Vangl2 suppresses NF-κB signaling and ameliorates sepsis by targeting p65 for NDP52-mediated autophagic degradation
Curation statements for this article:-
Curated by eLife

eLife assessment
This valuable manuscript describes a novel role of Vangl2, a core planar cell polarity protein, in linking the NF-kB pathway to selective autophagic protein degradation in myeloid cells. The mechanistic studies provide convincing evidence that Vangl2 targets p65 for NDP52-mediated autophagic degradation, limiting inflammatory NF-kB response, with functional significance of the proposed mechanism in sepsis. Additional future studies dissecting autophagic Vangl2 functions in various myeloid subsets in the context of inflammation could be informative, and additional Vangl2 targets in the inflammatory pathway, including IKK2, could also be explored. Overall, this exciting study can advance our understanding of NF-kB control, particularly in the context of inflammatory diseases.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Van Gogh-like 2 (Vangl2), a core planar cell polarity component, plays an important role in polarized cellular and tissue morphology induction, growth development, and cancer. However, its role in regulating inflammatory responses remains elusive. Here, we report that Vangl2 is upregulated in patients with sepsis and identify Vangl2 as a negative regulator of The nuclear factor-kappaB (NF-κB) signaling by regulating the protein stability and activation of the core transcription component p65. Mice with myeloid-specific deletion of Vangl2 ( Vangl2 ΔM ) are hypersusceptible to lipopolysaccharide (LPS)-induced septic shock. Vangl2-deficient myeloid cells exhibit enhanced phosphorylation and expression of p65, therefore, promoting the secretion of proinflammatory cytokines after LPS stimulation. Mechanistically, NF-κB signaling-induced-Vangl2 recruits E3 ubiquitin ligase PDLIM2 to catalyze K63-linked ubiquitination on p65, which serves as a recognition signal for cargo receptor NDP52-mediated selective autophagic degradation. Taken together, these findings demonstrate Vangl2 as a suppressor of NF-κB-mediated inflammation and provide insights into the crosstalk between autophagy and inflammatory diseases.
Article activity feed
-

-
-

eLife assessment
This valuable manuscript describes a novel role of Vangl2, a core planar cell polarity protein, in linking the NF-kB pathway to selective autophagic protein degradation in myeloid cells. The mechanistic studies provide convincing evidence that Vangl2 targets p65 for NDP52-mediated autophagic degradation, limiting inflammatory NF-kB response, with functional significance of the proposed mechanism in sepsis. Additional future studies dissecting autophagic Vangl2 functions in various myeloid subsets in the context of inflammation could be informative, and additional Vangl2 targets in the inflammatory pathway, including IKK2, could also be explored. Overall, this exciting study can advance our understanding of NF-kB control, particularly in the context of inflammatory diseases.
-
Reviewer #1 (Public Review):
The study shows a new mechanism of NFkB-p65 regulation mediated by Vangl2-dependent autophagic targeting. Autophagic regulation of p65 has been reported earlier; this study brings an additional set of molecular players involved in this important regulatory event, which may have implications for chronic and acute inflammatory conditions.
-
Reviewer #2 (Public Review):
Vangl2, a core planar cell polarity protein involved in Wnt/PCP signaling, cell proliferation, differentiation, homeostasis, and cell migration. Vangl2 malfunctioning has been linked to various human ailments, including autoimmune and neoplastic disorders. Interestingly, it was shown that Vangl2 interacts with the autophagy regulator p62, and autophagic degradation limits the activity of inflammatory mediators, such as p65/NF-κB. However, the possible role of Vangl2 in inflammation has not been investigated. In this manuscript, Lu et al. describe that Vangl2 expression is upregulated in human sepsis-associated PBMCs and that Vangl2 mitigates experimental sepsis in mice by negatively regulating p65/NF-κB signaling in myeloid cells. Their mechanistic studies further revealed that Vangl2 recruits the E3 …
Reviewer #2 (Public Review):
Vangl2, a core planar cell polarity protein involved in Wnt/PCP signaling, cell proliferation, differentiation, homeostasis, and cell migration. Vangl2 malfunctioning has been linked to various human ailments, including autoimmune and neoplastic disorders. Interestingly, it was shown that Vangl2 interacts with the autophagy regulator p62, and autophagic degradation limits the activity of inflammatory mediators, such as p65/NF-κB. However, the possible role of Vangl2 in inflammation has not been investigated. In this manuscript, Lu et al. describe that Vangl2 expression is upregulated in human sepsis-associated PBMCs and that Vangl2 mitigates experimental sepsis in mice by negatively regulating p65/NF-κB signaling in myeloid cells. Their mechanistic studies further revealed that Vangl2 recruits the E3 ubiquitin ligase PDLIM2 to promote K63-linked poly-ubiquitination of p65. Vangl2 also facilitated the recognition of ubiquitinated p65 by the cargo receptor NDP52. These molecular processes caused selective autophagic degradation of p65. Indeed, abrogation of PDLIM2 or NDP52 functions rescued p65 from autophagic degradation, leading to extended p65/NF-κB activity in myeloid cells. Overall, the manuscript presents convincing evidence for novel Vangl2-mediated control of inflammatory p65/NF-kB activity. The proposed pathway may expand interventional opportunities restraining aberrant p65/NF-kB activity in human ailments.
IKK is known to mediate p65 phosphorylation, which instructs NF-kB transcriptional activity. In this manuscript, Vangl2 deficiency led to an increased accumulation of phosphorylated p65 and IKK also at 30 minutes post-LPS stimulation; however, autophagic degradation of p-p65 may not have been initiated at this early time point. Therefore, this set of data put forward the exciting possibility that Vangl2 could also be regulating the immediate early phase of inflammatory response involving the IKK-p65 axis - a proposition that may be tested in future studies.
-
Reviewer #3 (Public Review):
Lu et al. describe Vangl2 as a negative regulator of inflammation in myeloid cells. The primary mechanism appears to be through binding p65 and promoting its degradation, albeit in an unusual autolysosome/autophagy dependent manner. Overall, these findings are novel, valuable and the crosstalk of PCP pathway protein Vangl2 with NF-kappaB is of interest.
Comments on latest version:
Lu et al. now address all my comments. All data included for the reviewers should be included in the main manuscript or Supplement and should be available to the readers. Please ensure that this criteria is met. I have no further comments.
