ARID1A governs the silencing of sex-linked transcription during male meiosis in the mouse
Curation statements for this article:-
Curated by eLife

eLife assessment
This study presents a valuable dataset regarding chromatin remodeling by the BAF complex in the context of meiotic sex chromosome inactivation. Solid data generally support the conclusions, although the partial deletion of the BAF complex in the germline could be considered limiting. This work will be of interest to researchers working on chromatin and reproductive biology.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
We present evidence implicating the BAF (BRG1/BRM Associated Factor) chromatin remodeler in meiotic sex chromosome inactivation (MSCI). By immunofluorescence (IF), the putative BAF DNA binding subunit, ARID1A (AT-rich Interaction Domain 1 a), appeared enriched on the male sex chromosomes during diplonema of meiosis I. Germ cells showing a Cre-induced loss of ARID1A arrested in pachynema and failed to repress sex-linked genes, indicating a defective MSCI. Mutant sex chromosomes displayed an abnormal presence of elongating RNA polymerase II coupled with an overall increase in chromatin accessibility detectable by ATAC-seq. We identified a role for ARID1A in promoting the preferential enrichment of the histone variant, H3.3, on the sex chromosomes, a known hallmark of MSCI. Without ARID1A, the sex chromosomes appeared depleted of H3.3 at levels resembling autosomes. Higher resolution analyses by CUT&RUN revealed shifts in sex-linked H3.3 associations from discrete intergenic sites and broader gene-body domains to promoters in response to the loss of ARID1A. Several sex-linked sites displayed ectopic H3.3 occupancy that did not co-localize with DMC1 (DNA meiotic recombinase 1). This observation suggests a requirement for ARID1A in DMC1 localization to the asynapsed sex chromatids. We conclude that ARID1A-directed H3.3 localization influences meiotic sex chromosome gene regulation and DNA repair.
Article activity feed
-

-
-

eLife assessment
This study presents a valuable dataset regarding chromatin remodeling by the BAF complex in the context of meiotic sex chromosome inactivation. Solid data generally support the conclusions, although the partial deletion of the BAF complex in the germline could be considered limiting. This work will be of interest to researchers working on chromatin and reproductive biology.
-
Reviewer #3 (Public review):
In this manuscript, Magnuson and colleagues investigate the meiotic functions of ARID1A, a putative DNA binding subunit of the SWI/SNF chromatin remodeler BAF. The authors develop a germ cell specific conditional knockout (cKO) mouse model using Stra8-cre and observe that ARID1A-deficient cells fail to progress beyond pachytene, although due to inefficiency of the Stra8-cre system the mice retain ARID1A-expressing cells that yield sperm and allow fertility. Because ARID1A was found to accumulate at the XY body late in Prophase I, the authors suspected a potential role in meiotic silencing and by RNAseq observe significant misexpression of sex-linked genes that typically are silenced at pachytene. They go on to show that ARID1A is required for exclusion of RNA PolII from the sex body and for limiting promoter …
Reviewer #3 (Public review):
In this manuscript, Magnuson and colleagues investigate the meiotic functions of ARID1A, a putative DNA binding subunit of the SWI/SNF chromatin remodeler BAF. The authors develop a germ cell specific conditional knockout (cKO) mouse model using Stra8-cre and observe that ARID1A-deficient cells fail to progress beyond pachytene, although due to inefficiency of the Stra8-cre system the mice retain ARID1A-expressing cells that yield sperm and allow fertility. Because ARID1A was found to accumulate at the XY body late in Prophase I, the authors suspected a potential role in meiotic silencing and by RNAseq observe significant misexpression of sex-linked genes that typically are silenced at pachytene. They go on to show that ARID1A is required for exclusion of RNA PolII from the sex body and for limiting promoter accessibility at sex-linked genes, consistent with a meiotic sex chromosome inactivation (MSCI) defect in cKO mice. The authors proceed to investigate the impacts of ARID1A on H3.3 deposition genome-wide. H3.3 is known be regulated by ARID1A and is linked to silencing, and here the authors find that upon loss of ARID1A, overall H3.3 enrichment at the sex body as measured by IF failed to occur, but H3.3 was enriched specifically at transcriptional start sites of sex-linked genes that are normally regulated by ARID1A. The results suggest that ARID1A normally prevents H3.3 accumulation at target promoters on sex chromosomes and based on additional data, restricts H3.3 to intergenic sites. Finally, the authors present data implicating ARID1A and H3.3 occupancy in DSB repair, finding that ARID1A cKO leads to a reduction in focus formation by DMC1, a key repair protein. Overall the paper provides new insights into the process of MSCI from the perspective of chromatin composition and structure and raises interesting new questions about the interplay between chromatin structure, meiotic silencing and DNA repair.
In general the data are convincing. The conditional KO mouse model has some inherent limitations due to incomplete recombination and the existence of 'escaper' cells that express ARID1A and progress through meiosis normally. This reviewer feels that the authors have addressed this point thoroughly and have demonstrated clear and specific phenotypes using the best available animal model. The data demonstrate that the mutant cells fail to progress past pachytene, although it is unclear whether this specifically reflects pachytene arrest, as accumulation in other stages of Prophase is also suggested by the data in Table 1.
The revised manuscript more appropriately describes the relationship between ARID1A and DNA damage response (DDR) signaling. The authors don't see defects in a few DDR markers in ARID1A CKO cells (including a low resolution assessment of ATR), suggesting that ARID1A may not be required for meiotic DDR signaling. However, as previously noted the data do not rule out the possibility that ARID1A is downstream of DDR signaling, and the authors note the possibility of a role for DDR signaling upstream of ARID1A.
A final comment relates to the impacts of ARID1A loss on DMC1 focus formation and the interesting observation of reduced sex chromosome association by DMC1. The authors additionally assess the related recombinase RAD51 and suggest that it is unaffected by ARID1A loss. However, only a single image of RAD51 staining in the cKO is provided (Fig. S11) and there are no associated quantitative data provided. The data are suggestive and conclusions about the impacts of ARID1A loss on RAD51 must be considered as preliminary until more rigorously assessed.
Comments on latest version:
The authors have effectively addressed the minor issues raised in the most recent round of non-public reviews. This reviewer has no additional recommendations.
-
Author response:
The following is the authors’ response to the previous reviews.
Reviewer 1:
I understand that the only spermatids observed in cKO testes are coming from cells that escaped the Cre system. However, I do think that the authors could provide sperm counts data also showing decreased sperm counts in the mutant, to make their claim stronger. This is a very common fertility assessment.
All round spermatids isolated from Arid1acKO testes appeared only to express the normal transcript associated with the floxed allele (Fig. S4A).
[New Data - Lines 154-159] Our evaluation of the first round of spermatid development based on DNA content (1C, 2C, and 4C), revealed a significantly reduced abundance of round spermatids (1C) in mutant testes compared to wild-type testes. This finding, obtained through flow cytometry, supports the …
Author response:
The following is the authors’ response to the previous reviews.
Reviewer 1:
I understand that the only spermatids observed in cKO testes are coming from cells that escaped the Cre system. However, I do think that the authors could provide sperm counts data also showing decreased sperm counts in the mutant, to make their claim stronger. This is a very common fertility assessment.
All round spermatids isolated from Arid1acKO testes appeared only to express the normal transcript associated with the floxed allele (Fig. S4A).
[New Data - Lines 154-159] Our evaluation of the first round of spermatid development based on DNA content (1C, 2C, and 4C), revealed a significantly reduced abundance of round spermatids (1C) in mutant testes compared to wild-type testes. This finding, obtained through flow cytometry, supports the observed meiotic block at the pachytene stage (new Fig. S5A-B).
Reviewer 3:
Lines 154-5: Currently read 'inefficient Stra8-cre inefficiency'. Should read 'inefficient Stra8-cre activity.' I see that this was noted in the first round of review but the original wording has persisted.
The nucleolin antibody used should be listed in Supplementary table 3.
'inefficient Stra8-cre inefficiency' now reads “inefficient Stra8-Cre activity” [Line 158]
Nucleolin antibody is now listed in Supplementary Table 3
-
-
Author response:
The following is the authors’ response to the previous reviews.
Public Reviews
Reviewer #1 (Public Review):
Comment: The fact that there are Arid1a transcripts that escape the Cre system in the Arid1a KO mouse model might difficult the interpretation of the data. The phenotype of the Arid1a knockout is probably masked by the fact that many of the sequencing techniques used here are done on a heterogeneous population of knockout and wild type spermatocytes. In relation to this, I think that the use of the term "pachytene arrest" might be overstated, since this is not the phenotype truly observed. Knockout mice produce sperm, and probably litters, although a full description of the subfertility phenotype is lacking, along with identification of the stage at which cell death is happening by detection of apoptosis.
