Systematic evaluation of intratumoral and peripheral BCR repertoires in three cancers
Curation statements for this article:-
Curated by eLife

eLife Assessment
This useful paper systematically evaluates B-cell receptor (BCR) repertoires across tumors, tumor-draining lymph nodes, and peripheral blood in patients with melanoma, lung adenocarcinoma, and colorectal cancer. It investigates the interplay between the tumor microenvironment and immune responses, revealing differences in BCR clonotype maturity, hypermutation, and spatial distribution. The study highlights the heterogeneity in immune responses and provides solid insights into the potential of tumor-infiltrating B cells for therapeutic applications, despite limitations in patient cohort size and sequencing methodology.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The current understanding of humoral immune response in cancer patients suggests that tumors may be infiltrated with diffuse B cells of extra-tumoral origin or may develop organized lymphoid structures, where somatic hypermutation and antigen-driven selection occur locally. These processes are believed to be significantly influenced by the tumor microenvironment through secretory factors and biased cell-cell interactions. To explore the manifestation of this influence, we used deep unbiased immunoglobulin profiling and systematically characterized the relationships between B cells in circulation, draining lymph nodes (draining LNs), and tumors in 14 patients with three human cancers. We demonstrated that draining LNs are differentially involved in the interaction with the tumor site, and that significant heterogeneity exists even between different parts of a single lymph node (LN). Next, we confirmed and elaborated upon previous observations regarding intratumoral immunoglobulin heterogeneity. We identified B cell receptor (BCR) clonotypes that were expanded in tumors relative to draining LNs and blood and observed that these tumor-expanded clonotypes were less hypermutated than non-expanded (ubiquitous) clonotypes. Furthermore, we observed a shift in the properties of complementarity-determining region 3 of the BCR heavy chain (CDR-H3) towards less mature and less specific BCR repertoire in tumor-infiltrating B-cells compared to circulating B-cells, which may indicate less stringent control for antibody-producing B cell development in tumor microenvironment (TME). In addition, we found repertoire-level evidence that B-cells may be selected according to their CDR-H3 physicochemical properties before they activate somatic hypermutation (SHM). Altogether, our work outlines a broad picture of the differences in the tumor BCR repertoire relative to non-tumor tissues and points to the unexpected features of the SHM process.
Article activity feed
-

-
-
-

eLife Assessment
This useful paper systematically evaluates B-cell receptor (BCR) repertoires across tumors, tumor-draining lymph nodes, and peripheral blood in patients with melanoma, lung adenocarcinoma, and colorectal cancer. It investigates the interplay between the tumor microenvironment and immune responses, revealing differences in BCR clonotype maturity, hypermutation, and spatial distribution. The study highlights the heterogeneity in immune responses and provides solid insights into the potential of tumor-infiltrating B cells for therapeutic applications, despite limitations in patient cohort size and sequencing methodology.
-
Reviewer #3 (Public Review):
In multiple cancers, the key roles of B cells are emerging in the tumor microenvironment (TME). The authors of this study appropriately introduce that B cells are relatively under-characterised in the TME and argue correctly that it is not known how the B cell receptor (BCR) repertoires across tumor, lymph node and peripheral blood relate. The authors therefore supply a potentially useful study evaluating the tumor, lymph node and peripheral blood BCR repertoires and site-to-site as well as intra-site relationships. The authors employ sophisticated analysis techniques, although the description of the methods is incomplete.
Major strengths:
(1) The authors provide a unique analysis of BCR repertoires across tumor, dLN, and peripheral blood. The work provides useful insights into inter- and intra-site BCR …
Reviewer #3 (Public Review):
In multiple cancers, the key roles of B cells are emerging in the tumor microenvironment (TME). The authors of this study appropriately introduce that B cells are relatively under-characterised in the TME and argue correctly that it is not known how the B cell receptor (BCR) repertoires across tumor, lymph node and peripheral blood relate. The authors therefore supply a potentially useful study evaluating the tumor, lymph node and peripheral blood BCR repertoires and site-to-site as well as intra-site relationships. The authors employ sophisticated analysis techniques, although the description of the methods is incomplete.
Major strengths:
(1) The authors provide a unique analysis of BCR repertoires across tumor, dLN, and peripheral blood. The work provides useful insights into inter- and intra-site BCR repertoire heterogeneity. While patient-to-patient variation is expected, the findings with regard to intra-tumor and intra-dLN heterogeneity with the use of fragments from the same tissue are of importance, contribute to the understanding of the TME, and will inform future study design.
(2) A particular strength of the study is the detailed CDR3 physicochemical properties analysis which leads the authors to observations that suggest a less-specific BCR repertoire of TIL-B compared to circulating B cells.
Comments on revisions:
Your efforts in addressing concerns related to methodological details, narrative clarity, and data representation are commendable. The expanded descriptions of Fig. 1A and the experimental design, as well as the restructuring of the discussion, have greatly enhanced the manuscript's clarity and coherence.
-
Author response:
The following is the authors’ response to the previous reviews.
Reviewer #3:
Concerns and comments on current version:
The revision has improved the manuscript but, in my opinion, remains inadequate. While most of my requested changes have been made, I do not see an expansion of Fig1A legend to incorporate more details about the analysis. Lacking details of methodology was a concern from all reviewers.
To address this concern, we expanded Fig.1A legend, and also significantly expanded the text describing experimental design, to also include the description of the data analysis approach.
“BCR repertoires libraries were obtained using the 5’-RACE (Rapid Amplification of cDNA Ends) protocol as previously described21 and sequenced with 150+150 bp read length. This approach allowed us to achieve high coverage for the …
Author response:
The following is the authors’ response to the previous reviews.
Reviewer #3:
Concerns and comments on current version:
The revision has improved the manuscript but, in my opinion, remains inadequate. While most of my requested changes have been made, I do not see an expansion of Fig1A legend to incorporate more details about the analysis. Lacking details of methodology was a concern from all reviewers.
To address this concern, we expanded Fig.1A legend, and also significantly expanded the text describing experimental design, to also include the description of the data analysis approach.
