCSs and the nuclear-vacuolar junction (NVJ). The study presents some solid data and proposes potential mechanisms underlying vacuolar fragmentation driven by this pathway. Although the manuscript is clear in what the data indicates and what is more hypothetical, the story would benefit from providing more conclusive evidence to support these hypothesis. Overall, the study provides valuable insights into the connection between MCSs, lipid …
Reviewer #1 (Public Review):
The manuscript investigates the role of membrane contact sites (MCSs) and sphingolipid metabolism in regulating vacuolar morphology in the yeast Saccharomyces cerevisiae. The authors show that tricalbin (1-3) deletion leads to vacuolar fragmentation and the accumulation of the sphingolipid phytosphingosine (PHS). They propose that PHS triggers vacuole division through MCSs and the nuclear-vacuolar junction (NVJ). The study presents some solid data and proposes potential mechanisms underlying vacuolar fragmentation driven by this pathway. Although the manuscript is clear in what the data indicates and what is more hypothetical, the story would benefit from providing more conclusive evidence to support these hypothesis. Overall, the study provides valuable insights into the connection between MCSs, lipid metabolism, and vacuole dynamics.
-
Reviewer #2 (Public Review):
This manuscript explores the mechanism underlying the accumulation of phytosphingosine (PHS) and its role in initiating vacuole fission. The study posits the involvement of membrane contact sites (MCSs) in two key stages of this process. Firstly, MCSs tethered by tricalbin between the endoplasmic reticulum (ER) and the plasma membrane (PM) or Golgi regulate the intracellular levels of PHS. Secondly, the amassed PHS triggers vacuole fission, most likely through the nuclear-vacuolar junction (NVJ). The authors propose that MCSs play a regulatory role in vacuole morphology via sphingolipid metabolism.
While some results in the manuscript are intriguing, certain broad conclusions occasionally surpass the available data. Despite the authors' efforts to enhance the manuscript, certain aspects remain unclear. It is …
Reviewer #2 (Public Review):
This manuscript explores the mechanism underlying the accumulation of phytosphingosine (PHS) and its role in initiating vacuole fission. The study posits the involvement of membrane contact sites (MCSs) in two key stages of this process. Firstly, MCSs tethered by tricalbin between the endoplasmic reticulum (ER) and the plasma membrane (PM) or Golgi regulate the intracellular levels of PHS. Secondly, the amassed PHS triggers vacuole fission, most likely through the nuclear-vacuolar junction (NVJ). The authors propose that MCSs play a regulatory role in vacuole morphology via sphingolipid metabolism.
While some results in the manuscript are intriguing, certain broad conclusions occasionally surpass the available data. Despite the authors' efforts to enhance the manuscript, certain aspects remain unclear. It is still uncertain whether subtle changes in PHS levels could induce such effects on vacuolar fission. Additionally, it is regrettable that the lipid measurements are not comparable with previous studies by the authors. Future advancements in methods for determining intracellular lipid transport and levels are anticipated to shed light on the remaining uncertainties in this study.