Res…
Author response:
The following is the authors’ response to the previous reviews.
Public Reviews
Reviewer #1 (Public Review):
Comment: The fact that there are Arid1a transcripts that escape the Cre system in the Arid1a KO mouse model might difficult the interpretation of the data. The phenotype of the Arid1a knockout is probably masked by the fact that many of the sequencing techniques used here are done on a heterogeneous population of knockout and wild type spermatocytes. In relation to this, I think that the use of the term "pachytene arrest" might be overstated, since this is not the phenotype truly observed. Knockout mice produce sperm, and probably litters, although a full description of the subfertility phenotype is lacking, along with identification of the stage at which cell death is happening by detection of apoptosis.
Response: As the reviewer indicates, we did not observe a complete arrest at Pachynema. In fact, the histology shows the presence of spermatids and sperm in seminiferous tubules and epididymides (Fig. Sup. 3). However, our data argue that the wild-type haploid gametes produced were derived from spermatocyte precursors that have likely escaped Cre mediated activity (Fig. Sup. 4). Furthermore, diplotene and metaphase-I spermatocytes lacking ARID1A protein by IF were undetectable in the Arid1acKO testes (Fig. S4B). Therefore, although we do not demonstrate a strict pachytene arrest, it is reasonable to conclude that ARID1A is necessary to progress beyond pachynema. We have revised the manuscript to reflect this point (Abstract lines 17,18; Results lines 153,154)
Comment: It is clear from this work that ARID1a is part of the protein network that contributes to silencing of the sex chromosomes. However, it is challenging to understand the timing of the role of ARID1a in the context of the well-known DDR pathways that have been described for MSCI.
Response: With respect to the comment on the lack of clarity as to which stage of meiosis we observe cell death, our data do suggest that it is reasonable to conclude that mutant spermatocytes (ARID1A-) undergo cell death at pachynema given their inability to execute MSCI, which is a well-established phenotype.
Comment: Staining of chromosome spreads with Arid1a antibody showed localization at the sex chromosomes by diplonema; however, analysis of gene expression in Arid1a KO was performed on pachytene spermatocytes. Therefore, is not very clear how the chromatin remodeling activity of Arid1a in diplonema is affecting gene expression of a previous stage. CUTnRUN showed that ARID1a is present at the sex chromatin in earlier stages, leading to hypothesize that immunofluorescence with ARID1a antibody might not reflect ARID1a real localization.
Response: It is unclear what the reviewer means about not understanding how ARID1A activity at diplonema affects gene expression at earlier stages. Our interpretations were not based solely on the observation of ARID1A associations with the XY body at diplonema. In fact, mRNA expression and CUT&RUN analyses were performed on pachytene-enriched populations. ARID1A's association with the XY body is not exclusive to diplonema. Based on both CUT&RUN and IF data, ARID1A associates with XY chromatin as early as pachynema. Only at late diplonema did we observe ARID1A hyperaccumulation on the XY body by IF.
Reviewer #2 (Public Review):
Comment: The inefficient deletion of ARID1A in this mouse model does not allow any detailed analysis in a quantitative manner.
Response: As explained in our response to these comments in the first revision, we respectfully disagree with this reviewer’s conclusions. We have been quantitative by co-staining for ARID1A, ensuring that we can score mutant pachytene spermatocytes from escapers. Additionally, we provide data to show the efficiency of ARID1A loss in the purified pachytene populations sampled in our genomic assays.
Reviewer #3 (Public Review):
Comment: The data demonstrate that the mutant cells fail to progress past pachytene, although it is unclear whether this specifically reflects pachytene arrest, as accumulation in other stages of Prophase also is suggested by the data in Table 1. The western blot showing ARID1A expression in WT vs. cKO spermatocytes (Fig. S2) is supportive of the cKO model but raises some questions. The blot shows many bands that are at lower intensity in the cKO, at MWs from 100-250kDa. The text and accompanying figure legend have limited information. Are the various bands with reduced expression different isoforms of ARID1A, or something else? What is the loading control 'NCL'? How was quantification done given the variation in signal across a large range of MWs?
Response: The loading control is Nucleolin. With respect to the other bands in the range of 100-250 kDa, it is difficult to say whether they represent ARID1A isoforms. The Uniprot entry for Mouse ARID1A only indicates a large mol. wt sequence of ~242 kDa; therefore, the band corresponding to that size was quantified. There is no evidence to suggest that lower molecular weight isoforms may be translated. Although speculative, it is possible that the lower molecular weight bands represent proteolytic/proteasomal degradation products or products of antibody non-specificity. These points are addressed in the revised manuscript (Legend to Fig S2, lines 926-931). Blots were scanned on a LI-COR Odyssey CLx imager and viewed and quantified using Image Studio Version 5.2.5 (Methods, lines 640-642).
Comment: An additional weakness relates to how the authors describe the relationship between ARID1A and DNA damage response (DDR) signaling. The authors don't see defects in a few DDR markers in ARID1A CKO cells (including a low-resolution assessment of ATR), suggesting that ARID1A may not be required for meiotic DDR signaling. However, as previously noted the data do not rule out the possibility that ARID1A is downstream of DDR signaling and the authors even indicate that "it is reasonable to hypothesize that DDR signaling might recruit BAF-A to the sex chromosomes (lines 509-510)." It therefore is difficult to understand why the authors continue to state that "...the mechanisms underlying ARID1A-mediated repression of the sex-linked transcription are mutually exclusive to DDR pathways regulating sex body formation" (p. 8) and that "BAF-A-mediated transcriptional repression of the sex chromosomes occurs independently of DDR signaling" (p. 16). The data provided do not justify these conclusions, as a role for DDR signaling upstream of ARID1A would mean that these mechanisms are not mutually exclusive or independent of one another.
Response: The reviewer’s argument is reasonable, and we have made the recommended changes (Results, lines 212-215; Discussion, lines 499-500).
Comment: A final comment relates to the impacts of ARID1A loss on DMC1 focus formation and the interesting observation of reduced sex chromosome association by DMC1. The authors additionally assess the related recombinase RAD51 and suggest that it is unaffected by ARID1A loss. However, only a single image of RAD51 staining in the cKO is provided (Fig. S11) and there are no associated quantitative data provided. The data are suggestive but it would be appropriate to add a qualifier to the conclusion regarding RAD51 in the discussion which states that "...loss of ARID1a decreases DMC1 foci on the XY chromosomes without affecting RAD51" given that the provided RAD51 data are not rigorous. In the long-term it also would be interesting to quantitatively examine DMC1 and RAD51 focus formation on autosomes as well.
Response: We agree with the reviewer’s comment and have made the recommended changes (Discussion, lines 518-519).
Response to non-public recommendations
Reviewer 2:
Comment: Meiotic arrest is usually judged based on testicular phenotypes. If mutant testes do not have any haploid spermatids, we can conclude that meiotic arrest is a phenotype. In this case, mutant testes have haploid spermatids and are fertile. The authors cannot conclude meiotic arrest. The mutant cells appear to undergo cell death in the pachytene stage, but the authors cannot say "meiotic arrest."
Response: We disagree with this comment. By IF, we see that ~70% of the spermatocytes have deleted ARID1A. Furthermore, we never observed diplotene spermatocytes that lacked ARID1A. The conclusion that the absence of ARID1A results in a pachynema arrest and that the escapers produce the haploid spermatids is firm.
Comment: Fig. S2 and S3 have wrong figure legends.
Response: The figure legends for Fig. S2 and S3 are correct.
Comment: The authors do not appear to evaluate independent mice for scoring (the result is about 74% deletion above, Table S1). Sup S2: how many independent mice did the authors examine?
Response:These were Sta-Put purified fractions obtained from 14-15 WT and mutant mice. It is difficult to isolate pachytene spermatocytes by Sta-Put at the required purity in sufficient yields using one mouse at a time. We used three technical replicates to quantify the band intensity, and the error bars represent the standard error of the mean (S.E.M) of the band intensity.
Comment: Comparison of cKO and wild-type littermate yielded nearly identical results (Avg total conc WT = 32.65 M/m; Avg total conc cKO = 32.06 M/ml)". This sounds like a negative result (i.e., no difference between WT and cKO).
Response: This is correct. There is no difference between Arid1aWT and Arid1aCKO sperm production. This is because wild-type haploid gametes produced were derived from spermatocyte precursors that have escaped Cre-mediated activity (Fig. S4). These data merely serve to highlight an inherent caveat of our conditional knockout model and are not intended to support the main conclusion that ARID1A is necessary for pachytene progression.