“BCR repertoires libraries were obtained using the 5’-RACE (Rapid Amplification of cDNA Ends) protocol as previously described21 and sequenced with 150+150 bp read length. This approach allowed us to achieve high coverage for the obtained libraries (Table S1) to reveal information on clonal composition, CDR-H3 properties, IgM/IgG/IgA isotypes and somatic hypermutation load within CDR-H3. For B cell clonal lineage reconstruction and phylogenetic analysis, however, 150+150 bp read length is suboptimal because it does not cover V-gene region outside CDR-H3, where hypermutations also occur. Therefore, to verify our conclusions based on the data obtained by 150+150 bp sequencing (“short repertoires”), for some of our samples we also generated BCR libraries by IG RNA Multiplex protocol (See Materials and Methods) and sequenced them at 250+250 bp read length (“long repertoires”). Libraries obtained by this protocol cover V gene sequence starting from CDR-H1 and capture most of the hypermutations in the V gene. Conclusions about clonal lineage phylogeny were drawn only when they were corroborated by “long repertoire” analysis.
For BCR repertoire reconstruction from sequencing data, we first performed unique molecular identifier (UMI) extraction and error correction (reads/UMI threshold = 3 for 5`RACE and 4 for IG Multiplex libraries). Then, we used MIXCR58 software to assemble reads into clonotypes, determine germline V, D, and J genes, isotypes, and find the boundaries of target regions, such as CDR-H3. Only
UMI counts, and not read counts, were used for quantitative analysis. Clonotypes derived from only one UMI were excluded from the analysis of individual clonotype features but were used to analyze clonal lineages and hypermutation phylogeny, where sample size was crucial. Samples with 50 or less clonotypes left after preprocessing were excluded from the analysis.”
Similarly, the 'fragmented' narrative was a concern of all reviewers. These matters have not been dealt with adequately enough - there are parts of the manuscript which remain fragmented and confusing.
Unfortunately, the reviewers do not give us a hint as to which parts of the text are the most problematic in their opinion. We identified the parts describing physicochemical properties of CDR3s, Intratumoral heterogeneity and Intra-LN heterogeneity as the most problematic, and edited these parts significantly. Also, we significantly edited the Discussion section (please see the Comparison file for details). Other parts sections were also edited to improve readability and clarity.
The narrative and analysis does not explain how the plasma cell bias has been dealt with adequately and in fact is simply just confusing. There is a paragraph at the beginning of the discussion re the plasma cell bias, which should be re-written to be clearer and moved to have a prominent place early in the results. Why are these results not properly presented? They are key for interpretation of the manuscript. Furthermore, the sorted plasma cell sequencing analysis also has only been performed on two patients.
In response to this concern, we moved the section describing plasma cell bias in the bulk BCR repertoires to the main text.
Another issue is that some disease cohorts are entirely composed of patients with metastasis, some without but metastasis is not mentioned. Metastasis has been shown to impact the immune landscape.
Intrinsic heterogeneity of the cohort is indeed one of the weaknesses of our work, which could negatively impact the statistical significance of our results and, as a consequence, mask certain observations or make them less statistically significant. We mention this in the discussion section. It should not, in our understanding, lead to any false conclusions. We did not, however, pool data from primary and metastatic tumor samples, and all tumor samples that we mention are primary tumors.
The following part of a sentence was added to the discussion:
“...which could negatively impact the statistical significance of our results and, as a consequence, mask certain observations or make them less statistically significant.”
A reviewer brought up a concern about the overlap analysis and I also asked for an explanation on why this F2 metric was chosen. Part of the rebuttal argues that another metric was explored showing similar results, thus the conclusion reached is reasonable. Remarkably, these data are not only omitted from the manuscript, but are not even provided for the reviewers.
We did not intend to conceal any data from the reviewers, and we now added the panel for D metric to the S1 figure. We would also like to point out that the panel describing R metric for repertoire overlaps (a measure of similarity of overlapping clonotype frequencies), was included in the first version of the S2 Figure (now S1 Figure), and it also showed a similar trend. We hope that now the data are fully conclusive.
This manuscript certainly includes some interesting and useful work. Unfortunately, a comprehensive re-write was required to make the work much clearer and easier to understand and this has not been realized.
Again, we thank the reviewers for their thorough evaluation, and hopefully we could make the text clearer in the second reviewed version.
-
-
Author response:
The following is the authors’ response to the original reviews.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
The authors attempt to fully characterize the immunoglobulin (Ig) heavy (H) chain repertoire of tumor-infiltrating B cells from three different cancer types by identifying the IgH repertoire overlap between these, their corresponding draining lymph nodes (DLNs), and peripheral B cells. The authors claim that B cells from tumors and DLNs have a closer IgH profile than those in peripheral blood and that DLNs are differentially involved with tumor B cells. The claim that tumor-resident B cells are more immature and less specific is made based on the characteristics of the CDR-H3 they express.
Strengths:
The authors show great expertise in developing in-house bioinformatics pipelines, as well as using tools …
Author response:
The following is the authors’ response to the original reviews.
Public Reviews:
Reviewer #1 (Public Review):
Summary:
The authors attempt to fully characterize the immunoglobulin (Ig) heavy (H) chain repertoire of tumor-infiltrating B cells from three different cancer types by identifying the IgH repertoire overlap between these, their corresponding draining lymph nodes (DLNs), and peripheral B cells. The authors claim that B cells from tumors and DLNs have a closer IgH profile than those in peripheral blood and that DLNs are differentially involved with tumor B cells. The claim that tumor-resident B cells are more immature and less specific is made based on the characteristics of the CDR-H3 they express.
Strengths:
The authors show great expertise in developing in-house bioinformatics pipelines, as well as using tools developed by others, to explore the IgH repertoire expressed by B cells as a means of better characterizing tumor-associated B cells for the future generation of tumor-reactive antibodies as a therapy.
Weaknesses:
This paper needs major editing, both of the text and the figures, because as it stands it is convoluted and extremely difficult to follow. The conclusions reached are often not obvious from the figures themselves. Sufficient a priori details describing the framework for their analyses are not provided, making the outcome of their results questionable and leaving the reader wondering whether the findings are on solid ground.
The authors are encouraged to explain in more detail the premises used in their algorithms, as well as the criteria they follow to define clonotypes, clonal groups, and clonal lineages, which are currently poorly defined and are crucial elements that may influence their results and conclusions.