-
Reviewer #3 (Public Review):
In this manuscript, the authors investigated the effects of deletion of the ER-plasma membrane/Golgi tethering proteins tricalbins (Tcb1-3) on vacuolar morphology to demonstrate the role of membrane contact sites (MCSs) in regulating vacuolar morphology in Saccharomyces cerevisiae. Their data show that tricalbin deletion causes vacuolar fragmentation possibly in parallel with TORC1 pathway. In addition, their data reveal that levels of various lipids including ceramides, long-chain base (LCB)-1P, and phytosphingosine (PHS) are increased in tricalbin-deleted cells. The authors find that exogenously added PHS can induce vacuole fragmentation and by performing analyses of genes involved in sphingolipid metabolism, they conclude that vacuolar fragmentation in tricalbin-deleted cells is due to the accumulated PHS …
Reviewer #3 (Public Review):
In this manuscript, the authors investigated the effects of deletion of the ER-plasma membrane/Golgi tethering proteins tricalbins (Tcb1-3) on vacuolar morphology to demonstrate the role of membrane contact sites (MCSs) in regulating vacuolar morphology in Saccharomyces cerevisiae. Their data show that tricalbin deletion causes vacuolar fragmentation possibly in parallel with TORC1 pathway. In addition, their data reveal that levels of various lipids including ceramides, long-chain base (LCB)-1P, and phytosphingosine (PHS) are increased in tricalbin-deleted cells. The authors find that exogenously added PHS can induce vacuole fragmentation and by performing analyses of genes involved in sphingolipid metabolism, they conclude that vacuolar fragmentation in tricalbin-deleted cells is due to the accumulated PHS in these cells. Importantly, exogenous PHS- or tricalbin deletion-induced vacuole fragmentation was suppressed by loss of the nucleus vacuole junction (NVJ), suggesting the possibility that PHS transported from the ER to vacuoles via the NVJ triggers vacuole fission. Of note, the authors find that hyperosmotic shock increases intracellular PHS levels, suggesting a general role of PHS in vacuole fission in response to physiological vacuolar division-inducing stimuli.
This work provides valuable insights into the relationship between MCS-mediated sphingolipid metabolism and vacuole morphology. The conclusions of this paper are mostly supported by their results, but inclusion of direct evidence indicating increased transport of PHS from the ER to vacuoles via NVJ in response to vacuolar division-inducing stimuli would have strengthened this study.
There is another weakness in their claim that the transmembrane domain of Tcb3 contributes to the formation of the tricalbin complex which is sufficient for tethering ER to the plasma membrane and the Golgi complex. Their claim is based only on the structural simulation, but not on by biochemical experiments such as co-immunoprecipitation and pull-down.
-
-
eLife assessment
This manuscript presents valuable findings about how sphingolipids and membrane contact sites are involved in promoting vacuole fission in S. cerevisiae. While a connection between the levels of sphingolipids and vacuole homeostasis is interesting, the data pertaining to this issue are incomplete and do not fully support the claim for causality between altered lipid composition and organelle dynamics. The work will be of interest to cell biologists working on organelle biogenesis and lipid metabolism.
-
Reviewer #1 (Public Review):
The manuscript investigates the role of membrane contact sites (MCSs) and sphingolipid metabolism in regulating vacuolar morphology in the yeast Saccharomyces cerevisiae. The authors show that tricalbin (1-3) deletion leads to vacuolar fragmentation and the accumulation of the sphingolipid phytosphingosine (PHS). They propose that PHS triggers vacuole division through MCSs and the nuclear-vacuolar junction (NVJ). The study presents some solid data and proposes potential mechanisms underlying vacuolar fragmentation driven by this pathway. However, there are some concerns regarding the strength and interpretation of their lipid data, and the robustness of some conclusions. The manuscript would benefit from addressing these concerns and providing more conclusive evidence to support the proposed conclusions. …
Reviewer #1 (Public Review):
The manuscript investigates the role of membrane contact sites (MCSs) and sphingolipid metabolism in regulating vacuolar morphology in the yeast Saccharomyces cerevisiae. The authors show that tricalbin (1-3) deletion leads to vacuolar fragmentation and the accumulation of the sphingolipid phytosphingosine (PHS). They propose that PHS triggers vacuole division through MCSs and the nuclear-vacuolar junction (NVJ). The study presents some solid data and proposes potential mechanisms underlying vacuolar fragmentation driven by this pathway. However, there are some concerns regarding the strength and interpretation of their lipid data, and the robustness of some conclusions. The manuscript would benefit from addressing these concerns and providing more conclusive evidence to support the proposed conclusions. Overall, the study provides valuable insights into the connection between MCSs, lipid metabolism, and vacuole dynamics, but further clarification will be highly valuable to strengthen the conclusions.