Comment: The authors now admit ~ 70 % efficiency in deletion, and the authors did not show the purity of these samples. If the purity of pachytene spermatocytes is ~ 80%, the real proportion of mutant cells can be ~ 56%. It is very difficult to interpret the data.
Response: The original submission did refer to inefficient Cre-induced recombination. The reviewer asked for the % efficiency, which was provided in the revised version. Also, please refer to Fig. S2, where Western blot analysis demonstrates a significant loss of ARID1A protein levels in CKO relative to WT pachytene spermatocyte populations that were used for CUT&RUN data generation.
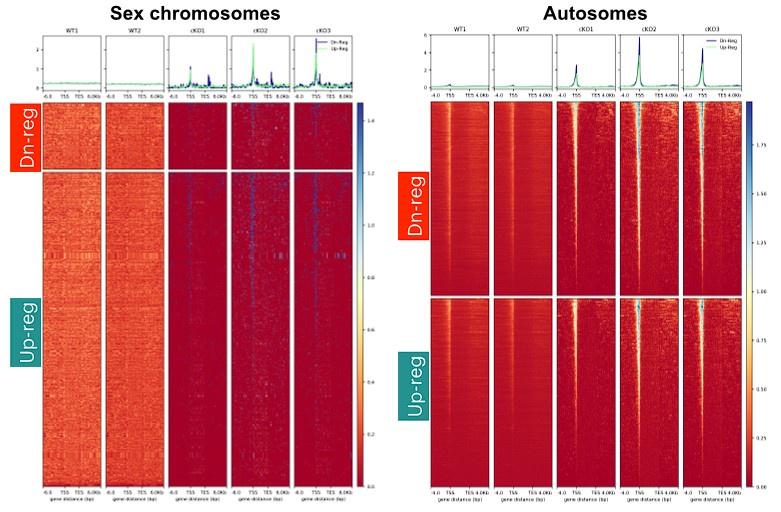
Comment: The authors should not use the other study to justify their own data. The H3.3 ChIP-seq data in the NAR paper detected clear peaks on autosomes. However, in this study, as shown in Fig. S7A, the authors detected only 4 peaks on autosomes based on MACS2 peak calling. This must be a failed experiment. Also, S7A appears to have labeling errors.
Response: I believe the reviewer is referring to supplementary figure 8A. Here, it is not clear which labeling errors the reviewer is referring to. In the wild type, the identified peaks were overwhelmingly sex-linked intergenic sites. This is consistent with the fact that H3.3 is hyper-accumulated on the sex chromosomes at pachynema.
The authors of the NAR paper did not perform a peak-calling analysis using MACS2 or any other peak-calling algorithm. They merely compared the coverage of H3.3 relative to input. Therefore, it is not clear on what basis the reviewer says that the NAR paper identified autosomal peaks. Their H3.3 signal appears widely distributed over a 6 kb window centered at the TSS of autosomal genes, which, compared to input, appears enriched. Our data clearly demonstrates a less noisy and narrower window of H3.3 enrichment at autosomal TSSs in WT pachytene spermatocytes, albeit at levels lower than that seen in CKO pachytene spermatocytes (Fig S8B and see data copied below for each individual replicate). Moreover, the lack of peaks does not mean that there was an absence of H3.3 at these autosomal TSSs (Supp. Fig. S8B). Therefore, we disagree with the reviewer’s comment that the H3.3 CUT&RUN was a failed experiment.
Author response image 1.
H3.3 Occupancy at genes mis-regulated in the absence of ARID1A
Comment: If the author wishes to study the function of ARID2 in spermatogenesis, they may need to try other cre-lines to have more robust phenotypes, and all analyses must be redone using a mouse model with efficient deletion of ARID2.
Response: As noted, we chose Stra8-Cre to conditionally knockout Arid1a because ARID1A is haploinsufficient during embryonic development. The lack of Cre expression in the maternal germline allows for transmission of the floxed allele, allowing for the experiments to progress.
Comment: The inefficient deletion of ARID1A in this mouse model does not allow any detailed analysis in a quantitative manner.
Response: In many experiments, we have been quantitative when possible by co-staining for ARID1A, ensuring that we can score mutant pachytene spermatocytes from escapers. Additionally, we provide data to show the efficiency of ARID1A loss in the purified pachytene populations sampled in our genomic assays.
Reviewer 3:
Comment: The Methods section refers to antibodies as being in Supplementary Table 3, but the table is labeled as Supplementary Table 2.
Response: This has been corrected
-
eLife assessment
This study presents a valuable dataset regarding chromatin remodeling by the BAF complex in the context of meiotic sex chromosome inactivation. Solid data generally support the conclusions, although there is room for improvement. This work will be of interest to researchers working on chromatin and reproductive biology.
-
Reviewer #1 (Public Review):
The work by Debashish U. Menon, Noel Murcia, and Terry Magnuson brings important knowledge about histone H3.3 dynamics involved in meiotic sex chromosome inactivation (MSCI). MSCI is unique to gametes and failure during this process can lead to infertility. Classically, MSCI has been studied in the context of DNA Damage repair pathways and little is known about the epigenetic mechanisms behind maintenance of the sex body as a silencing platform during meiosis. One of the major strengths of this work is the evidence provided on the role of ARID1A, a BAF subunit, in MSCI through the regulation of H3.3 occupancy in specific genic regions.
Using RNA seq and CUT&RUN and ATAC-seq, the authors show that ARID1A regulates chromatin accessibility of the sex chromosomes and XY gene expression. Loss of ARID1A increases …
Reviewer #1 (Public Review):
The work by Debashish U. Menon, Noel Murcia, and Terry Magnuson brings important knowledge about histone H3.3 dynamics involved in meiotic sex chromosome inactivation (MSCI). MSCI is unique to gametes and failure during this process can lead to infertility. Classically, MSCI has been studied in the context of DNA Damage repair pathways and little is known about the epigenetic mechanisms behind maintenance of the sex body as a silencing platform during meiosis. One of the major strengths of this work is the evidence provided on the role of ARID1A, a BAF subunit, in MSCI through the regulation of H3.3 occupancy in specific genic regions.
Using RNA seq and CUT&RUN and ATAC-seq, the authors show that ARID1A regulates chromatin accessibility of the sex chromosomes and XY gene expression. Loss of ARID1A increases promoter accessibility of XY linked genes with concomitant influx of RNA pol II to the sex body and up regulation of XY-linked genes. This work suggests that ARID1A regulates chromatin composition of the sex body since in the absence of ARID1A, spermatocytes show less enrichment of H3.3 in the sex chromosomes and stable levels of the canonical histones H3.1/3.2. By overlapping CUT&RUN and ATAC-seq data, authors show that changes in chromatin accessibility in the absence of ARID1A are given by redistribution of occupancy of H3.3. Gained open chromatin in mutants corresponds to up regulation of H3.3 occupancy at transcription start sites of genes mediated by ARID1A.
Interestingly, ARID1A loss caused increased promoter occupancy by H3.3 in regions usually occupied by PRDM9. PRDM9 catalyzes histone H3 lysine 4 trimethylation during meiotic prophase I, and positions double strand break (DSB) hotspots. Lack of ARID1A causes reduction in occupancy of DMC1, a recombinase involved in DSB repair, in non-homologous sex regions. These data suggest that ARID1A might indirectly influence DNA DSB repair on the sex chromosomes by regulating the localization of H3.3. This is very interesting given the recently suggested role for ARID1A in genome instability in cancer cells. It raises the question of whether this role is also involved in meiotic DSB repair in autosomes and/or how this mechanism differs in sex chromosomes compared to autosomes.
The fact that there are Arid1a transcripts that escape the Cre system in the Arid1a KO mouse model might difficult the interpretation of the data. The phenotype of the Arid1a knockout is probably masked by the fact that many of the sequencing techniques used here are done on a heterogeneous population of knockout and wild type spermatocytes. In relation to this, I think that the use of the term "pachytene arrest" might be overstated, since this is not the phenotype truly observed. Nonetheless, the authors provide evidence showing that the spermatids observed in cKO testes that progress in spermatogenesis are the ones expressing Arid1a. This work presents enough evidence to include the BAF complex as part of the MSCI process, which increases our knowledge on specific regulation of the sex chromatin during meiosis.
-
Reviewer #2 (Public Review):
The authors tried to characterize the function of the SWI/SNF remodeler family, BAF, in spermatogenesis. The authors focused on ARID1A, a BAF-specific putative DNA binding subunit, based on gene expression profiles.
The authors disagreed with my previous assessments. I disagree with their response.