In response to this comment, we significantly expanded the paragraph dedicated to the tumor and non-tumor repertoire overlap and isotype composition. The following sections were added:
First, we characterized the relative similarity of IGH repertoires derived from tumors, DLN, and PBMC on the individual CDR-H3 clonotype level. We define clonotype as an instance with an identical CDR-H3 nucleotide sequence and identical V- and J- segment attribution (isotype attribution may be different). Unlike other authors, here we do not pool together similar CDR-H3 sequences to account for hypermutation. (Hypermutation analysis is done separately and defined as clonal group analysis. )
As overlap metrics are dependent on overall repertoire richness, we normalized the comparison using the same number of top most frequent clonotypes of each isotype from each sample (N = 109). Repertoire data for each sample were split according to the immunoglobulin isotype, and the F2 metric was calculated for each isotype separately and plotted as an individual point.
We also analyzed D metric, which represents the relative overlap diversity uninfluenced by clonotype frequency (Dij=dij/(di*dj), where dij is the number of clonotypes present in both samples, while di and dj are the diversities of samples i and j respectively). The results for D metric are not shown, as they indicate a similar trend to that of F2 metric. This observation allows us to conclude that tumor IGH repertoires are more similar to the repertoires of lymph nodes than to those of peripheral blood, both if clonotype frequency is taken into account, and when it is not.
Having excluded the IGHD gene segment from some of their analyses (at least those related to clonal lineage inference and phylogenetic trees), it is not well explained which region of CDR-H3 is responsible for the charge, interaction strength, and Kidera factors, since in some cases the authors mention that the central part of CDR-H3 consists of five amino acids and in others of seven amino acids.
We considered different ways of calculating amino acid properties of CDR3 and used different parameters for sample-average and individual-sequence CDR3s. Now plots for Fig S6 C are updated for consistency and the parameters depicted there are now calculated using 5 central amino acids, as in other sections.
How can the authors justify that the threshold for CDR-H3 identity varies according to individual patient data?
Ideal similarity threshold may depend on several factors, such as sampling, sequencing depth etc. For example, imagine a sample picking up 100% of the clonal lineage sequences which differ only 1 amino acid from each other, and a worse quality sample/sequencing picking up only every other sequence. Obviously, the minimal threshold required to accumulate these into a cluster/clonal group would be different for these two cases (1aa for the former, and ~2 aa for the latter for single-linkage clustering). Or, in other words, the more the sequencing depth, the more dense the clusters will be. The method of individual threshold tailoring relies on the following: https://changeo.readthedocs.io/en/latest/examples/cloning.html
Although individual kidera factors that are significant in the context of our analysis are described in the text one by one on their first appearance, we now also added a sentence to describe Kidera factor analysis in general (page 8):
Kidera factors are a set of scores which quantify physicochemical properties of protein sequences (Nakai et al. 1988). 188 physical properties of the 20 amino acids are encoded using dimension reduction techniques.
Throughout the analyses, the reasons for choosing one type of cancer over another sometimes seem subjective and are not well justified in the text.
Whenever possible, we pooled all patients with all cancer types together, because the number of available samples did not allow us to draw any significant conclusions comparing between individual cancer types. When analyzing and showing individual patient data, we also did not attempt to depict any cancer-type-specific findings, but it is inevitable that we name a specific cancer type when labelling a sample coming from a specific tumor.
Overall, the narrative is fragmented. There is a lack of well-defined conclusions at the end of the results subheadings.
In addition to the described above, a conclusion was added to the paragraph describing hypermutation analysis:
IGHG clonotypes from lung cancer samples show higher number of hypermutations, possibly reflecting high mutational load found in lung cancer tissue. For melanoma, another cancer known for high mutational load, no statistically significant difference was found. This may be due to higher variance between melanoma samples, which hinders the analysis, or due to the small sample size.
The exact same paragraph is repeated twice in the results section.
Corrected.
The authors have also failed to synchronise the actual number of main figures with the text, and some panels are included in the main figures that are neither described nor mentioned in the text (Venn diagram Fig. 2A and phylogenetic tree Fig. 5D). Overall, the manuscript appears to have been rushed and not thoroughly read before submission.
Corrected.
Reviewers are forced to wade through, unravel, and validate poorly explained algorithms in order to understand the authors' often bold conclusions.
We hope that the aforementioned additions to the text and also addition to the Figure 1 make the narrative more easily understandable.
Reviewer #2 (Public Review):
Summary:
The authors sampled the B cell receptor repertoires of Cancers, their draining lymph nodes, and blood. They characterized the clonal makeup of all B cells sampled and then analyzed these clones to identify clonal overlap between tissues and clonal activation as expressed by their mutation level and CDR3 amino acid characteristics and length. They conclude that B cell clones from the Tumor interact more with their draining lymph node than with the blood and that there is less mutation/expansion/activation of B cell clones in Tumors. These conclusions are interesting but hard to verify due to the under-sampling and short sequencing reads as well as confusion as to when analysis is across all individuals or of select individuals.
Strengths:
The main strength of their analysis is that they take into account multiple characteristics of clonal expansion and activation and their different modes of visualization, especially of clonal expansion and overlap. The triangle plots once one gets used to them are very nice.
Weaknesses:
The data used appears inadequate for the conclusions reached. The authors' sample size of B cells is small and they do not address how it could be sufficient. At such low sampling rates, compounded by the plasmablast bias they mention, it is unclear if the overlap trends they observe show real trends. Analyzing only top clones by size does not solve this issue. As it could be that the top 100 clones of one tissue are much bigger than those of another and that all overlap trends are simply because the clones are bigger in one tissue or the other. i.e there is equal overlap of clones with blood but blood is not sufficiently sampled given its greater diversity and smaller clones.
Regarding the number of clonotypes to be taken into account, we were limited by the B cell infiltration of tumor samples and our ability to capture their repertoire. However, we use technical replicates on the level of cell suspension to ensure that at least top clonotypes are consistently sampled. So, this is how the data should be interpreted - as describing the most abundant clones in the repertoire (which also may be considered the most functionally relevant in case of tumor infiltrating lymphocytes).
To analyze the repertoire overlap, we generally use the F2 metric that takes clone size into account - because we think that clone size is an important functional factor. However, we have now added the description of using D metric (does not include clone frequency as a parameter) - which shows exactly the same trend as F2 metric. So, both F2 and D overlap metrics support our conclusion of higher overlap between tumor and LN.