-
Reviewer #2 (Public Review):
This manuscript investigates the mechanism behind the accumulation of phytosphingosine (PHS) and its role in triggering vacuole fission. The study proposes that membrane contact sites (MCSs) are involved in two steps of this process. First, tricalbin-tethered MCSs between the endoplasmic reticulum (ER) and the plasma membrane (PM) or Golgi modulate the intracellular amount of PHS. Second, the accumulated PHS induces vacuole fission, most likely via the nuclear-vacuolar junction (NVJ). The authors suggest that MCSs regulate vacuole morphology through sphingolipid metabolism.
While some of the results in the manuscript are interesting the overall logic is hard to follow. In my assessment of the manuscript, my primary concern lies in its broad conclusions which, in my opinion, exceed the available data and …Reviewer #2 (Public Review):
This manuscript investigates the mechanism behind the accumulation of phytosphingosine (PHS) and its role in triggering vacuole fission. The study proposes that membrane contact sites (MCSs) are involved in two steps of this process. First, tricalbin-tethered MCSs between the endoplasmic reticulum (ER) and the plasma membrane (PM) or Golgi modulate the intracellular amount of PHS. Second, the accumulated PHS induces vacuole fission, most likely via the nuclear-vacuolar junction (NVJ). The authors suggest that MCSs regulate vacuole morphology through sphingolipid metabolism.
While some of the results in the manuscript are interesting the overall logic is hard to follow. In my assessment of the manuscript, my primary concern lies in its broad conclusions which, in my opinion, exceed the available data and raise doubts. Here are some instances where this comes into play for this manuscript:2.) Major points for revision
1.) The rationale to start investigating a vacuolar fission phenotype in the beginning is very weak. It is basically based on a negative genetic interaction with NVJ1. Based on this vacuolar fragmentation is quantified. The binning for the quantifications is already problematic as, in my experience, WT cells often harbor one to three vacuoles. How are quantifications looking when 1-3 vacuoles are counted as "normal" and more than 3 vacuoles as "fragmented"? The observed changes seem to be relatively small and the various combinations of TCB mutants do not yield a clear picture.
2.) The analysis of the structural requirements of the Tcb3 protein is interesting but does not seem to add any additional value to this study. While it was used to quantify the mild vacuolar fragmentation phenotype it does not reoccur in any following analysis. Is the tcb3Δ sufficient to yield the lipid phenotype that is later proposed to cause the vacuolar fragmentation phenotype?
3.) The quantified lipid data also has several problems. i) The quantified effects are very small. The relative change in lipid levels does not allow any conclusion regarding the phenotypes. What is the change in absolute PHS in the cell. This would be important to know for judging the proposed effects. ii) It seems as if the lipid data is contradictory to the previous study from the lab regarding the role of tricalbins in ceramide transfer. Previously it was shown that ceramides remain unchanged and IPC levels were reduced. This was the rationale for proposing the tricalbins as ceramide transfer proteins between the ER and the mid-Golgi. What could be an explanation for this discrepancy? Does the measurement of PHS after labelling the cells with DHS just reflect differences in the activity of the Sur2 hydroxylase or does it reflect different steady state levels.
4.) Determining the vacuole fragmentation phenotype of a lag1Δlac1Δ double mutant does not allow the conclusion that elevated PHS levels are responsible for the observed phenotype. This just shows that lag1Δlac1Δ cells have fragmented vacuoles. Can the observed phenotype be rescued by treating the cells with myriocin? What is the growth rate of a LAG1 LAC1 double deletion as this strain has been previously reported to be very sick. Similarly, what is the growth phenotype of the various LCB3 LCB4 and LCB5 deletions and its combinations.
5.) The model in Figure 3 E proposes that treatment with PHS accumulates PHS in the endoplasmic reticulum. How do the authors know where exogenously added PHS ends up in the cell? It would also be important to determine the steady state levels of sphingolipids after treatment with PHS. Or in other words, how much PHS is taken up by the cells when 40 µM PHS is added?