-
Author response:
The following is the authors’ response to the previous reviews.
Responses to Reviewer #1:
Reviewer #1: The study shows a new mechanism of NFkB-p65 regulation mediated by Vangl2-dependent autophagic targeting. Autophagic regulation of p65 has been reported earlier; this study brings an additional set of molecular players involved in this important regulatory event, which may have implications for chronic and acute inflammatory conditions.
Comments on the revised version:
The authors have addressed the earlier concerns and I am satisfied with the revised version. I have no additional comments to make.
We appreciate the reviewer’s comments on our revised manuscript.
Responses to Reviewer #2:
Reviewer #2: Vangl2, a core planar cell polarity protein involved in Wnt/PCP signaling, cell proliferation, differentiation, …
Author response:
The following is the authors’ response to the previous reviews.
Responses to Reviewer #1:
Reviewer #1: The study shows a new mechanism of NFkB-p65 regulation mediated by Vangl2-dependent autophagic targeting. Autophagic regulation of p65 has been reported earlier; this study brings an additional set of molecular players involved in this important regulatory event, which may have implications for chronic and acute inflammatory conditions.
Comments on the revised version:
The authors have addressed the earlier concerns and I am satisfied with the revised version. I have no additional comments to make.
We appreciate the reviewer’s comments on our revised manuscript.
Responses to Reviewer #2:
Reviewer #2: Vangl2, a core planar cell polarity protein involved in Wnt/PCP signaling, cell proliferation, differentiation, homeostasis, and cell migration. Vangl2 malfunctioning has been linked to various human ailments, including autoimmune and neoplastic disorders. Interestingly, it was shown that Vangl2 interacts with the autophagy regulator p62, and autophagic degradation limits the activity of inflammatory mediators, such as p65/NF-κB. However, the possible role of Vangl2 in inflammation has not been investigated. In this manuscript, Lu et al. describe that Vangl2 expression is upregulated in human sepsis-associated PBMCs and that Vangl2 mitigates experimental sepsis in mice by negatively regulating p65/NF-κB signaling in myeloid cells. Their mechanistic studies further revealed that Vangl2 recruits the E3 ubiquitin ligase PDLIM2 to promote K63-linked poly-ubiquitination of p65. Vangl2 also facilitated the recognition of ubiquitinated p65 by the cargo receptor NDP52. These molecular processes caused selective autophagic degradation of p65. Indeed, abrogation of PDLIM2 or NDP52 functions rescued p65 from autophagic degradation, leading to extended p65/NF-κB activity in myeloid cells. Overall, the manuscript presents convincing evidence for novel Vangl2-mediated control of inflammatory p65/NF-kB activity. The proposed pathway may expand interventional opportunities restraining aberrant p65/NF-kB activity in human ailments.
IKK is known to mediate p65 phosphorylation, which instructs NF-kB transcriptional activity. In this manuscript, Vangl2 deficiency led to an increased accumulation of phosphorylated p65 and IKK also at 30 minutes post-LPS stimulation; however, autophagic degradation of p-p65 may not have been initiated at this early time point. Therefore, this set of data put forward the exciting possibility that Vangl2 could also be regulating the immediate early phase of inflammatory response involving the IKK-p65 axis - a proposition that may be tested in future studies.
We appreciate the reviewer’s comments on our manuscript, and we have added the discussion about IKK-p65 axis in revised version. (Page 15, lines 467-474)
Responses to Reviewer #3:
Reviewer #3: Lu et al. describe Vangl2 as a negative regulator of inflammation in myeloid cells. The primary mechanism appears to be through binding p65 and promoting its degradation, albeit in an unusual autolysosome/autophagy dependent manner. Overall, these findings are novel, valuable and the crosstalk of PCP pathway protein Vangl2 with NF-kappaB is of interest. While generally solid, some concerns still remain about the rigor and conclusions drawn.
Comments on the revised version:
(1) Lu et al. address my comments through responses and new experimental data. However, some of the explanations provided are inadequate.
However, in response to my enquiry regarding directly exploring PCP effects, the authors simply assert "Our study revealed that Vangl2 recruits the E3 ubiquitin ligase PDLIM2 to facilitate K63-linked ubiquitination of p65, which is subsequently recognized by autophagy receptor NDP52 and then promotes the autophagic degradation of p65. Our findings by using autophagy inhibitors and autophagic-deficient cells indicate that Vangl2 regulates NFkB signaling through a selective autophagic pathway, rather than affecting the PCP pathway, WNT, HH/GLI, Fat-Dachsous or even mechanical tension."
I do not agree that the use of autophagy inhibitors and autophagy-deficient cells can rule out the contributions of PCP or any other pathways. Only experimentally inhibiting the pathway(s) with adequate demonstration of target inhibition/abolition of well-known effector function and documenting unaltered p65 regulation under these conditions can be considered proof. Autophagy inhibitors and autophagy-deficient cells only prove that this particular pathway is necessary. Nonetheless, I do not want to dwell on proving a negative and agree that Vangl2 is a novel regulator of p65 through its role in promoting p65 degradation. The inclusion of a statement discussing the limitations of their approach would have sufficed. The response from the authors could have been better.
We thank the reviewer for helping us improve the quality of the manuscript. We provided new data and revised the Discussion as suggested.
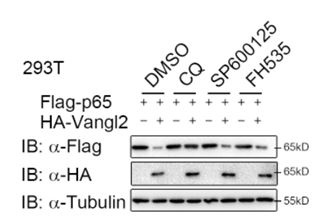
To ascertain whether Vangl2 degrades p65 through a selective autophagic pathway or the PCP pathway, 293T cells were transfected with p65, together with or without the Vangl2 plasmids, and treated with different pharmacological inhibitors. We found the degradation of p65 induced by Vangl2 was blocked by autolysosome inhibitor (CQ), but not by the JNK inhibitor (SP600125) or Wnt/β-catenin inhibitor (FH535) (New Figure. 1). These data suggest that Vangl2 primarily degrades p65 through a selective autophagic pathway, rather than through the JNK or Wnt signaling pathway. Nevertheless, additional pathway inhibitions, such as those of the HH/GLI and Fat-Dachsous pathways, should also be employed to further elucidate the function of Vangl2 in p65 degradation. As suggested, we have added a statement about the limitation of the approach in the discussion (Page 12, lines 378-385).