The following text was added:
We also analyzed D metric, which represents the relative overlap diversity uninfluenced by clonotype frequency (Dij=dij/(di*dj), where dij is the number of clonotypes present in both samples, while di and dj are the diversities of samples i and j respectively). The results for D metric are not shown, as they indicate a similar trend to that of F2 metric. This observation allows us to conclude that tumor IGH repertoires are more similar to the repertoires of lymph nodes than to those of peripheral blood, both if clonotype frequency is taken into account, and when it is not.
All in all, of course, the deeper the better, but given the data we were able to generate from the samples, this was the best approach to normalization that could be used.
Similarly, the read length (150bp X2) is too short, missing FWR1 and CDR1 and often parts of FWR2 if CDR3 is long. As the authors themselves note (and as was shown in (Zhang 2015 - PMC4811607) this makes mutation analysis difficult.
Indeed, we are aware of this problem, and therefore only a small part of the manuscript is dedicated to the hypermutation analysis. However, as the CDR-H3 region is the most mutated part, we still can capture significant diversity of mutations. To address the question of applicability of our data for the hypermutation phylogeny analysis, we compare the distribution of physico-chemical properties along the trees of hypermutation using the 150+150 and 300+300 data from the same donor and the same set of samples. The main conclusion is that neither for long, nor for short datasets could any correlation of physicochemical properties of the CDR-H3 region with the rank of the clonotype on the tree be found.
It also makes the identification of V genes and thus clonal identification ambiguous. This issue becomes especially egregious when clones are mutated.
Again, this would be important for clonotype phylogeny analysis. However, for the simple questions that we address with our clonal group analysis, such as clonal group overlap between tissues etc, we consider this data acceptable, because if any mislabelling of V segment occurs, it is a) rare and b) is equally frequent in all types of samples. Therefore, any conclusions made are still valid despite this technical drawback.
To directly address the question of mislabelling of V-genes in our data, we looked at the average number of different V-genes attributed to the same nucleotide sequence of CDR-H3 region in the short (150+150) and long (300+300) datasets from the same donor. Indeed, some ambiguity of V-gene labelling is observed (see below), but we think that it is unlikely to influence any of our cautious conclusions.
Author response image 1.
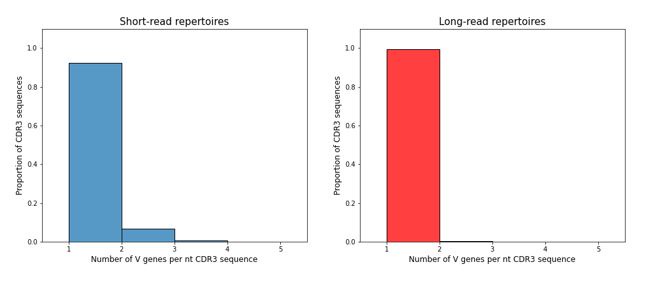
Finally, it is not completely clear when the analysis is of single individuals or across all individuals. If it is the former the authors did not explain how they chose the individuals analyzed and if the latter then it is not clear from the figures which measurements belong to which individual (i.e they are mixing measurements from different people).
We addressed this issue by adding a comment to each figure caption, describing whether a particular figure or panel describes individual or pooled data, and also whether the analysis is done on individual clonotype or clonal group level.
Also, in case pooled data were used, we added the number of patients that was pooled for a particular type of analysis. This number differs from one type of analysis to the other, because not all the patients had a complete set of tissues, and also not all samples passed a quality check for a particular analysis.
Here are the numbers listed:
Fig 2A: N=6 (we were only considering those who had all three tissues)
Fig 2C, N=14 (all)
2D: N=14 (all)
2E N=7 (have both tum and PBMC).
2F N=9 (have both tum and PBMC).
2G N=9 (have both tum and PBMC)
2H N=7 (have both tum and LN)
3A N=14 (all)
3B N=11 (only those with tumor)
3E - N=14
7F N=11 (all that have tumor)
Reviewer #3 (Public Review):
In multiple cancers, the key roles of B cells are emerging in the tumor microenvironment (TME). The authors of this study appropriately introduce that B cells are relatively under-characterised in the TME and argue correctly that it is not known how the B cell receptor (BCR) repertoires across tumors, lymph nodes, and peripheral blood relate. The authors therefore supply a potentially useful study evaluating the tumor, lymph node, and peripheral blood BCR repertoires and site-to-site as well as intra-site relationships. The authors employ sophisticated analysis techniques, although the description of the methods is incomplete. Among other interesting observations, the authors argue that the tumor BCR repertoire is more closely related to that of draining lymph node (dLN) than the peripheral blood in terms of clonal and isotype composition. Furthermore, the author's findings suggest that tumor-infiltrating B cells (TIL-B) exhibit a less mature and less specific BCR repertoire compared with circulating B cells. Overall, this is a potentially useful work that would be of interest to both medical and computational biologists working across cancer. However, there are aspects of the work that would have benefitted from further analysis and areas of the manuscript that could be written more clearly and proofread in further detail.
Major Strengths:
(1) The authors provide a unique analysis of BCR repertoires across tumor, dLN, and peripheral blood. The work provides useful insights into inter- and intra-site BCR repertoire heterogeneity. While patient-to-patient variation is expected, the findings with regard to intra-tumor and intra-dLN heterogeneity with the use of fragments from the same tissue are of importance, contribute to the understanding of the TME, and will inform future study design.
(2) A particular strength of the study is the detailed CDR3 physicochemical properties analysis which leads the authors to observations that suggest a less-specific BCR repertoire of TIL-B compared to circulating B cells.
Major Weaknesses:
The study would have benefitted from a deeper biological interpretation of the data. While given the low number of patients one can plausibly understand a reluctance to speculate about clinical details, there is limited discussion about what may contribute to observed heterogeneity.