-
Reviewer #3 (Public Review):
In this manuscript, Magnuson and colleagues investigate the meiotic functions of ARID1A, a putative DNA binding subunit of the SWI/SNF chromatin remodeler BAF. The authors develop a germ cell specific conditional knockout (cKO) mouse model using Stra8-cre and observe that ARID1A-deficient cells fail to progress beyond pachytene, although due to inefficiency of the Stra8-cre system the mice retain ARID1A-expressing cells that yield sperm and allow fertility. Because ARID1A was found to accumulate at the XY body late in Prophase I, the authors suspected a potential role in meiotic silencing and by RNAseq observe significant misexpression of sex-linked genes that typically are silenced at pachytene. They go on to show that ARID1A is required for exclusion of RNA PolII from the sex body and for limiting promoter …
Reviewer #3 (Public Review):
In this manuscript, Magnuson and colleagues investigate the meiotic functions of ARID1A, a putative DNA binding subunit of the SWI/SNF chromatin remodeler BAF. The authors develop a germ cell specific conditional knockout (cKO) mouse model using Stra8-cre and observe that ARID1A-deficient cells fail to progress beyond pachytene, although due to inefficiency of the Stra8-cre system the mice retain ARID1A-expressing cells that yield sperm and allow fertility. Because ARID1A was found to accumulate at the XY body late in Prophase I, the authors suspected a potential role in meiotic silencing and by RNAseq observe significant misexpression of sex-linked genes that typically are silenced at pachytene. They go on to show that ARID1A is required for exclusion of RNA PolII from the sex body and for limiting promoter accessibility at sex-linked genes, consistent with a meiotic sex chromosome inactivation (MSCI) defect in cKO mice. The authors proceed to investigate the impacts of ARID1A on H3.3 deposition genome-wide. H3.3 is known be regulated by ARID1A and is linked to silencing, and here the authors find that upon loss of ARID1A, overall H3.3 enrichment at the sex body as measured by IF failed to occur, but H3.3 was enriched specifically at transcriptional start sites of sex-linked genes that are normally regulated by ARID1A. The results suggest that ARID1A normally prevents H3.3 accumulation at target promoters on sex chromosomes and based on additional data, restricts H3.3 to intergenic sites. Finally, the authors present data implicating ARID1A and H3.3 occupancy in DSB repair, finding that ARID1A cKO leads to a reduction in focus formation by DMC1, a key repair protein. Overall the paper provides new insights into the process of MSCI from the perspective of chromatin composition and structure, and raises interesting new questions about the interplay between chromatin structure, meiotic silencing and DNA repair.
In general the data are convincing. The conditional KO mouse model has some inherent limitations due to incomplete recombination and the existence of 'escaper' cells that express ARID1A and progress through meiosis normally. This reviewer feels that the authors have addressed this point thoroughly and have demonstrated clear and specific phenotypes using the best available animal model. The data demonstrate that the mutant cells fail to progress past pachytene, although it is unclear whether this specifically reflects pachytene arrest, as accumulation in other stages of Prophase also is suggested by the data in Table 1.
The revised manuscript more appropriately describes the relationship between ARID1A and DNA damage response (DDR) signaling. The authors don't see defects in a few DDR markers in ARID1A CKO cells (including a low resolution assessment of ATR), suggesting that ARID1A may not be required for meiotic DDR signaling. However, as previously noted the data do not rule out the possibility that ARID1A is downstream of DDR signaling, and the authors note the possibility of a role for DDR signaling upstream of ARID1A.
A final comment relates to the impacts of ARID1A loss on DMC1 focus formation and the interesting observation of reduced sex chromosome association by DMC1. The authors additionally assess the related recombinase RAD51 and suggest that it is unaffected by ARID1A loss. However, only a single image of RAD51 staining in the cKO is provided (Fig. S11) and there are no associated quantitative data provided. The data are suggestive and conclusions about the impacts of ARID1A loss on RAD51 must be considered as preliminary until more rigorously assessed.
-
-
Author Response
The following is the authors’ response to the original reviews.
Reviewer 1
Comment 1: It is worth mentioning that the authors show that there are Arid1a transcripts that escape the Cre system. This might mask the phenotype of the Arid1a knockout, given that many sequencing techniques used here are done on a heterogeneous population of knockout and wild-type spermatocytes.
Response: The proportions of undifferentiated spermatogonia (PLZF+) with detectable (ARID1A+) and non-detectable (ARID1A=) levels of ARID1A protein by immunostaining on testes cryosections obtained from 1-month old Arid1afl/fl (control) and Arid1acKO (CKO) males were 74% ARID1A negative (CKO) and 26% ARID1A positive (CKO) as compared to 95% ARID1A positive and 5% ARID1A negative in WT controls. The manuscript includes these data (page 5, lines …
Author Response
The following is the authors’ response to the original reviews.
Reviewer 1
Comment 1: It is worth mentioning that the authors show that there are Arid1a transcripts that escape the Cre system. This might mask the phenotype of the Arid1a knockout, given that many sequencing techniques used here are done on a heterogeneous population of knockout and wild-type spermatocytes.
Response: The proportions of undifferentiated spermatogonia (PLZF+) with detectable (ARID1A+) and non-detectable (ARID1A=) levels of ARID1A protein by immunostaining on testes cryosections obtained from 1-month old Arid1afl/fl (control) and Arid1acKO (CKO) males were 74% ARID1A negative (CKO) and 26% ARID1A positive (CKO) as compared to 95% ARID1A positive and 5% ARID1A negative in WT controls. The manuscript includes these data (page 5, lines 114-116). Furthermore, Western blot analysis of STA-Put purified pachytene WT and mutant spermatocytes showed significantly reduced levels of ARID1A protein in mutant cells (95% reduction). The manuscript has added these data (page 5, line 116 and Fig. S2).
Comment 2: In relation to this, I think that the use of the term "pachytene arrest" might be overstated, since this is not the phenotype truly observed (these mice produce sperm).
Response: Based on the profiling of prophase-I spermatocytes by co-staining for SYCP3 and ARID1A, we observed a marked reduction in mid-late pachytene spermatocytes that lacked ARID1A, indicating a failure to progress beyond pachynema in the absence of ARID1A (Table 1 in manuscript). Furthermore, we were unable to detect diplotene spermatocytes lacking ARID1A protein. Haploid spermatid populations isolated from Arid1acKO males appeared normal, expressing the wild-type allele, suggesting that they originated from spermatocytes that failed to undergo efficient Cre recombination (Fig. S3). Arid1acKO also produces viable sperm at a level equal to their wild-type controls (see page 5, lines 123-126). It is reasonable to conclude that the absence of ARID1A results in a pachynema arrest and that the viable sperm are from escapers. We cannot make any conclusions regarding the requirement of ARID1A for progression beyond pachynema.
Comment 3: ARID1A is present throughout prophase I, and it might have pre-MSCI roles that impact earlier stages of Meiosis I, and cell death might be happening in these earlier stages too.
Response: We did not observe an effect on the frequency of leptotene and zygotene spermatocytes lacking ARID1A. There appeared to be an accumulation of these prophase-I populations in response to the loss of ARID1A, consistent with a failure in progression beyond pachynema in the mutants (Table 1 in the manuscript).
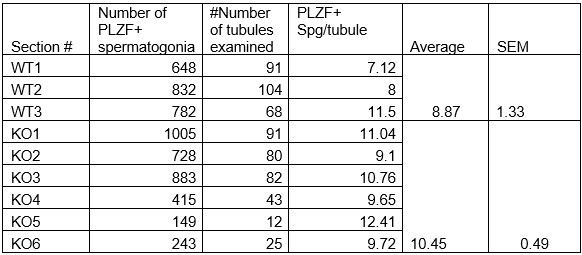
Additionally, we did not detect any significant difference in the numbers of undifferentiated spermatogonia expressing PLZF (also known as ZBTB16) in 1-month-old Arid1acKO relative to Arid1afl/fl males (see Table below, now included in the manuscript as supplemental Table 1). Therefore, the Arid1a conditional knockouts generated with a Stra8-Cre did not appear to impact earlier stages of spermatogenesis. However, potential roles of ARID1A early in spermatogenesis might be revealed using a more efficient and earlier-acting germline Cre transgene. In this case, an inducible Cre transgene would be needed, given the haploinsufficiency associated with Arid1a. Such haploinsufficiency was why we used the Stra8-Cre. The lack of Cre expression in the female germline allowed the transmission of the floxed allele maternally.
Author response table 1.
Comment 4: Overall, the research presented here is solid, adds new knowledge on how sex chromatin is silenced during meiosis, and has generated relevant databases for the field.
Response: We thank the reviewer for this comment.
Reviewer 2
Comment 1: The conditional deletion mouse model of ARIDA using Stra8-cre showed inefficient deletion; spermatogenesis did not appear to be severely compromised in the mutants. Using this data, the authors claimed that meiotic arrest occurs in the mutants. This is obviously a misinterpretation.