Author response image 1.
Vangl2 degrades p65 through a selective autophagic pathway, but not by the PCP pathway. HEK293T cells were transfected with Flag-p65 and HA-Vangl2 plasmids, and treated with DMSO, CQ (50 mM) for 6 h, SP600125 (20 mM) for 1 h or FH535 (30 mM) for 6 h. The cell lysates were analyzed by immunoblot.
(2) I am also not satisfied with the explanation that "immune cells represent a minor fraction of the lungs and liver". There are lots of resident immune cells in the lungs and liver (alveolar macrophages in the lung and Kuppfer cells in the liver). For example, it may be so that Vangl2 is important in monocytes and not in the resident population. This might be a potential explanation. But this is not explored. The restricted tissue-specificity of the interaction between two ubiquitously present proteins is still a challenge to understand. The response from the authors is not satisfactory. There is plenty of Vangl2 in the liver in their western blot.
We thank the reviewer for this question. We added this explanation in the Discussion. (Page 13, lines 398-404)
(3) I had also simply pointed out PMID: 34214490 with reference to the findings described in the manuscript. There were no suggestions of contradiction. In fact, I would refer to the publication in discussion to support the findings and stress the novelty. The response from the authors could have been better.
Thank you for the reviewer's insightful comments. We have modified this discussion as suggested. (Page 13, lines 410-415; Page 14, lines 419-421)
(4) The response to my enquiry regarding homo- or heterozygosity is unsupported by any reference or data.
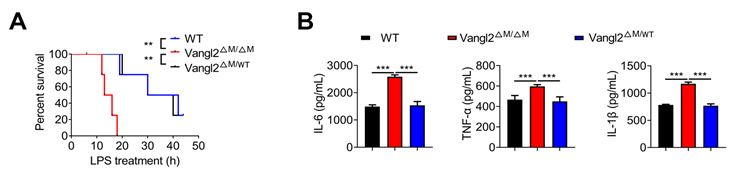
As suggested, we provided the data that only Vangl2 deficient homozygous showed inhibition of the activation of NF-kB in New Figure. 2.
Author response image 2.
Vangl2 deficiency promotes NF-kB activation. (A) The survival rates of WT, Vangl2ΔM/ΔM and Vangl2ΔM/WT mice treated with high-dosage of LPS (30 mg/kg, i.p.) (n≥4). (B) IL-6 and TNF-a secretion by WT and Vangl2-deficient BMDMs treated with LPS for 6 h was measured by ELISA. IL-1β secretion by WT, Vangl2ΔM/ΔM and Vangl2ΔM/WT BMDMs treated with LPS for 6 h and ATP for 30 min was measured by ELISA.
(5) The listing of 8 patients and healthy controls are also appreciated. The body temperature of #6 doesn't fall in the <36 or >38 degree C SIRS criteria. The inclusion of CRP, PCT, heart rate and respiratory rate, and other lab values would have further improved the inclusion criteria. Moreover, it is difficult to understand why there are 16 value points for healthy and sepsis cohorts in Fig 1 when there are 8 patients.
We thank the reviewer for this valuable suggestion. We are sorry for our mistake that we entered data from two repeated experiments in Figure. 1 A and we have revised this data in the updated version (Figure. 1 A, Pages 12 Lines 146). As suggested, we have added CRP, WBC and heart rate in sepsis patients’ information. (Supplementary Materials and Methods)
Recommendations for the authors:
Reviewer #2 (Recommendations For The Authors):
The proposition that Vangl2 may target additional mediators of inflammation could be indicated in the text.
We thank the reviewer for this valuable suggestion. We had added discussion in modified version. (Page 15, lines 467-474)
Reviewer #3 (Recommendations For The Authors):
It is advised that some of the deficiencies pointed out by Reviewer #3 are textually addressed. Additionally, there could be some inconsistency in the number of healthy controls and patients (see Fig S1A and FIg 1A and Supplementary table, also see comments from Reviewer #3) - this should be carefully scrutinised and revised, if necessary.
We thank the reviewer for this valuable suggestion. We are sorry for our mistake that we entered data from two repeated experiments in Figure. 1 A and we have revised this data in the updated version (Figure. 1 A, Pages 12 Lines 146).
-
-
Author response:
The following is the authors’ response to the original reviews.
Public Reviews:
Reviewer #1 (Public Review):
In the manuscript titled "Vangl2 suppresses NF-κB signaling and ameliorates sepsis by targeting p65 for NDP52-mediated autophagic degradation" by Lu et al, the authors show that Vangl2, a planner cell polarity component, plays a direct role in autophagic degradation of NFkB-p65 by facilitating its ubiquitination via PDLIM2 and subsequent recognition and autophagic targeting via the autophagy adaptor protein NDP52. Conceptually it is a wonderful study with excellent execution of experiments and controls. The concerns with the manuscript are mainly on two counts - First issue is the kinetics of p65 regulation reported here, which does not fit into the kinetics of the mechanism proposed here, i.e., …
Author response:
The following is the authors’ response to the original reviews.
Public Reviews:
Reviewer #1 (Public Review):
In the manuscript titled "Vangl2 suppresses NF-κB signaling and ameliorates sepsis by targeting p65 for NDP52-mediated autophagic degradation" by Lu et al, the authors show that Vangl2, a planner cell polarity component, plays a direct role in autophagic degradation of NFkB-p65 by facilitating its ubiquitination via PDLIM2 and subsequent recognition and autophagic targeting via the autophagy adaptor protein NDP52. Conceptually it is a wonderful study with excellent execution of experiments and controls. The concerns with the manuscript are mainly on two counts - First issue is the kinetics of p65 regulation reported here, which does not fit into the kinetics of the mechanism proposed here, i.e., Vangl2-mediated ubiquitination followed by autophagic degradation of p65. The second issue is more technical- an absolute lack of quantitative analyses. The authors rely mostly on visual qualitative interpretation to assess an increase or decrease in associations between partner molecules throughout the study. While the overall mechanism is interesting, the authors should address these concerns as highlighted below:
Major points:
(1) Kinetics of p65 regulation by Vangl2: As mentioned above, authors report that LPS stimulation leads to higher IKK and p65 activation in the absence of Vangl2. The mechanism of action authors subsequently work out is that- Vangl2 helps recruit E3 ligase PDLIM to p65, which causes K63 ubiquitination, which is recognised by NDP52 for autophagic targeting. Curiously, peak p65 activation is achieved within 30 minutes of LPS stimulation. The time scale of all other assays is way longer. It is not clear that in WT cells, p65 could be targeted to autophagic degradation in Vangl2 dependent manner within 30 minutes. The HA-Myc-Flag-based overexpression and Co-IP studies do confirm the interactions as proposed. However, they do not prove that this mechanism was responsible for the Vangl2-mediated modulation of p65 activation upon LPS stimulation. Moreover, the Vangl2 KO line also shows increased IKK activation. The authors do not show the cause behind increased IKK activation, which in itself can trigger increased p65 phosphorylation.