We indeed do not want to overinterpret our data, especially where it comes to the difference between types of cancer. On the other hand, extracting similar patterns between different cancer types allows to pinpoint mechanisms that are more general and do not depend on cancer type. As for the potential source of intratumoral heterogeneity that we observe, we think that it may be coming from the selective sampling of tertiary lymphoid structures. We include IHC data for TLS detection in the supplementary Fig.5. Also, tumor mutation clonality may correlate with differential antibody response (i.e. different IGH clonotypes developing to recognize different antigens) – as has been previously described for TCRs by the lab of B.Chain in https://www.ncbi.nlm.nih.gov/pmc/articles/PMC6890490/.
For example, for the analysis of three lymph nodes taken per patient which were examined for inter-LN heterogeneity, there is a lack of information regarding these lymph nodes.
Unfortunately no clinical information about the lymph nodes was available.
'LN3' is deemed as exhibiting the most repertoire overlap with the tumor but there is no discussion as to why this may be the case.
The following phrases describes this in the “LN-to-LN heterogeneity in colorectal cancer” paragraph:
Similarly, an unequal interaction of tumors with DLNs was observed at the level of hypermutating clonal groups.
Functionally, this may again indicate that within a group of DLNs, nodes are unequal in terms of access to tumor antigens, and this inequality shapes the BCR repertoires within these lymph nodes.
(2) At times the manuscript is difficult to follow. In particular, the 'Intra-LN heterogeneity' section follows the 'LN-LN heterogeneity in colorectal cancer' section and compares the overlap of LN fragments (LN11, LN21, LN31) with the tumor in two separate patients (Fig 6A). In the previous section (LN-LN), LN11, LN21, LN31 are names given to separate lymph nodes from the same patient. The fragments are referred to as 'LN2' and the nodes in the previous section are referred to similarly. This conflation of naming for nodes and fragments is confusing.
We corrected this.
(3) There is a duplicated paragraph in 'Short vs long trees' and the following section 'Productive involvement in hypermutation lineages depends on CDR3 characteristics.
Corrected.
Reviewer #1 (Recommendations For The Authors):
- Figures:
Figure 1A lacks resolution
Corrected
Figure 2A, Venn diagram: What do the colors indicate?
Corrected
Figure 5D, why include this tree when there is no mention of it in the text?
Described
Figures 8, 9, and 10 are not to be found. One should not have to figure out that they became supplementary in the end.
Corrected
Regarding the physicochemical properties of CDR-H3, what do the authors mean by "the central part"? Do the authors refer to the CDR-H3 loop, and if so, how is that defined when the IGHD gene segment is excluded from the analyses? Is it 5 amino acids (Productive involvement in hypermutating lineages depends on CDR3 characteristics, Page 21/39 in merged document) and (CDR3 properties, Page 8/39 in merged document), or 7 amino acids (Short vs long trees phylogeny analysis, Page 19/39 in merged document)? Please clarify.
We considered different ways of calculating amino acid properties of CDR3 and used different parameters for sample-average and individual-sequence CDR3s. Now plots for Fig S6 C are updated for consistency. IGHD segment was not excluded from the analysis. The reviewer might be confused by our description of phylogenetic inference, when an artificial outgroup with D segment deleted is added to the clonal group to facilitate the inference process. All other sequences were analyzed in their original form with the D segment. This way, we could avoid biases in phylogeny introduced by misassignment of D gene germline to the outgroup.
What was the threshold for CDR-H3 identity in their analyses? How can the authors justify that this value changes according to individual patient datasets? (Materials & methods, Clonal lineage inference Page 29/39 in merged document).
As described earlier, ideal similarity threshold may depend on several factors, such as sampling, sequencing depth etc. For example, imagine a sample picking up 100% of the clonal lineage sequences which differ only 1 amino acid from each other, and a worse quality sample/sequencing picking up only every other sequence. Obviously, the minimal threshold required to accumulate these into a clonotype would be different for these two cases (1aa for the former, and ~2 aa for the latter for single-linkage clustering). The method of individual threshold tailoring relies on this: https://changeo.readthedocs.io/en/latest/examples/cloning.html
What is the difference between tumor-induced and tumor-infiltrating B cells? How can the authors discriminate between the two? Page 6/39 in the merged document.
corrected to tumor-infiltrating
"Added nucleotides" meaning N additions? Page 3/39 in the merged document.
yes
How many cancer patients were enrolled? 17 or 14(Materials & methods page 27/39 in the merged document)? Please clarify.
In the current project 14 patients were enrolled. The appropriate changes have been introduced in the final text. Supplementary table 2 has been added with the patient data.
Abbreviations are used without full descriptions.
According to reviewer’s recommendation, a list of abbreviations was added in the manuscript, and also full descriptions were added in the text upon first mentioning of the term.
Use either CDR3 or CDR-H3
We corrected the text to use CDR-H3 abbreviation throughout the text.
Reviewer #2 (Recommendations For The Authors):
I would like to start by apologizing for the time it took me to review.
As I mentioned above there are issues with the clonal sampling of the sequencing length and the statistics in this paper. From reading the paper I am not sure if they are fixable but there are some things that could be tried.
(1) The authors mention the diversity of their individual analysis - 17 individuals across 3 cancer types, but do not then systematically show us how the different things they measure track across the different individuals and cancer types. it is possible that some trends would be more convincing if we saw them happening again and again across all individuals. But, as I said above, the authors do not identify individuals clearly across all their types of analysis nor do they explain why sometimes they show analysis of specific individuals.
For overlap analysis (Fig. 2 except panel B), CDR3 properties analysis (Fig. 3, Fig. S7), clonal group analysis (Fig. 4) we used pooled data on all cancers, unless it is indicated otherwise on the panel. For overlap analysis, we used Cytoscape graph (Fig. 2B) for one patient, mp3, to illustrate the findings that were made on pooled data. For other types of analysis, such as overlap between individual lymph nodes, or tumor fragments (Fig. 5, 6, 7 except panel F) pooled analysis is not possible due to the individual nature of the processes in question.
(2) The authors do not address how lacking their sampling is nor the distribution of clone sizes in different tissues/ individuals/ subsets. Without such a discussion it is not clear how tenuous or convincing their conclusions are.
(3) The short sequencing lengths limit the ability to exactly identify V and thus the germline root of clones, whose positions are mutated and clonal association of sequences. The authors appear to be aware of this as they often use the most common ancestor as the start of their analysis... however, again there are inconsistencies that are not clearly described in the text. in creating trees with change they defined roots as the putative germline and at least in most cases also in clone association although in some analyses potentially similar clones were collapsed into clonotypes. Again it is not clear when one method was used or the other and how the choice was made what to choose.