Response: As stated in response to Reviewer 1, testes cryosections obtained from 1-month-old control and mutant males showed that 74% are ARID1A negative (CKO) and 26% ARID1A positive (CKO) as compared to 95% ARID1A positive and 5% ARID1A negative in WT controls (page 5, lines 114-116). This difference is dramatic. Western blot analysis of STA-Put purified pachytene WT and mutant spermatocytes also showed a significant reduction of ARID1A protein in mutant cells (Fig. S2). We observed a marked decrease in mid-late pachytene spermatocytes that lacked ARID1A, indicating a failure to progress beyond pachynema without ARID1A (Table 1 from the manuscript). Furthermore, we were unable to detect any diplotene spermatocytes lacking ARID1A protein. These data suggest that the haploid spermatids originated from spermatocytes that failed to undergo efficient Cre recombination (Fig. S3). Comparison of cKO and wild-type littermate yielded nearly identical results (Avg total conc WT = 32.65 M/m; Avg total conc cKO = 32.06 M/ml), indicating that the cKO’s produce viable sperm at a level equal to their wild-type controls. Taken together, the conclusion that the absence of ARID1A results in a pachynema arrest and that the escapers produce the haploid spermatids is firm. By IF, we see that ~70% of the spermatocytes have deleted ARID1A. Therefore, we disagree with the reviewer’s comments that “spermatogenesis did not appear to be severely compromised in the mutants”.
Comment 2: In the later parts, the authors performed next-gen analyses, including ATAC-seq and H3.3 CUT&RUN, using the isolated cells from the mutant mice. However, with this inefficient deletion, most cells isolated from the mutant mice appeared not to undergo Cre-mediated recombination. Therefore, these experiments do not tell any conclusion pertinent to the Arid1a mutation.
Response: We agree that the ATAC-seq and CUT&RUN data were derived from a mixed population of pachytene spermatocytes consisting of mutants and, to a much lesser extent, escapers. As stated, based on our previous study (Menon et al., 2021, Nat. Commun., PMID: 34772938) and additional analyses in this current work, the undifferentiated spermatogonia lacking ARID1A indicates that Stra8-Cre is ~ 70% efficient. With this efficiency, we can detect striking changes in H3.3 occupancy and chromatin accessibility in the mutants relative to wild-type spermatocytes.
Comment 3: Furthermore, many of the later parts of this study focus on the analysis of H3.3 CUT&RUN. However, Fig. S7 clearly suggests that the H3.3 CUT&RUN experiment in the wild-type simply failed. Thus, none of the analyses using the H3.3 CUT&RUN data can be interpreted.
Response: We would like to draw the attention of the reviewer to a recent study (Fointane et al., 2022, NAR, PMID: 35766398) where the authors observed an identical X chromosome-wide spreading of H3.3 in mouse meiotic cells by ChIP-seq. The genomic distribution matches the microscopic observation of H3.3 coating of the sex chromosomes. Therefore, in normal spermatocytes, H3.3 distribution is pervasive across the X chromosome, with very few peaks observed in intergenic regions. Additionally, we detected H3.3 enrichment at TSSs of ARID1A-regulated autosomal genes in wild-type pachytene spermatocytes, albeit reduced relative to the mutants, indicating that the H3.3 CUT&RUN worked. For these reasons, we do not agree with the reviewer’s assessment that the H3.3 CUT&RUN experiment failed in the wild type.
Comment 4: If the author wishes to study the function of ARID2 in spermatogenesis, they may need to try other cre-lines to have more robust phenotypes, and all analyses must be redone using a mouse model with efficient deletion of ARID2.
Response: As noted, we chose Stra8-Cre to conditionally knockout Arid1a because ARID1A is haploinsufficient during embryonic development. The lack of Cre expression in the maternal germline allows for transmission of the floxed allele, allowing for the experiments to progress.
Reviewer 3
Comment 1: A challenge with the author's CKO model is the incomplete efficiency of ARID1A loss, due to incomplete CRE-mediated deletion. The authors effectively work around this issue, but they don't state specifically what percentage of CKO cells lack ARID1A staining. This information should be added.
Response: Our data indicate that Stra8-Cre is ~ 70% efficient. This information has been added.
Comment 2: They refer to cells that retain ARID1A staining in CKO testes as 'internal controls' but this reviewer finds that label inappropriate.
Response: We have dropped ‘internal controls’ and used ‘escapers’ instead.
Comment 3: Although some cells that retain ARID1A won't have undergone CRE-mediated excision, others may have excised but possibly have delayed kinetics of deletion or ARID1A RNA/protein turnover and loss. Such cells likely have partial ARID1A depletion to different extents and, therefore, in some cases, are no longer wild-type. In subsequent figures in which co-staining for ARID1A is done, it would be appropriate for the authors to specify if they are quantifying all cells from CKO testes, or only those that lack ARID1A staining.
Response: We were unable to detect any diplotene spermatocytes lacking ARID1A protein. The data suggest that the haploid spermatids originated from spermatocytes that failed to undergo efficient Cre recombination (Fig. S3). Thus, we conclude that the absence of ARID1A results in a pachynema arrest and that the escapers produce haploid spermatids. In figures displaying quantification data, we indicate whether the quantification was performed on spermatocytes lacking or containing ARID1A from cKO testes. By IF, we see that ~70% of the spermatocytes have deleted ARID1A.
Comment 4: The authors don't see defects in a few DDR markers in ARID1A CKO cells and conclude that the role of ARID1A in silencing is 'mutually exclusive to DDR pathways' (p 12) and 'occurs independently of DDR signaling' (p30). The data suggest that ARID1A may not be required for DDR signaling, but do not rule out the possibility that ARID1A is downstream of DDR signaling (and the authors even hypothesize this on p30). The data provided do not justify the conclusion that ARID1A acts independently of DDR signaling.
associated DDR factors such as: H2Ax; ATR; and MDC1. We observed an abnormal persistence of elongating RNA polymerase II on the mutant XY body in response to the loss of ARID1A, emphasizing its role in the transcriptional repression of the XY during pachynema. The loss of ARID1A results in a failure to silence sex-linked genes and does so in the presence of DDR signaling factors in the XY body. As the reviewer notes, we highlighted the possibility that DDR pathways might influence ARID1A recruitment to the XY, evidenced by the hyperaccumulation of ARID1A on the sex body late in diplonema. Therefore, whether ARID1A is dependent on DDR signaling remains an open question.
Comment 5: After observing no changes in levels or localization of H3.3 chaperones, the authors conclude that 'ARID1A impacts H3.3 accumulation on the sex chromosomes without affecting its expression or incorporation during pachynema.' It's not clear to this reviewer what the authors mean by this. Aside from the issue of not having tested DAXX or HIRA activity, are they suggesting that some other process besides altered incorporation leads to H3.3 accumulation, and if so, what process would that be?
Response: The loss of ARID1A might result in an abnormal redistribution of DAXX or HIRA on the XY, potentially contributing to the defects in H3.3 accumulation and canonical H3.1/3.2 eviction on the XY. While speculative at this point, it is also possible that the persistence of elongating RNAPII in response to the loss of ARID1A might prevent the sex chromosome-wide coating of H3.3. Addressing the mechanism underlying ARID1A-governed H3.3 accumulation on the XY body remains a topic for future investigation.
Comment 6: The authors find an interesting connection between certain regions that gained chromatin accessibility after ARID1A loss (clusters G1 and G3) and the presence of the PRDM9 sequence motif. The G1 and G3 clusters also show DMC1 occupancy and H3K4me3 enrichment. However, an additional cluster with gained accessibility (G4) also shows DMC1 occupancy and H3K4me3 enrichment but has modest H3.3 accumulation. The paper would benefit for additional discussion about the G4 cluster (which encompasses 960 peak calls). Is there any enrichment of PRDM9 sites in G4? If H3.3 exclusion governs meiotic DSBs, how does cluster G4 fit into the model?
Response: We agree that, compared to G1+G3, cluster G4 shows an insignificant increase in H3.3 occupancy in the absence of ARID1A (Figure 6B). The plot profile associated with the heatmap confirms this result (Figure 6B). Therefore, cluster G4 is very distinct in its chromatin composition from G1+G3 upon the loss of ARID1A and, as such, is not inconsistent with our model of H3.3 antagonism with DSB sites. Additionally, we did not observe an enrichment of PRDM9 sites in G4. Since G4 does not display similar dynamics in H3.3 occupancy to G1+G3, DMC1 association might not be perturbed at G4 in response to the loss of ARID1A. Future studies will be required to determine the genomic associations of DMC1 and H3K4me3 in response to the loss of ARID1A.