We thank the reviewer for this valuable suggestion.
Indeed, we agreed with the reviewer that peak p65 activation is achieved within 30 minutes of LPS stimulation in vitro, and p65 could not be targeted to autophagic degradation in a Vangl2 dependent manner within 30 minutes. Given that the protein and mRNA levels of Vangl2 were elevated at 3-6 h of LPS stimulation (Fig. S1 C-E), we extended the stimulation time scale in the revised manuscript. The data (Fig. 2A-D in the revised manuscript) demonstrated that IKK phosphorylation was enhanced in Vangl2 KO myeloid cells during the early phase (within 3 h) of LPS stimulation, but not for the prolonged period of LPS stimulation. The underlying mechanism may be complex. Only p65 phosphorylation was continuously enhanced after long-term LPS stimulation in Vangl2 KO cells, compared to WT cells. Furthermore, the overexpression of Vangl2 in A549 cells also demonstrated a reduction of phosphorylation and total endogenous p65 (Fig. 2 I, J in the revised manuscript). These findings were corroborated by overexpression and Co-IP experiments, which collectively indicated that Vangl2 regulates the stability of p65 by promoting its interaction with NDP52 and autophagic degradation. (Page 7; Line 183-185).
(2) The other major concern is regarding the lack of quantitative assessments. For Co-IP experiments, I can understand it is qualitative observation. However, when the authors infer that there is an increase or decrease in the association through co-IP immunoblots, it should also be quantified, especially since the differences are quite marginal and could be easily misinterpreted.
We are grateful to the reviewer for this suggestion. The quantitative analysis has been updated in the revised version.
(3) Figure 4E and F: It is evident that inhibiting Autolysosome (CQ or BafA1) or autophagy (3MA) led to the recovery of p65 levels and inducing autophagy by Rapamycin led to faster decay in p65 levels. Did the authors also note/explore the possibility that Vangl2 itself may be degraded via the autophagy pathway? IB of WCL upon CQ/BAF/3MA or upon Rapa treatment does indicate the same. If true, how would that impact the dynamics of p65 activation?
We thank the reviewer for this question. Previous studies have shown that Vangl2 is primarily degraded by the proteasome pathway, rather than by the autolysosomal pathway (doi: 10.1126/sciadv.abg2099; doi: 10.1038/s41598-019-39642-z). In our experiments, Vangl2 recruits E3 ligase PDLIM2 to enhance K63-linked ubiquitination on p65, which serves as a recognition signal for cargo receptor NDP52-mediated selective autophagic degradation. Vangl2 facilitated the interaction between p65 and NDP52, yet itself did not undergo significant autophagic degradation.
(4) Autophagic targeting of p65 should also be shown through alternate evidence, like microscopy etc., in the LPS-stimulated WT cells.
We thank the reviewer for this suggestion. We have added the data (co-localization of p65 and LC3 was detected by immunofluorescence) in the revised version (Fig. S4 H in the revised manuscript). (Page 9, lines 267-268)
Reviewer #2 (Public Review):
Vangl2, a core planar cell polarity protein involved in Wnt/PCP signaling, mediates cell proliferation, differentiation, homeostasis, and cell migration. Vangl2 malfunctioning has been linked to various human ailments, including autoimmune and neoplastic disorders. Interestingly, Vangl2 was shown to interact with the autophagy regulator p62, and indeed, autophagic degradation limits the activity of inflammatory mediators such as p65/NF-κB. However, if Vangl2, per se, contributes to restraining aberrant p65/NF-kB activity remains unclear.
In this manuscript, Lu et al. describe that Vangl2 expression is upregulated in human sepsis-associated PBMCs and that Vangl2 mitigates experimental sepsis in mice by negatively regulating p65/NF-κB signaling in myeloid cells. Vangl2 recruits the E3 ubiquitin ligase PDLIM2 to promote K63-linked poly-ubiquitination of p65. Vangl2 also facilitates the recognition of ubiquitinated p65 by the cargo receptor NDP52. These molecular processes cause selective autophagic degradation of p65. Indeed, abrogation of PDLIM2 or NDP52 functions rescued p65 from autophagic degradation, leading to extended p65/NF-κB activity.
As such, the manuscript presents a substantial body of interesting work and a novel mechanism of NF-κB control. If found true, the proposed mechanism may expand therapeutic opportunities for inflammatory diseases. However, the current draft has significant weaknesses that need to be addressed.
We appreciate the reviewer’s comments on our manuscript, and we have further improved the manuscript as suggested.
Specific comments
(1) Vangl2 deficiency did not cause a discernible increase in the cellular level of total endogenous p65 (Fig 2A and Fig 2B) but accumulated also phosphorylated IKK.
Even Fig 4D reveals that Vangl2 exerts a rather modest effect on the total p65 level and the figure does not provide any standard error for the quantified data. Therefore, these results do not fully support the proposed model (Figure 7) - this is a significant draw back. Instead, these data provoke an alternate hypothesis that Vangl2 could be specifically mediating autophagic removal of phosphorylated IKK and phosphorylated IKK, leading to exacerbated inflammatory NF-κB response in Vangl2-deficient cells. One may need to use phosphorylation-defective mutants of p65, at least in the over-expression experiments, to dissect between these possibilities.
We appreciate the reviewer’s comments on our manuscript, and we have further improved the manuscript as suggested.