Here we can only state that we consistently used the approach described in the Methods section, which was the following:
First, the repertoires were clustered into clonal lineages using the criteria described in “Methods: Clonal lineage inference” Assuming that each clonotype sequence in the clonal lineage originated from the same ancestor, we try to recover the phylogeny. Please note that we refer to the individual BCR sequences as “clonotypes”, and to a group of clonotypes that presumably share a common ancestor - as “clonal lineage” or “clonal group”.
The phylogeny of B-cell hypermutations was inferred for each clonal lineage of size five or more using the maximum likelihood method and the GTR GAMMA nucleotide substitution model. To find the most recent common ancestor (MRCA) or “root” of the tree, we used an artificial outgroup constructed as a conjugate of germline segments V and J defined by MIXCR and added it to the clonal lineage. The D segment was excluded from the outgroup formation, as there was insufficient confidence in the germline annotations due to its short length and high level of mutations. The rest of the clonotypes were still analyzed in their original form with D segment in place. Deleting D segment from the outgroup simply eliminates the risk of biasing the phylogeny by missasigning D segment germline sequence to the outgroup. The MUSCLE tool was used for multiple sequence alignment and RAxML software was used to build and root phylogenetic trees.
(4) Beyond the statistical issues mentioned above: the unclear selection of individual examples for comparison and significance testing, the mixing of individuals and cancer types without clear identification, etc. there is in general a lack of coherence in the statistical analysis performed. specifically:
(a) the authors should choose one cutoff for significance (0.01 for instance) and then just mention when things are significant and when not. There is no need and it is confusing to add the p-value for every comparison. P-values are not good measures of effect size.
We corrected the figures and left p-values only where they are below significance threshold.
(b) the Bonferroni correction used is not well characterized. For an alpha of 0.01 in Figures 3 C and D how many tests were performed?
The number of tests performed that was used for Bonferroni-Holm correction equals the number of comparisons on the heatmap which makes it 39 for each heatmap on Fig 3C and 13 for Fig 3D.
Finally some minor issues -
(1) Not all acronyms are described, for instance, TME and TIL. The first time any acronym is used it should be spelled out. -> Katya B- список сокращений
(2) The figure captions are not all there...
(a) there is no caption for Figure 3E.
corrected
(b) there are Figure 7 F and G panels but no Figure 7E panel and Figure F is described after Figure G.
corrected
(3) A few problems with wording -
(a) bottom paragraph of page 3 - instead of :
"different lymph nodes from one draining lymph node pool may be more or less involved"
Corrected to "different lymph nodes from one draining lymph node pool may be differentially involved"
(b) figure caption for figure 3a: instead of:
"CDR3 are on average significantly higher in tumor"
Corrected to "CDR3 are on average significantly longer in tumor"
Reviewer #3 (Recommendations For The Authors):
- FIG1A - Suggest expanding the legend to include more information on the computational analyses.
added
- PAGE SIX: Suggest adding a table or some text on patient characteristics. Numbers of unique clonotypes per sample etc. Are there differences in age/sex that need to be considered? Some clonotype information is available in S1 but some summary and statistics would be appreciated.
Added patient information as Supplementary table 2.
- PAGE SIX: F2 Metric, suggestion to explain why this was used vs. other metrics.
We expanded the following paragraph to include information about F2 metric and D metric, and the reason why we are using F2.
Repertoire data for each sample were split according to the immunoglobulin isotype, and the F2 metric was calculated for each isotype separately and plotted as an individual point. We used the repertoire overlap metric F2 (Сlonotype-wise sum of geometric mean frequencies of overlapping clonotypes), which accounts for both the number and frequency of overlapping clonotypes (Fig. 2A). As expected, significantly lower overlaps were observed between the IGH repertoires of peripheral blood and tumors compared to LN/tumor overlaps. The LN/PBMC overlap also tended to be lower, but the difference was not statistically significant. We also analyzed D metric, which represents the relative overlap diversity uninfluenced by clonotype frequency (Dij=dij/(di*dj), where dij is the number of clonotypes present in both samples, while di and dj are the diversities of samples i and j respectively). The results for D metric are not shown, as they indicate a similar trend to that of F2 metric. This observation allows us to conclude that tumor IGH repertoires are more similar to the repertoires of tumor-draining LNs than to those of peripheral blood, both if clonotype frequency is taken into account, and when it is not.
- PAGE SIX: Make clear in the text that mp3 is a patient.
Added “melanoma patient mp3”
- PAGE EIGHT: Suggest explaining kidera factors at first use - not all readers will know what they are.
We expanded the following paragraph to add more information about Kidera factors:
To explore CDR-H3 physicochemical properties, we calculated the mean charge, hydropathy, predicted interaction strength, and Kidera factors 1 - 9 (kf1-kf9) for five central amino acids of the CDR-H3 region for the 100 most frequent clonotypes of each sample using VDJtools. Kidera factors are a set of scores which quantify physicochemical properties of protein sequences 61. 188 physical properties of the 20 amino acids are encoded using dimension reduction techniques, to yield 9 factors which are used to quantitatively characterize physicochemical properties of amino acid sequences.
- Fig 5D is not referred to.
Corrected
-
eLife assessment
This study provides useful insights into inter- and intra-site B cell receptor repertoire heterogeneity, noting that B cell clones from the tumour interact more with their draining lymph node than with the blood and that there is less mutation/expansion/activation of B cell clones in tumours. Unfortunately, the main claims are incomplete and only partially supported. The work could be of interest to an audience including medical biologists/immunologists and computational biologists across cancer specialities.
-
Reviewer #3 (Public Review):
In multiple cancers, the key roles of B cells are emerging in the tumor microenvironment (TME). The authors of this study appropriately introduce that B cells are relatively under-characterised in the TME and argue correctly that it is not known how the B cell receptor (BCR) repertoires across tumor, lymph node and peripheral blood relate. The authors therefore supply a potentially useful study evaluating the tumor, lymph node and peripheral blood BCR repertoires and site-to-site as well as intra-site relationships. The authors employ sophisticated analysis techniques, although the description of the methods is incomplete.