Comment 7: The impacts of ARID1A loss on DMC1 focus formation (reduced sex chromosome association) are very interesting and also raise additional questions. Are DMC1 foci on autosomes also affected during pachynema? The corresponding lack of apparent effect on RAD51 implies that breaks are still made and resected, enabling RAD51 filament formation. A more thorough quantitative assessment of RAD51 focus formation will be interesting in the long run, enabling determination of the number of break sites and the kinetics of repair, which the authors suggest is perturbed by ARID1A loss but doesn't directly test. It isn't clear how a nucleosomal factor (H3.3) would influence loading of recombinases onto ssDNA, especially if the alteration is not at the level of resection and ssDNA formation. Additional discussion of this point is warranted. Lastly, there currently are various notions for the interplay between RAD51 and DMC1 in filament formation and break repair, and brief discussion of this area and the implications of the new findings from the ARID1A CKO would strengthen the paper further.
Response: The impact of H3.3 on the loading of recombinases might be an indirect consequence of ARID1A-governed sex-linked transcriptional repression. In a recent study, Alexander et al. (Nat. Commun, 2023, PMID: 36990976) showed that transcriptional activity and meiotic recombination are spatially compartmentalized during meiosis. Therefore, the persistence of elongating RNA polymerase II on a sex body depleted for H3.3 in the absence of ARID1A might contribute to the defect in DMC1 association. RAD51 and DMC1 are known to bind ssDNA at PRDM9/SPO11 designated DSB hotspots. However, these recombinases occupy unique domains. DMC1 localizes nearest the DSB breakpoint, promoting strand exchange, whereas RAD51 is further away (Hinch et al., PMID32610038). We show that loss of Arid1a decreases DMC1 foci on the XY chromosomes without affecting RAD51. These findings indicate that BAF-A plays a role in the loading and/or retention of DMC1 to the XY chromosomes. This information has been added to the discussion.
-
eLife assessment
This solid study presents a useful dataset regarding chromatin remodeling by the BAF complex in the context of meiotic sex chromosome inactivation. Using knockouts of the BAF complex subunit ARID1A, there appears to be pachynema arrest and a failure to repress sex-linked genes, which is supported by an increase in chromatin accessibility, as assessed by ATAC-seq.
-
Reviewer #1 (Public Review):
The work by Debashish U. Menon, Noel Murcia, and Terry Magnuson brings important knowledge about histone H3.3 dynamics involved in meiotic sex chromosome inactivation (MSCI). MSCI is unique to gametes and failure during this process can lead to infertility. Classically, MSCI has been studied in the context of DNA Damage repair pathways and little is known about the epigenetic mechanisms behind maintenance of the sex body as a silencing platform during meiosis. One of the major strengths of this work is the evidence provided on the role of ARID1A, a BAF subunit, in MSCI through the regulation of H3.3 occupancy in specific genic regions.
Using RNA seq and CUT&RUN and ATAC-seq, the authors show that ARID1A regulates chromatin accessibility of the sex chromosomes and XY gene expression. Loss of ARID1A increases …
Reviewer #1 (Public Review):
The work by Debashish U. Menon, Noel Murcia, and Terry Magnuson brings important knowledge about histone H3.3 dynamics involved in meiotic sex chromosome inactivation (MSCI). MSCI is unique to gametes and failure during this process can lead to infertility. Classically, MSCI has been studied in the context of DNA Damage repair pathways and little is known about the epigenetic mechanisms behind maintenance of the sex body as a silencing platform during meiosis. One of the major strengths of this work is the evidence provided on the role of ARID1A, a BAF subunit, in MSCI through the regulation of H3.3 occupancy in specific genic regions.
Using RNA seq and CUT&RUN and ATAC-seq, the authors show that ARID1A regulates chromatin accessibility of the sex chromosomes and XY gene expression. Loss of ARID1A increases promoter accessibility of XY linked genes with concomitant influx of RNA pol II to the sex body and up regulation of XY-linked genes. This work suggests that ARID1A regulates chromatin composition of the sex body since in the absence of ARID1A, spermatocytes show less enrichment of H3.3 in the sex chromosomes and stable levels of the canonical histones H3.1/3.2. By overlapping CUT&RUN and ATAC-seq data, authors show that changes in chromatin accessibility in the absence of ARID1A are given by redistribution of occupancy of H3.3. Gained open chromatin in mutants corresponds to up regulation of H3.3 occupancy at transcription start sites of genes mediated by ARID1A.
Interestingly, ARID1A loss caused increased promoter occupancy by H3.3 in regions usually occupied by PRDM9. PRDM9 catalyzes histone H3 lysine 4 trimethylation during meiotic prophase I, and positions double strand break (DSB) hotspots. Lack of ARID1A causes reduction in occupancy of DMC1, a recombinase involved in DSB repair, in non-homologous sex regions. These data suggest that ARID1A might indirectly influence DNA DSB repair on the sex chromosomes by regulating the localization of H3.3. This is very interesting given the recently suggested role for ARID1A in genome instability in cancer cells. It raises the question of whether this role is also involved in meiotic DSB repair in autosomes and/or how this mechanism differs in sex chromosomes compared to autosomes.
The fact that there are Arid1a transcripts that escape the Cre system in the Arid1a KO mouse model might difficult the interpretation of the data. The phenotype of the Arid1a knockout is probably masked by the fact that many of the sequencing techniques used here are done on a heterogeneous population of knockout and wild type spermatocytes. In relation to this, I think that the use of the term "pachytene arrest" might be overstated, since this is not the phenotype truly observed. Knockout mice produce sperm, and probably litters, although a full description of the subfertility phenotype is lacking, along with identification of the stage at which cell death is happening by detection of apoptosis.
It is clear from this work that ARID1a is part of the protein network that contribute to silencing of the sex chromosomes. However, it is challenging to understand the timing of the role of ARID1a in the context of the well-known DDR pathways that have been described for MSCI. Staining of chromosome spreads with Arid1a antibody showed localization at the sex chromosomes by diplonema, however, analysis of gene expression in Arid1a ko was performed on pachytene spermatocytes. Therefore, is not very clear how the chromatin remodeling activity of Arid1a in diplonema is affecting gene expression of a previous stage. CUTnRUN showed that ARID1a is present at the sex chromatin in earlier stages, leading to hypothesize that immunofluorescence with ARID1a antibody might not reflect ARID1a real localization. -
Reviewer #2 (Public Review):
The authors tried to characterize the function of the SWI/SNF remodeler family, BAF, in spermatogenesis. The authors focused on ARID1A, a BAF-specific putative DNA binding subunit, based on gene expression profiles. The study has several serious issues with the data and interpretation. The conditional deletion mouse model of ARIDA using Stra8-cre showed inefficient deletion; spermatogenesis did not appear to be severely compromised in the mutants. Using this data, the authors claimed that meiotic arrest occurs in the mutants. This is obviously a misinterpretation. In the later parts, the authors performed next-gen analyses, including ATAC-seq and H3.3 CUT&RUN, using the isolated cells from the mutant mice. However, with this inefficient deletion, most cells isolated from the mutant mice appeared not to …
Reviewer #2 (Public Review):
The authors tried to characterize the function of the SWI/SNF remodeler family, BAF, in spermatogenesis. The authors focused on ARID1A, a BAF-specific putative DNA binding subunit, based on gene expression profiles. The study has several serious issues with the data and interpretation. The conditional deletion mouse model of ARIDA using Stra8-cre showed inefficient deletion; spermatogenesis did not appear to be severely compromised in the mutants. Using this data, the authors claimed that meiotic arrest occurs in the mutants. This is obviously a misinterpretation. In the later parts, the authors performed next-gen analyses, including ATAC-seq and H3.3 CUT&RUN, using the isolated cells from the mutant mice. However, with this inefficient deletion, most cells isolated from the mutant mice appeared not to undergo Cre-mediated recombination. Therefore, these experiments do not tell any conclusion pertinent to the Arid1a mutation. Furthermore, many of the later parts of this study focus on the analysis of H3.3 CUT&RUN. However, Fig. S7 clearly suggests that the H3.3 CUT&RUN experiment in the wild-type simply failed. Thus, none of the analyses using the H3.3 CUT&RUN data can be interpreted. Overall, I found that the study does not have rigorous data, and the study is not interpretable. If the author wishes to study the function of ARID2 in spermatogenesis, they may need to try other cre-lines to have more robust phenotypes, and all analyses must be redone using a mouse model with efficient deletion of ARID2.
In this revised manuscript, the authors did not make any efforts to address my major criticisms, and I do not see any improvement. I only found the responses to 4 points, but I do not see any response to other major and minor comments. I understand the challenge (~70 deletion efficiency in the mutants) in this study. However, the inefficient deletion of ARID1A in this mouse model does not allow any detailed analysis in a quantitative manner.