(1) Indeed, we agreed with the reviewer that Vangl2 deficiency did not cause a discernible increase in the cellular level of total p65 after a short time of LPS stimulation in vitro, and p65 could not be targeted to autophagic degradation in a Vangl2 dependent manner within 30 minutes. Given that the protein and mRNA levels of Vangl2 were elevated at 3-6 h of LPS stimulation (Fig. S1 C-E), we extended the stimulation time scale in the revised manuscript. The data (Fig. 2A-D in the revised manuscript) demonstrated that IKK phosphorylation was enhanced in Vangl2 KO myeloid cells during the early phase (within 3 h) of LPS stimulation, but not for the prolonged period of LPS stimulation. The underlying mechanism may be complex. Only phosphorylation of p65 and total endogenous p65 was continuously enhanced after long-term LPS stimulation in Vangl2 KO cells, compared to WT cells. Furthermore, the overexpression of Vangl2 in A549 cells also demonstrated a reduction of phosphorylation and total endogenous p65 (Fig. 2 I, J in the revised manuscript). These findings were corroborated by overexpression and Co-IP experiments, which collectively indicated that Vangl2 regulates the stability of p65 by promoting its interaction with NDP52 and autophagic degradation. (Page 7; Line 183-185).
(2) Similarly, the stimulation time scale in Fig 4D was extended, and it was demonstrated that p65 was more stable in Vangl2-deficient cells.
- Moreover, we constructed phosphorylation-defective mutants of p65 (S536A), and found that Vangl2 could also promote the degradation of the p65 phosphorylation mutants (Fig. S4 A, B in the revised manuscript). Thus, Vangl2 promote the degradation of the basal/unphosphorylated p65. (Page 8, lines 237-240)
(2) Fig 1A: The data indicates the presence of two subgroups within the sepsis cohort - one with high Vangl2 expressions and the other with relatively normal Vangl2 expression. Was there any difference with respect to NF-κB target inflammatory gene expressions between these subgroups?
As suggested, we conducted an analysis of NF-kB target inflammatory gene expressions between the high and relatively low Vangl2 expression groups in sepsis patients. The results showed that the serum of the high Vangl2 expression group exhibited lower levels of IL-6, WBC, and CRP than the low Vangl2 expression group, which suggested an inverse correlation between Vangl2 and the inflammatory response (Fig. S1 A in the revised manuscript) (Page 5, lines 126-128).
(3) The effect of Vangl2 deficiency was rather modest in the neutrophil. Could it be that Vangl2 mediates its effect mostly in macrophages?
As showed in Fig. S1C-E, the induction of Vangl2 by LPS stimulation is more rapid in macrophages than in neutrophils. This may contribute to its dominant effect in macrophages. Consequently, we primarily focused our investigation on the role of Vangl2 in macrophages.
(4) Fig 1D and Figure 1E: Data for unstimulated Vangl2 cells should be provided. Also, the source of the IL-1β primary antibody has not been mentioned.
Thank you for the suggestion. We have updated the data for unstimulated cells in the revised manuscript (Fig. 1 D, E in the revised manuscript). Also, IL-1β primary antibody was purchased from Cell Signaling Technology and the information has been included in the Materials and Methods section (Table S1).
(5) The relevance and the requirement of RNA-seq analysis are not clear in the present draft. Figure 1E already reveals upregulation of the signature NF-κB target inflammatory genes upon Vangl2 deficiency.
We agreed with the reviewer that the data presented in Figure 1E demonstrated the upregulation of the signature NF-kB target inflammatory genes upon Vangl2 deficiency in a murine model of LPS induced sepsis. Subsequently, we proceeded to investigate the mechanism by which Vangl2 regulates NF-kB target inflammatory genes at the cellular level in Figure 2. To this end, we performed RNA-seq analysis to screen signal pathways involved in LPS-induced septic shock by comparing LPS-stimulated BMDMs from Vangl2ΔM and WT mice, and identified that TNF signaling pathway and cytokine-cytokine receptor interaction were found to be significantly enriched in Vangl2ΔM BMDMs upon LPS stimulation. This analysis provides further evidence that Vangl2 plays a role in regulating NF-kB signaling pathways and the release of related inflammatory cytokines.
(6) Fig 2A reveals an increased accumulation of phosphorylated p65 and IKK in Vangl2-deficient macrophages upon LPS stimulation within 30 minutes. However, Vangl2 accumulates at around 60 minutes post-stimulation in WT cells. Similar results were obtained for neutrophils (Fig 2B). There appears to be a temporal disconnect between Vangl2 and phosphorylated p65 accumulation - this must be clarified.
This concern has been addressed above (see response to questions 1 from reviewer #2).
(7) Figure 2E and 2F do not have untreated controls. Presentations in Fig 2E may be improved to more clearly depict IL6 and TNF data, preferably with separate Y-axes.
Thank you for the suggestion. We have added untreated controls and separated Y-axes for IL-6 and TNF data in the revised manuscript (Fig. 2 E, F in the revised manuscript).
(8) Line 219: "strongly with IKKα, p65 and MyD88, and weak" - should be revised.
We have improved the manuscript as suggested in the revised manuscript (Page 7; Line 203).
(9) It is not clear why IKKβ was excluded from interaction studies in Fig S3G.
We added the Co-IP experiment and showed that HA-tagged Vangl2 only interacted with Flag-tagged p65, but not with Flag-tagged IKKb in 293T cells (Fig S3H). Furthermore, endogenous co-IP immunoblot analyses showed that Vangl2 did not associate with IKKb (Fig. S3I)
(10) Fig 3F- In the text, authors mentioned that Vangl2 strongly associates with p65 upon LPS stimulation in BMDM. However, no controls, including input or another p65-interacting protein, were used.
As reviewer suggested, we have added input and positive control (IkBa) in this experiment (Fig. 3F in the revised manuscript). The results demonstrated that the interaction between p65 and IkBa was attenuated, although the total IkBa did not undergo significant degradation over long-term course of LPS stimulation.
(11) Figure 4D - Authors claim that Vangl2-deficient BMDMs stabilized the expression of endogenous p65 after LPS treatment. However, p65 levels were particularly constitutively elevated in knockout cells, and LPS signaling did not cause any further upregulation. This again indicates the role of Vangl2 in the basal state. The authors need to explain this and revise the test accordingly.