Major strengths:
(1) The authors provide a unique analysis of BCR repertoires across tumor, dLN, and peripheral blood. The work provides useful insights into inter- and intra-site BCR …
Reviewer #3 (Public Review):
In multiple cancers, the key roles of B cells are emerging in the tumor microenvironment (TME). The authors of this study appropriately introduce that B cells are relatively under-characterised in the TME and argue correctly that it is not known how the B cell receptor (BCR) repertoires across tumor, lymph node and peripheral blood relate. The authors therefore supply a potentially useful study evaluating the tumor, lymph node and peripheral blood BCR repertoires and site-to-site as well as intra-site relationships. The authors employ sophisticated analysis techniques, although the description of the methods is incomplete.
Major strengths:
(1) The authors provide a unique analysis of BCR repertoires across tumor, dLN, and peripheral blood. The work provides useful insights into inter- and intra-site BCR repertoire heterogeneity. While patient-to-patient variation is expected, the findings with regard to intra-tumor and intra-dLN heterogeneity with the use of fragments from the same tissue are of importance, contribute to the understanding of the TME, and will inform future study design.
(2) A particular strength of the study is the detailed CDR3 physicochemical properties analysis which leads the authors to observations that suggest a less-specific BCR repertoire of TIL-B compared to circulating B cells.
Concerns and comments on current version:
The revision has improved the manuscript but, in my opinion, remains inadequate. While most of my requested changes have been made, I do not see an expansion of Fig1A legend to incorporate more details about the analysis. Lacking details of methodology was a concern from all reviewers. Similarly, the 'fragmented' narrative was a concern of all reviewers. These matters have not been dealt with adequately enough - there are parts of the manuscript which remain fragmented and confusing. The narrative and analysis does not explain how the plasma cell bias has been dealt with adequately and in fact is simply just confusing. There is a paragraph at the beginning of the discussion re the plasma cell bias, which should be re-written to be clearer and moved to have a prominent place early in the results. Why are these results not properly presented? They are key for interpretation of the manuscript. Furthermore, the sorted plasma cell sequencing analysis also has only been performed on two patients. Another issue is that some disease cohorts are entirely composed of patients with metastasis, some without but metastasis is not mentioned. Metastasis has been shown to impact the immune landscape.
A reviewer brought up a concern about the overlap analysis and I also asked for an explanation on why this F2 metric chosen. Part of the rebuttal argues that another metric was explored showing similar results, thus conclusion reached is reasonable. Remarkably, these data are not only omitted from the manuscript, but is not even provided for the reviewers.
This manuscript certainly includes some interesting and useful work. Unfortunately, a comprehensive re-write was required to make the work much clearer and easier to understand and this has not been realised.
-
-
eLife assessment
This study provides useful insights into inter- and intra-site B cell receptor repertoire heterogeneity, noting that B cell clones from the tumour interact more with their draining lymph node than with the blood and that there is less mutation/expansion/activation of B cell clones in tumours. Unfortunately, the main claims are incomplete and only partially supported. This work could be of interest to an audience including medical biologists/immunologists and computational biologists across cancer specialities.
-
Reviewer #1 (Public Review):
Summary:
The authors attempt to fully characterise the immunoglobulin (Ig) heavy (H) chain repertoire of tumor-infiltrating B cells from three different cancer types by identifying the IgH repertoire overlap between these, their corresponding draining lymph nodes (DLNs), and peripheral B cells. The authors claim that B cells from tumors and DLNs have a closer IgH profile than those in peripheral blood and that DLNs are differentially involved with tumor B cells. The claim that tumor-resident B cells are more immature and less specific is made based on the characteristics of the CDR-H3 they express.
Strengths:
The authors show great expertise in developing in-house bioinformatics pipelines, as well as using tools developed by others, to explore the IgH repertoire expressed by B cells as a means of better …
Reviewer #1 (Public Review):
Summary:
The authors attempt to fully characterise the immunoglobulin (Ig) heavy (H) chain repertoire of tumor-infiltrating B cells from three different cancer types by identifying the IgH repertoire overlap between these, their corresponding draining lymph nodes (DLNs), and peripheral B cells. The authors claim that B cells from tumors and DLNs have a closer IgH profile than those in peripheral blood and that DLNs are differentially involved with tumor B cells. The claim that tumor-resident B cells are more immature and less specific is made based on the characteristics of the CDR-H3 they express.
Strengths:
The authors show great expertise in developing in-house bioinformatics pipelines, as well as using tools developed by others, to explore the IgH repertoire expressed by B cells as a means of better characterising tumour-associated B cells for the future generation of tumour-reactive antibodies as a therapy.
Weaknesses:
This paper needs major editing, both of the text and the figures, because as it stands it is convoluted and extremely difficult to follow. The conclusions reached are often not obvious from the figures themselves. Sufficient a priori details describing the framework for their analyses are not provided, making the outcome of their results questionable and leaving the reader wondering whether the findings are on solid ground. The authors are encouraged to explain in more detail the premises used in their algorithms, as well as the criteria they follow to define clonotypes, clonal groups, and clonal lineages, which are currently poorly defined and are crucial elements that may influence their results and conclusions. Having excluded the IGHD gene segment from some of their analyses (at least those related to clonal lineage inference and phylogenetic trees), it is not well explained which region of CDR-H3 is responsible for the charge, interaction strength, and Kidera factors, since in some cases the authors mention that the central part of CDR-H3 consists of five amino acids and in others of seven amino acids. How can the authors justify that the threshold for CDR-H3 identity varies according to individual patient data?
Throughout the analyses, the reasons for choosing one type of cancer over another sometimes seem subjective and are not well justified in the text.
Overall, the narrative is fragmented. There is a lack of well-defined conclusions at the end of the results subheadings. The exact same paragraph is repeated twice in the results section. The authors have also failed to synchronise the actual number of main figures with the text, and some panels are included in the main figures that are neither described nor mentioned in the text (Venn diagram Fig. 2A and phylogenetic tree Fig. 5D). Overall, the manuscript appears to have been rushed and not thoroughly read before submission.
Reviewers are forced to wade through, unravel, and validate poorly explained algorithms in order to understand the authors' often bold conclusions.