-
Reviewer #3 (Public Review):
In this manuscript, Magnuson and colleagues investigate the meiotic functions of ARID1A, a putative DNA binding subunit of the SWI/SNF chromatin remodeler BAF. The authors develop a germ cell specific conditional knockout (cKO) mouse model using Stra8-cre and observe that ARID1A-deficient cells fail to progress beyond pachytene, although due to inefficiency of the Stra8-cre system the mice retain ARID1A-expressing cells that yield sperm and allow fertility. Because ARID1A was found to accumulate at the XY body late in Prophase I, the authors suspected a potential role in meiotic silencing and by RNAseq observe significant misexpression of sex-linked genes that typically are silenced at pachytene. They go on to show that ARID1A is required for exclusion of RNA PolII from the sex body and for limiting promoter …
Reviewer #3 (Public Review):
In this manuscript, Magnuson and colleagues investigate the meiotic functions of ARID1A, a putative DNA binding subunit of the SWI/SNF chromatin remodeler BAF. The authors develop a germ cell specific conditional knockout (cKO) mouse model using Stra8-cre and observe that ARID1A-deficient cells fail to progress beyond pachytene, although due to inefficiency of the Stra8-cre system the mice retain ARID1A-expressing cells that yield sperm and allow fertility. Because ARID1A was found to accumulate at the XY body late in Prophase I, the authors suspected a potential role in meiotic silencing and by RNAseq observe significant misexpression of sex-linked genes that typically are silenced at pachytene. They go on to show that ARID1A is required for exclusion of RNA PolII from the sex body and for limiting promoter accessibility at sex-linked genes, consistent with a meiotic sex chromosome inactivation (MSCI) defect in cKO mice. The authors proceed to investigate the impacts of ARID1A on H3.3 deposition genome-wide. H3.3 is known be regulated by ARID1A and is linked to silencing, and here the authors find that upon loss of ARID1A, overall H3.3 enrichment at the sex body as measured by IF failed to occur, but H3.3 was enriched specifically at transcriptional start sites of sex-linked genes that are normally regulated by ARID1A. The results suggest that ARID1A normally prevents H3.3 accumulation at target promoters on sex chromosomes and based on additional data, restricts H3.3 to intergenic sites. Finally, the authors present data implicating ARID1A and H3.3 occupancy in DSB repair, finding that ARID1A cKO leads to a reduction in focus formation by DMC1, a key repair protein. Overall the paper provides new insights into the process of MSCI from the perspective of chromatin composition and structure, and raises interesting new questions about the interplay between chromatin structure, meiotic silencing and DNA repair.
In general the data are convincing. The conditional KO mouse model has some inherent limitations due to incomplete recombination and the existence of 'escaper' cells that express ARID1A and progress through meiosis normally. This reviewer feels that the authors have addressed this point thoroughly and have demonstrated clear and specific phenotypes using the best available animal model. The data demonstrate that the mutant cells fail to progress past pachytene, although it is unclear whether this specifically reflects pachytene arrest, as accumulation in other stages of Prophase also is suggested by the data in Table 1. The western blot showing ARID1A expression in WT vs. cKO spermatocytes (Fig. S2) is supportive of the cKO model but raises some questions. The blot shows many bands that are at lower intensity in the cKO, at MWs from 100-250kDa. The text and accompanying figure legend have limited information. Are the various bands with reduced expression different isoforms of ARID1A, or something else? What is the loading control 'NCL'? How was quantification done given the variation in signal across a large range of MWs?
An additional weakness relates to how the authors describe the relationship between ARID1A and DNA damage response (DDR) signaling. The authors don't see defects in a few DDR markers in ARID1A CKO cells (including a low resolution assessment of ATR), suggesting that ARID1A may not be required for meiotic DDR signaling. However, as previously noted the data do not rule out the possibility that ARID1A is downstream of DDR signaling and the authors even indicate that "it is reasonable to hypothesize that DDR signaling might recruit BAF-A to the sex chromosomes." It therefore is difficult to understand why the authors continue to state that "...the mechanisms underlying ARID1A-mediated repression of the sex-linked transcription are mutually exclusive to DDR pathways regulating sex body formation" (p. 8) and that "BAF-A-mediated transcriptional repression of the sex chromosomes occurs independently of DDR signaling" (p. 16). The data provided do not justify these conclusions, as a role for DDR signaling upstream of ARID1A would mean that these mechanisms are not mutually exclusive or independent of one another.
A final comment relates to the impacts of ARID1A loss on DMC1 focus formation and the interesting observation of reduced sex chromosome association by DMC1. The authors additionally assess the related recombinase RAD51 and suggest that it is unaffected by ARID1A loss. However, only a single image of RAD51 staining in the cKO is provided (Fig. S11) and there are no associated quantitative data provided. The data are suggestive but it would be appropriate to add a qualifier to the conclusion regarding RAD51 in the discussion which states that "...loss of ARID1a decreases DMC1 foci on the XY chromosomes without affecting RAD51" given that the provided RAD51 data are not rigorous. In the long-term it also would be interesting to quantitatively examine DMC1 and RAD51 focus formation on autosomes as well.
-
-
eLife assessment
This paper presents a useful study regarding the meiotic functions of ARID1A, the DNA-binding component of the SWI/SNF chromatin remodeler BAF. Whereas this work suggests that ARID1A regulates chromatin composition of the sex body relative to the autosomes, the reviewers raised substantial issues with the data and interpretation. The efficiency of the conditional deletion allele seems low and the CUT&RUN experiments are inadequate.
-
Reviewer #1 (Public Review):
The work by Debashish U. Menon, Noel Murcia, and Terry Magnuson brings important knowledge about histone H3.3 dynamics involved in meiotic sex chromosome inactivation (MSCI). MSCI is unique to gametes and failure during this process can lead to infertility. Classically, MSCI has been studied in the context of DNA Damage repair pathways and little is known about the epigenetic mechanisms behind maintenance of the sex body as a silencing platform during meiosis. One of the major strengths of this work is the evidence provided on the role of ARID1A, a BAF subunit, in MSCI through the regulation of H3.3 occupancy in specific genic regions. This is well supported by a combination of immunofluorescence, RNA seq, CUT&RUN and ATAC-seq.
The mouse model in this study is a conditional Stra8 Cre mouse. Loss of ARID1A in …Reviewer #1 (Public Review):
The work by Debashish U. Menon, Noel Murcia, and Terry Magnuson brings important knowledge about histone H3.3 dynamics involved in meiotic sex chromosome inactivation (MSCI). MSCI is unique to gametes and failure during this process can lead to infertility. Classically, MSCI has been studied in the context of DNA Damage repair pathways and little is known about the epigenetic mechanisms behind maintenance of the sex body as a silencing platform during meiosis. One of the major strengths of this work is the evidence provided on the role of ARID1A, a BAF subunit, in MSCI through the regulation of H3.3 occupancy in specific genic regions. This is well supported by a combination of immunofluorescence, RNA seq, CUT&RUN and ATAC-seq.
The mouse model in this study is a conditional Stra8 Cre mouse. Loss of ARID1A in this mouse, caused up regulation of XY linked genes in prophase I spermatocytes and ingression of RNA pol II to the sex body, indicating a role for this chromatin remodeller in MSCI. Using RNA seq and CUT&RUN and ATAC-seq, the authors show that ARID1A regulates chromatin accessibility of the sex chromosomes. ARID1A interacts with gene transcription start sites of sex-linked genes, and loss of ARID1A increased promoter accessibility of XY linked genes with concomitant gene up regulation.This work suggests that ARID1A regulates chromatin composition of the sex body relative to the autosomes. In the absence of ARID1A, spermatocytes show less enrichment of H3.3 in the sex chromosomes and stable levels of the canonical histones H3.1/3.2. By overlapping CUT&RUN and ATAC-seq data, authors show that changes in chromatin accessibility in the absence of ARID1A are given by redistribution of occupancy of H3.3. Gained open chromatin in mutants corresponds to up regulation of H3.3 occupancy at transcription start sites of genes regulated by ARID1A.
Interestingly, ARID1A loss caused increased promoter occupancy by H3.3 in regions usually occupied by PRDM9. PRDM9 is a protein with histone methyltransferase activity that catalyzes histone H3 lysine 4 trimethylation during meiotic prophase I, and positions double strand break (DSB) hotspots. Lack of ARID1A causes reduction in occupancy of DMC1, a recombinase involved in DSB repair, in non-homologous sex regions. These data suggest that ARID1A might indirectly influence DNA DSB repair on the sex chromosomes by regulating the localization of H3.3. This is very interesting given the suggested role for ARID1A in genome instability in cancer cells (Nacarelli et al 2020: 10.1080/23723556.2019.1690923, Zhang et al. 2023: 10.1093/carcin/bgad011 and others). It raises the question of whether this role is also involved in meiotic DSB repair in autosomes and/or how this mechanism differs in sex chromosomes compared to autosomes.