Thank you for the reviewer's comments. We repeated the experiment to ascertain whether Vangl2 could stabilize the expression of endogenous p65 before and after LPS treatment. It was found that, due to the extremely low expression of Vangl2 in WT cells in the absence of stimulation, there was no observable difference on the basal level of p65 between WT and Vangl2DM cells. However, upon prolonged LPS stimulation, Vangl2 expression was induced, resulting in p65 degradation in WT cells. In contrast, p65 protein was more stable in Vangl2 deficient cells after LPS stimulation (Fig. 4D in the revised manuscript).
Reviewer #3 (Public Review):
Lu et al. describe Vangl2 as a negative regulator of inflammation in myeloid cells. The primary mechanism appears to be through binding p65 and promoting its degradation, albeit in an unusual autolysosome/autophagy dependent manner. Overall, the findings are novel and the crosstalk of PCP pathway protein Vangl2 with NF-kappaB is of interest. …….Regardless, Vangl2 as a negative regulator of NF-kappaB is an important finding. There are, however, some concerns about methodology and statistics that need to be addressed.
Thank you for your comments on our manuscript, and we have further improved the manuscript as suggested.
(1) Whether PCP is anyway relevant or if this is a PCP-independent function of Vangl2 is not directly explored (the later appears more likely from the manuscript/discussion). PCP pathways intersect often with developmentally important pathways such as WNT, HH/GLI, Fat-Dachsous and even mechanical tension. It might be of importance to investigate whether Vangl2-dependent NF-kappaB is influenced by developmental pathways.
Thank you for the reviewer's insightful comments. Our study revealed that Vangl2 recruits the E3 ubiquitin ligase PDLIM2 to facilitate K63-linked ubiquitination of p65, which is subsequently recognized by autophagy receptor NDP52 and then promotes the autophagic degradation of p65. Our findings by using autophagy inhibitors and autophagic-deficient cells indicate that Vangl2 regulates NF-kB signaling through a selective autophagic pathway, rather than affecting the PCP pathway, WNT, HH/GLI, Fat-Dachsous or even mechanical tension. Moreover, a discussion section has been added to the revised version. (Page 12, lines 377-393)
(2) Are Vangl2 phosphorylations (S5, S82 and S84) in anyway necessary for the observed effects on NF-kappaB or would a phospho-mutant (alanine substitution mutant) Vangl2 phenocopy WT Vangl2 for regulation of NF-kappaB?
As suggested, we generated phospho-mutants of Vangl2 (S82/84A) and observed that Vangl2 (S82/84A) could still facilitate the degradation of p65 (Fig. S4 B in the revised manuscript), suggesting that Vangl2 regulates the NF-kB pathway independently of its phosphorylation.
(3) Another area to strengthen might be with regards to specificity of cell types where this phenomenon may be observed. LPS treatment in mice resulted in Vangl2 upregulation in spleen and lymph nodes, but not in lung and liver. What explains the specificity of organ/cell-type Vangl2 upregulation and its consequences observed here? Why is NF-kappaB signaling not more broadly or even ubiquitously affected in all cell types in a Vangl2-dependent manner, rather than being restricted to macrophages, neutrophils and peritoneal macrophages, or, for that matter, in spleen and LN and not liver and lung? After all, one may think that the PCP proteins, as well as NF-kappaB, are ubiquitous.
Thank you for the reviewer's comments.
(1) LPS is an important mediator to trigger sepsis with excessive immune activation. As is well known, the spleen and lymph nodes are important peripheral immune organs, where immune cells (e.g., macrophages) are abundant and respond sensitively to LPS stimulation. Nevertheless, immune cells represent a minor fraction of the lungs and liver. Consequently, Vangl2 represents a pivotal regulator of immune function, exhibiting a more pronounced increase in the immune organs and cells.
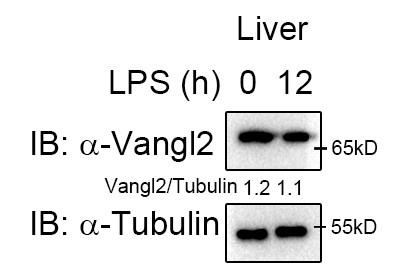
- Induction of Vangl2 expression by LPS stimulation is cell specific. Given that different cells exhibit varying protein abundances, the molecular events involved may also differ. Moreover, we observed high Vangl2 expression in the liver at the basal state (Author response image 1), whereas it was not induced after 12 h of LPS stimulation. Therefore, the functional role of Vangl2 exhibits significant phenotype in macrophages and neutrophils/spleen and LN, rather than in liver or lung cells.
Author response image 1.
Vangl2 showed no significant changes in the liver after LPS treatment. Mice (n≥3) were treated with LPS (30 mg/kg, i.p.). Livers were collected at 12 h after LPS treatment. Immunoblot analysis of Vangl2.
Recommendations For The Authors:
Reviewer #1 (Recommendations For The Authors):
General points:
Figure 4G- panels appear mislabeled. Pl correct.
We have corrected this mislabeling as you suggested.
The dynamics of Vangl2 interaction with p65 and autophagy adaptors is not clear/apparent. For example, Vangl2 expression destabilises p65 levels (as in Fig. 4), but in Fig. 5, it seems there is no decline in the p65 protein level, and a large fraction of it coprecipitates with NDP52.
We appreciate the reviewer’s comments. In the co-IP assay, we used the lysosomal inhibitor CQ to inhibit p65 degradation to observe the interaction between p65 and NDP52 or Vangl2.
Fig 5E- I would expect p65 levels to be lower in WT cells than Vangl2 KO cells. But as such, there is no difference between the two.
We appreciate the reviewer’s comments. We repeated the experiments and updated the data. Firstly, Vangl2 was not induced in WT cells in the absence of LPS stimulation, thus there was no difference in p65 expression between the two groups at the basal level. Secondly, we used CQ/Baf-A1 to inhibit the degradation of Vangl2 in the co-IP assay to observe the interaction between p65 and other molecule.
Reviewer #2 (Recommendations For The Authors):
A few points that can be looked at and revised.
(1) Quantification of the presented data is needed for Fig 4D and Fig 4E.
We added the quantification analysis as suggested.
(2) The labeling of Fig 4G should be scrutinized.
We have corrected this mislabeling as you suggested.
(3) Fig 6B and Fig 6C should be explained in the result section more elaborately.
We thank the reviewer for the suggestion, and we have rephrased this sentence to better describe the results. (Page 10, lines 306-313)
(4) Line 85: "Vangl2 mediated downstream of Toll-like or interleukin (IL)-1" - unclear.