-
Reviewer #2 (Public Review):
Summary:
The authors sampled the B cell receptor repertoires of Cancers, their draining lymph nodes, and blood. They characterized the clonal makeup of all B cells sampled and then analyzed these clones to identify clonal overlap between tissues and clonal activation as expressed by their mutation level and CDR3 amino acid characteristics and length. They conclude that B cell clones from the Tumor interact more with their draining lymph node than with the blood and that there is less mutation/expansion/activation of B cell clones in Tumors. These conclusions are interesting but hard to verify due to the under-sampling and short sequencing reads as well as confusion as to when analysis is across all individuals or of select individuals.
Strengths:
The main strength of their analysis is that they take into …
Reviewer #2 (Public Review):
Summary:
The authors sampled the B cell receptor repertoires of Cancers, their draining lymph nodes, and blood. They characterized the clonal makeup of all B cells sampled and then analyzed these clones to identify clonal overlap between tissues and clonal activation as expressed by their mutation level and CDR3 amino acid characteristics and length. They conclude that B cell clones from the Tumor interact more with their draining lymph node than with the blood and that there is less mutation/expansion/activation of B cell clones in Tumors. These conclusions are interesting but hard to verify due to the under-sampling and short sequencing reads as well as confusion as to when analysis is across all individuals or of select individuals.
Strengths:
The main strength of their analysis is that they take into account multiple characteristics of clonal expansion and activation and their different modes of visualization, especially of clonal expansion and overlap. The triangle plots once one gets used to them are very nice.
Weaknesses:
The data used appears inadequate for the conclusions reached. The authors' sample size of B cells is small and they do not address how it could be sufficient. at such low sampling rates, compounded by the palsmablast bias they mention, it is unclear if the overlap trends they observe show real trends. Analyzing only top clones by size does not solve this issue. As it could be that the top 100 clones of one tissue are much bigger than those of another and that all overlap trends are simply because the clones are bigger in one tissue or the other. i.e there is equal overlap of clones with blood but blood is not sufficiently sampled given its greater diversity and smaller clones. Similarly, the read length (150bp X2) is too short missing FWR1 and CDR1 and often parts of FWR2 if CDR3 is long. As the authors themselves note (and as was shown in (Zhang 2015 - PMC4811607) this makes mutation analysis difficult. It also makes the identification of V genes and thus clonal identification ambiguous. This issue becomes especially egregious when clones are mutated. Finally, it is not completely clear when the analysis is of single individuals or across all individuals. If it is the former the authors did not explain how they chose the individuals analyzed and if the latter then it is not clear from the figures which measurements belong to which individual (i.e they are mixing measurements from different people). For all these reasons while the authors make many interesting suggestions about the potential relationships of B cell repertoires in cancer tissues and their draining lymph nodes and how to characterize and visualize them, it is hard to assess any of their conclusions and specific results.
-
Reviewer #3 (Public Review):
In multiple cancers, the key roles of B cells are emerging in the tumor microenvironment (TME). The authors of this study appropriately introduce that B cells are relatively under-characterised in the TME and argue correctly that it is not known how the B cell receptor (BCR) repertoires across tumors, lymph nodes, and peripheral blood relate. The authors therefore supply a potentially useful study evaluating the tumor, lymph node, and peripheral blood BCR repertoires and site-to-site as well as intra-site relationships. The authors employ sophisticated analysis techniques, although the description of the methods is incomplete. Among other interesting observations, the authors argue that the tumor BCR repertoire is more closely related to that of draining lymph node (dLN) than the peripheral blood in terms of …
Reviewer #3 (Public Review):
In multiple cancers, the key roles of B cells are emerging in the tumor microenvironment (TME). The authors of this study appropriately introduce that B cells are relatively under-characterised in the TME and argue correctly that it is not known how the B cell receptor (BCR) repertoires across tumors, lymph nodes, and peripheral blood relate. The authors therefore supply a potentially useful study evaluating the tumor, lymph node, and peripheral blood BCR repertoires and site-to-site as well as intra-site relationships. The authors employ sophisticated analysis techniques, although the description of the methods is incomplete. Among other interesting observations, the authors argue that the tumor BCR repertoire is more closely related to that of draining lymph node (dLN) than the peripheral blood in terms of clonal and isotype composition. Furthermore, the author's findings suggest that tumor-infiltrating B cells (TIL-B) exhibit a less mature and less specific BCR repertoire compared with circulating B cells. Overall, this is a potentially useful work that would be of interest to both medical and computational biologists working across cancer. However, there are aspects of the work that would have benefitted from further analysis and areas of the manuscript that could be written more clearly and proofread in further detail.
Major Strengths:
1. The authors provide a unique analysis of BCR repertoires across tumor, dLN, and peripheral blood. The work provides useful insights into inter- and intra-site BCR repertoire heterogeneity. While patient-to-patient variation is expected, the findings with regard to intra-tumor and intra-dLN heterogeneity with the use of fragments from the same tissue are of importance, contribute to the understanding of the TME, and will inform future study design.
2. A particular strength of the study is the detailed CDR3 physicochemical properties analysis which leads the authors to observations that suggest a less-specific BCR repertoire of TIL-B compared to circulating B cells.
Major Weaknesses:
1. The study would have benefitted from a deeper biological interpretation of the data. While given the low number of patients one can plausibly understand a reluctance to speculate about clinical details, there is limited discussion about what may contribute to observed heterogeneity. For example, for the analysis of three lymph nodes taken per patient which were examined for inter-LN heterogeneity, there is a lack of information regarding these lymph nodes. 'LN3' is deemed as exhibiting the most repertoire overlap with the tumor but there is no discussion as to why this may be the case.
2. At times the manuscript is difficult to follow. In particular, the 'Intra-LN heterogeneity' section follows the 'LN-LN heterogeneity in colorectal cancer' section and compares the overlap of LN fragments (LN11, LN21, LN31) with the tumor in two separate patients (Fig 6A). In the previous section (LN-LN), LN11, LN21, LN31 are names given to separate lymph nodes from the same patient. The fragments are referred to as 'LN2' and the nodes in the previous section are referred to similarly. This conflation of naming for nodes and fragments is confusing.
3. There is a duplicated paragraph in 'Short vs long trees' and the following section 'Productive involvement in hypermutation lineages depends on CDR3 characteristics.
-