It is worth mentioning that authors show that there are Arid1a transcripts that escape the Cre system. This might mask the phenotype of the Arid1a knockout, given that many of the sequencing techniques used here are done on a heterogeneous population of knockout and wild type spermatocytes. In relation to this, I think that the use of the term "pachytene arrest" might be overstated, since this is not the phenotype truly observed (these mice produce sperm). ARID1A is present throughout prophase I and it might have pre-MSCI roles that impact earlier stages of Meiosis I and cell death might be happening in these earlier stages too.
Overall the research presented here is solid, adds new knowledge on how the sex chromatin is silenced during meiosis and has generated relevant databases for the field.
-
Reviewer #2 (Public Review):
The authors tried to characterize the function of the SWI/SNF remodeler family, BAF, in spermatogenesis. The authors focused on ARID1A, a BAF-specific putative DNA binding subunit, based on gene expression profiles. The study has several serious issues with the data and interpretation. The conditional deletion mouse model of ARIDA using Stra8-cre showed inefficient deletion; spermatogenesis did not appear to be severely compromised in the mutants. Using this data, the authors claimed that meiotic arrest occurs in the mutants. This is obviously a misinterpretation. In the later parts, the authors performed next-gen analyses, including ATAC-seq and H3.3 CUT&RUN, using the isolated cells from the mutant mice. However, with this inefficient deletion, most cells isolated from the mutant mice appeared not to …
Reviewer #2 (Public Review):
The authors tried to characterize the function of the SWI/SNF remodeler family, BAF, in spermatogenesis. The authors focused on ARID1A, a BAF-specific putative DNA binding subunit, based on gene expression profiles. The study has several serious issues with the data and interpretation. The conditional deletion mouse model of ARIDA using Stra8-cre showed inefficient deletion; spermatogenesis did not appear to be severely compromised in the mutants. Using this data, the authors claimed that meiotic arrest occurs in the mutants. This is obviously a misinterpretation. In the later parts, the authors performed next-gen analyses, including ATAC-seq and H3.3 CUT&RUN, using the isolated cells from the mutant mice. However, with this inefficient deletion, most cells isolated from the mutant mice appeared not to undergo Cre-mediated recombination. Therefore, these experiments do not tell any conclusion pertinent to the Arid1a mutation. Furthermore, many of the later parts of this study focus on the analysis of H3.3 CUT&RUN. However, Fig. S7 clearly suggests that the H3.3 CUT&RUN experiment in the wild-type simply failed. Thus, none of the analyses using the H3.3 CUT&RUN data can be interpreted. Overall, I found that the study does not have rigorous data, and the study is not interpretable. If the author wishes to study the function of ARID2 in spermatogenesis, they may need to try other cre-lines to have more robust phenotypes, and all analyses must be redone using a mouse model with efficient deletion of ARID2.
-
Reviewer #3 (Public Review):
In this manuscript, Magnuson and colleagues investigate the meiotic functions of ARID1A, a putative DNA binding subunit of the SWI/SNF chromatin remodeler BAF. The authors develop a germ cell specific knockout mouse model using Stra8-cre and observe that ARID1A-deficient cells undergo pachytene arrest, although due to inefficiency of the Stra8-cre system the mice retain ARID1A-expressing cells that yield sperm and allow fertility. Because ARID1A was found to accumulate at the XY body late in Prophase I, the authors suspected a potential role in meiotic silencing and by RNAseq observe significant misexpression of sex-linked genes that typically are silenced at pachytene. They go on to show that ARID1A is required for exclusion of RNA PolII from the sex body, consistent with a meiotic sex chromosome …
Reviewer #3 (Public Review):
In this manuscript, Magnuson and colleagues investigate the meiotic functions of ARID1A, a putative DNA binding subunit of the SWI/SNF chromatin remodeler BAF. The authors develop a germ cell specific knockout mouse model using Stra8-cre and observe that ARID1A-deficient cells undergo pachytene arrest, although due to inefficiency of the Stra8-cre system the mice retain ARID1A-expressing cells that yield sperm and allow fertility. Because ARID1A was found to accumulate at the XY body late in Prophase I, the authors suspected a potential role in meiotic silencing and by RNAseq observe significant misexpression of sex-linked genes that typically are silenced at pachytene. They go on to show that ARID1A is required for exclusion of RNA PolII from the sex body, consistent with a meiotic sex chromosome inactivation (MSCI) defect. The authors proceed to investigate the impacts of ARID1A on chromatin accessibility and H3.3 deposition genome-wide. H3.3 is known be regulated by ARID1A and is linked to silencing, and here the authors find that upon loss of ARID1A, overall H3.3 enrichment at the sex body as measured by IF failed to occur, but H3.3 was enriched specifically at transcriptional start sites of sex-linked genes that are normally regulated by ARID1A. The results suggest that ARID1A normally prevents H3.3 accumulation at target promoters on sex chromosomes and based on additional data, restricts H3.3 to intergenic sites. Finally, the authors present data implicating ARID1A and H3.3 occupancy in DSB repair, finding that ARID1A KO leads to a reduction in focus formation by DMC1, a key repair protein. Overall the paper covers a lot of ground, provides important new insights into the process of MSCI from the perspective of chromatin composition and structure, and raises many interesting questions. In general the paper is well written and the data are clear. Specific points to address are as follows:
1. A challenge with the author's CKO model is the incomplete efficiency of ARID1A loss, due to incomplete CRE-mediated deletion. The authors effectively work around this issue, but they don't state specifically what percentage of CKO cells lack ARID1A staining. This information should be added. They refer to cells that retain ARID1A staining in CKO testes as 'internal controls' but this reviewer finds that label inappropriate. Although some cells that retain ARID1A won't have undergone CRE-mediated excision, others may have excised but possibly have delayed kinetics of deletion or ARID1A RNA/protein turnover and loss. Such cells likely have partial ARID1A depletion to different extents and therefore in some cases are no longer wild-type. In subsequent figures in which co-staining for ARID1A is done, it would be appropriate for the authors to specify if they are quantifying all cells from CKO testes, or only those that lack ARID1A staining.
2. The authors don't see defects in a few DDR markers in ARID1A CKO cells and conclude that the role of ARID1A in silencing is 'mutually exclusive to DDR pathways' (p 12) and 'occurs independently of DDR signaling' (p30). The data suggest that ARID1A may not be required for DDR signaling, but do not rule out the possibility that ARID1A is downstream of DDR signaling (and the authors even hypothesize this on p30). The data provided do not justify the conclusion that ARID1A acts independently of DDR signaling.
3. After observing no changes in levels or localization of H3.3 chaperones, the authors conclude that 'ARID1A impacts H3.3 accumulation on the sex chromosomes without affecting its expression or incorporation during pachynema.' It's not clear to this reviewer what the authors mean by this. Aside from the issue of not having tested DAXX or HIRA activity, are they suggesting that some other process besides altered incorporation leads to H3.3 accumulation and if so what process would that be?
4. The authors find an interesting connection between certain regions that gained chromatin accessibility after ARID1A loss (clusters G1 and G3) and presence of the PRDM9 sequence motif. The G1 and G3 clusters also show DMC1 occupancy and H3K4me3 enrichment. However, an additional cluster with gained accessibility (G4) also shows DMC1 occupancy and H3K4me3 enrichment but unlike clusters G1 and G3 has modest H3.3 accumulation. The paper would benefit for additional discussion about the G4 cluster (which encompasses 960 peak calls). Is there any enrichment of PRDM9 sites in G4? If H3.3 exclusion governs meiotic DSBs, how does cluster G4 fit into the model?
5. The impacts of ARID1A loss on DMC1 focus formation (reduced sex chromosome association) are very interesting and also raise additional questions. Are DMC1 foci on autosomes also affected during pachynema? The corresponding lack of apparent effect on RAD51 implies that breaks are still made and resected, enabling RAD51 filament formation. A more thorough quantitative assessment of RAD51 focus formation will be interesting in the long run, enabling determination of the number of break sites and the kinetics of repair, which the authors suggest is perturbed by ARID1A loss but don't directly test. It isn't clear how a nucleosomal factor (H3.3) would influence loading of recombinases onto ssDNA, especially if the alteration is not at the level of resection and ssDNA formation. Additional discussion of this point is warranted. Lastly, there currently are various notions for the interplay between RAD51 and DMC1 in filament formation and break repair, and brief discussion of this area and the implications of the new findings from the ARID1A CKO would strengthen the paper further.
-