We appreciate the reviewer’s comments on our manuscript, and we have further improved the manuscript as suggested in the revised manuscript. (Page 3, lines 68)
(5) Line 181: "mice. Differentially expression analysis" - this should be revised.
We appreciate the reviewer’s comments on our manuscript, and we have further improved the manuscript as suggested in the revised manuscript. (Page 11, lines 323)
(6) Line 261-264- CHX-chase assay showed the degradation rate of p65 in Vangl2-deficient BMDM was slower compared with WT cells. However, Vangl2 is not induced in WT BMDMs upon CHX treatment (Fig. S4B).
We appreciate the reviewer’s comments on our manuscript, and we have further improved the manuscript as suggested in the revised manuscript (Fig. S4D).
(7) Finally, some editing to provide data only critical for the conclusions could improve the ease of reading.
We have further improved the manuscript as suggested in the revised manuscript.
Reviewer #3 (Recommendations For The Authors):
Comments (general, please address at least in Discussion. Some experimental data, for example the role, if any, of Vangl2 phosphorylations will be very useful):
(1) It might be interesting to explore whether there are any potential effects of developmental pathways on the observed effect mediated by Vangl2 or if the effects are entirely a PCP-independent function of Vangl2. Please see above public review.
Thank you for the reviewer's insightful comments. Our study revealed that Vangl2 recruits the E3 ubiquitin ligase PDLIM2 to facilitate K63-linked ubiquitination of p65, which is subsequently recognized by autophagy receptor NDP52 and then promotes the autophagic degradation of p65. Our findings by using autophagy inhibitors and autophagic-deficient cells indicate that Vangl2 regulates NF-kB signaling through a selective autophagic pathway, rather than affecting the PCP pathway, WNT, HH/GLI, Fat-Dachsous or even mechanical tension. Furthermore, we generated phospho-mutants of Vangl2 (S82/84A) and observed that Vangl2 (S82/84A) could still facilitate the degradation of p65 (Fig. S4 B), suggesting that Vangl2 regulates the NF-kB pathway independently of its phosphorylation. In addition, a discussion section has been added to the revised version. (Page 12, lines 377-393)
(2) What explains the specificity of organ/cell-type Vangl2 upregulation and its consequences observed here? Why is NF-kappaB signaling not more broadly or even ubiquitously affected in all cell types in a Vangl2-dependent manner, rather than being restricted to macrophages, neutrophils and peritoneal macrophages, or, for that matter, in spleen and LN and not liver and lung? Afterall, one may think that the PCP proteins, as well as NF-kappaB, are ubiquitous.
Thank you for the reviewer's comments. A similar question has been addressed above (refer to the response to question 3 of reviewer 3).
(3) Another specificity-related question that comes to mind is whether the Vangl2 function in autolysomal/autophagic degradation is restricted to p65 as the exclusive substrate? The cytosolic targeting of p65 as opposed to the more well-known nuclear-targeting is interesting.
Our previous finding demonstrated that Vangl2 inhibits antiviral IFN-I signaling by targeting TBK1 for autophagic degradation (doi: 10.1126/sciadv.adg2339), thereby indicating that p65 is not the sole substrate for Vangl2. However, in the NF-kB pathway, p65 is a specific substrate for Vangl2. Moreover, our findings indicate that the interaction between Vangl2 and p65 occurs predominantly in the cytoplasm, rather than in the nucleus (Fig. S4 C).
(4) Pharmacological approach is used to tease apart autolysosome versus proteasome pathway. What is the physiological importance of autophagic degradation? It is interesting to note that Vangl2 was already previously implicated in degrading LAMP-2A and increasing chaperon-mediated autophagy (CMA)-lysosome numbers (PMID: 34214490).
Previous literature has domonstrated that Vangl2 can inhibit CMA degradation (PMID: 34214490). However, in our study, we found that Vangl2 can promote the selective autophagic degradation of p65. It is important to note that CMA degradation and selective autophagic degradation are two distinct degradation modes, which is not contradictory.
(5) Are these phenotypes discernable in heterozygotes or only when ablated in homozygosity? Any phenotypes recapitulated in the looptail heterozygote mice?
We found that these phenotypes discernable only in homozygosity.
(6) What is the conservation of the Vangl2 p65-interaction site between Vangl2 and Vangl1? PDLIM2 recruitment between Vangl2 and Vangl1?
We appreciate the reviewer’s comments on our manuscript. Previous studies have shown that human Vangl1 and Vangl2 exhibit only 72% identity and exhibit distinct functional properties (doi: 10.1530/ERC-14-0141).Thus, the interaction of Vangl2 with p65 and PDLIM2 recruitment may not necessarily occur in Vangl1.
Comments (specific to experiments and data analyses. Please address the following):
(7) The patient population used in Fig 1 is not described in the Methods. This is a critical omission. Were age, sex etc. controlled for between healthy and disease? How was the diagnosis made? What times during sepsis were the samples collected? As presented, this data is impossible to evaluate and interpret.
We appreciate the reviewer’s comments on our manuscript, and we have further improved the manuscript as suggested in the revised supplement materials. (Supplementary information, Page 12, lines 146-147)
(8) In general, the statistical method should be described for each experiment presented in the figures. Comparisons should not be made only at the time point with maximal difference (such as in Fig 1F or Fig 2C, but at all time points using appropriate statistical methods). The sample size should also be included to allow determination appropriateness of parametric or non-parametric tests.
We appreciate the reviewer’s comments on our manuscript, and we have further improved the manuscript as suggested in the revised manuscript (Figures 1F and 2C).
(9) PCP pathways can activate p62/SQSTM1 or JNK via RhoA. JNK activation should be tested experimentally.