6.) Previous studies have observed that myriocin treatment itself results in vacuolar fragmentation (e.g. Hepowit et al. biorXivs 2022, Fröhlich et al. eLife 2015). Why does both, depletion and accumulation of PHS lead to vacuolar fragmentation?
7.) The experiments regarding the NVJ genes are not conclusive. While the authors mention that a NVJ1/2/3 MDM1 mutant was shown to result in a complete loss of the NVJ the observed effects cannot be simply correlated. It is also not clear why PHS would be transported towards the vacuole. In the cited study (Girik et al.) the authors show PHS transport from the vacuole towards the ER. Here the authors claim that PHS is transported via the NVJ towards the vacuole. Also, the origin of the rationale of this study is the negative genetic interaction of tcb1/2/3Δ with nvj1. This interaction appears to result in a strong growth defect according to the Developmental Cell paper. What are the phenotypes of the mutants used here? Does the additional deletion of NVJ genes or MDM1 results in stronger growth phenotypes?
8.) As a consequence of the above points, several results are over-interpreted in the discussion. Most important, it is not clear that indeed the accumulation of PHS causes the observed phenotypes. -
Reviewer #3 (Public Review):
In this manuscript, the authors investigated the effects of deletion of the ER-plasma membrane/Golgi tethering proteins tricalbins (Tcb1-3) on vacuolar morphology to demonstrate the role of membrane contact sites (MCSs) in regulating vacuolar morphology in Saccharomyces cerevisiae. Their data show that tricalbin deletion causes vacuolar fragmentation possibly in parallel with TORC1 pathway. In addition, their data reveal that levels of various lipids including ceramides, long-chain base (LCB)-1P and phytosphingosine (PHS) are increased in tricalbin-deleted cells. The authors find that exogenously added PHS can induce vacuole fragmentation and by performing analyses of genes involved in sphingolipid metabolism, they conclude that vacuolar fragmentation in tricalbin-deleted cells is due to the accumulated PHS …
Reviewer #3 (Public Review):
In this manuscript, the authors investigated the effects of deletion of the ER-plasma membrane/Golgi tethering proteins tricalbins (Tcb1-3) on vacuolar morphology to demonstrate the role of membrane contact sites (MCSs) in regulating vacuolar morphology in Saccharomyces cerevisiae. Their data show that tricalbin deletion causes vacuolar fragmentation possibly in parallel with TORC1 pathway. In addition, their data reveal that levels of various lipids including ceramides, long-chain base (LCB)-1P and phytosphingosine (PHS) are increased in tricalbin-deleted cells. The authors find that exogenously added PHS can induce vacuole fragmentation and by performing analyses of genes involved in sphingolipid metabolism, they conclude that vacuolar fragmentation in tricalbin-deleted cells is due to the accumulated PHS in these cells. Importantly, exogenous PHS- or tricalbin deletion-induced vacuole fragmentation was suppressed by loss of the nucleus vacuole junction (NVJ), suggesting the possibility that PHS transported from the ER to vacuoles via the NVJ triggers vacuole fission.
This work provides valuable insights into the relationship between MCS-mediated sphingolipid metabolism and vacuole morphology. The conclusions of this paper are mostly supported by their results, but there is concern about physiological roles of tricalbins and PHS in regulating vacuole morphology under known vacuole fission-inducing conditions. That is, in this paper it is not addressed whether the functions of tricalbins and PHS levels are controlled in response to osmotic shock, nutrient status, or ER stress.
There is another weakness in their claim that the transmembrane domain of Tcb3 contributes to the formation of the tricalbin complex which is sufficient for tethering ER to the plasma membrane and the Golgi complex. Their claim is based only on the structural simulation, but not on biochemical experiments such as co-immunoprecipitation and pull-down.
-