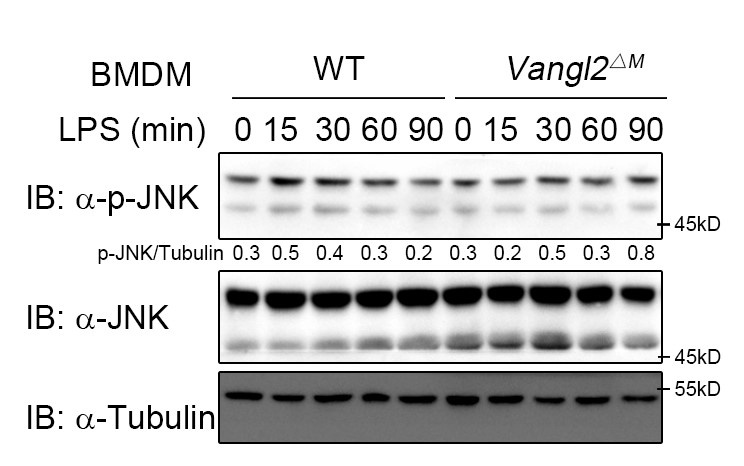
According to the reviewer's comments, we further examined the effect of Vangl2 on the JNK pathway. The results showed that Vangl2 did not affect the JNK pathway (Author response image 2). This suggests that Vangl2 functions independently of the PCP pathway.
Author response image 2.
Vangl2 did not affect the JNK pathway. WT and Vangl2-deficient (n≥3) BMDMs were stimulated with LPS (100 ng/ml) for the indicated times. Immunoblot analysis of total and phosphorylated JNK.
(10) Why are different cells such as A549, HEK293, CHO, 293T, THP-1 used during the studies for different experiments? Consistency would improve rigor. At least, logical explanation driving the cell type of choice for each experiment should be included in the manuscript. Nonetheless, one aspect of using a panel of cell lines indicate that the effect of Vangl2 on NF-kappa B is pleiotropic.
We are grateful to the reviewer for their comments on our manuscript. A549, HEK293, CHO, and 293T cells are commonly utilized in protein-protein interaction studies. The selection of cell lines for overexpression (exogenous) experiment is dependent on their transfection efficiency and the ability to express TLR4 (the receptor for LPS). Additionally, we conducted endogenous experiments by using THP-1 and BMDMs, which are human macrophage cell lines and murine primary macrophages, respectively. Moreover, we generated Vangl2f/f lyz-cre mice by specifically knocking out Vangl2 in myeloid cells, and investigated the effect of Vangl2 on NF-kB signaling in vivo.
-
eLife assessment
This valuable manuscript describes a novel role of Vangl2, a core planar cell polarity protein, in linking the NF-kB pathway to selective autophagic protein degradation in myeloid cells. The mechanistic studies suggest that Vangl2 targets p65 for NDP52-mediated autophagic degradation, limiting inflammatory NF-kB response, with functional significance of the proposed mechanism in sepsis. The presented evidence is convincing. Additional studies dissecting autophagic Vangl2 functions in various myeloid subsets in the context of inflammation could be informative, and additional Vangl2 targets in the inflammatory pathway, including IKK2, could also be explored. Overall, this exciting study will likely advance our understanding of NF-kB control, particularly in the context of inflammatory diseases.
-
Reviewer #1 (Public Review):
The study shows a new mechanism of NFkB-p65 regulation mediated by Vangl2-dependent autophagic targeting. Autophagic regulation of p65 has been reported earlier; this study brings an additional set of molecular players involved in this important regulatory event, which may have implications for chronic and acute inflammatory conditions.
Comments on the revised version:
The authors have addressed the earlier concerns and I am satisfied with the revised version. I have no additional comments to make.
-
Reviewer #2 (Public Review):
Vangl2, a core planar cell polarity protein involved in Wnt/PCP signaling, cell proliferation, differentiation, homeostasis, and cell migration. Vangl2 malfunctioning has been linked to various human ailments, including autoimmune and neoplastic disorders. Interestingly, it was shown that Vangl2 interacts with the autophagy regulator p62, and autophagic degradation limits the activity of inflammatory mediators, such as p65/NF-κB. However, the possible role of Vangl2 in inflammation has not been investigated. In this manuscript, Lu et al. describe that Vangl2 expression is upregulated in human sepsis-associated PBMCs and that Vangl2 mitigates experimental sepsis in mice by negatively regulating p65/NF-κB signaling in myeloid cells. Their mechanistic studies further revealed that Vangl2 recruits the E3 …
Reviewer #2 (Public Review):
Vangl2, a core planar cell polarity protein involved in Wnt/PCP signaling, cell proliferation, differentiation, homeostasis, and cell migration. Vangl2 malfunctioning has been linked to various human ailments, including autoimmune and neoplastic disorders. Interestingly, it was shown that Vangl2 interacts with the autophagy regulator p62, and autophagic degradation limits the activity of inflammatory mediators, such as p65/NF-κB. However, the possible role of Vangl2 in inflammation has not been investigated. In this manuscript, Lu et al. describe that Vangl2 expression is upregulated in human sepsis-associated PBMCs and that Vangl2 mitigates experimental sepsis in mice by negatively regulating p65/NF-κB signaling in myeloid cells. Their mechanistic studies further revealed that Vangl2 recruits the E3 ubiquitin ligase PDLIM2 to promote K63-linked poly-ubiquitination of p65. Vangl2 also facilitated the recognition of ubiquitinated p65 by the cargo receptor NDP52. These molecular processes caused selective autophagic degradation of p65. Indeed, abrogation of PDLIM2 or NDP52 functions rescued p65 from autophagic degradation, leading to extended p65/NF-κB activity in myeloid cells. Overall, the manuscript presents convincing evidence for novel Vangl2-mediated control of inflammatory p65/NF-kB activity. The proposed pathway may expand interventional opportunities restraining aberrant p65/NF-kB activity in human ailments.
IKK is known to mediate p65 phosphorylation, which instructs NF-kB transcriptional activity. In this manuscript, Vangl2 deficiency led to an increased accumulation of phosphorylated p65 and IKK also at 30 minutes post-LPS stimulation; however, autophagic degradation of p-p65 may not have been initiated at this early time point. Therefore, this set of data put forward the exciting possibility that Vangl2 could also be regulating the immediate early phase of inflammatory response involving the IKK-p65 axis - a proposition that may be tested in future studies.
-
Reviewer #3 (Public Review):
Lu et al. describe Vangl2 as a negative regulator of inflammation in myeloid cells. The primary mechanism appears to be through binding p65 and promoting its degradation, albeit in an unusual autolysosome/autophagy dependent manner. Overall, these findings are novel, valuable and the crosstalk of PCP pathway protein Vangl2 with NF-kappaB is of interest. While generally solid, some concerns still remain about the rigor and conclusions drawn.
Comments on the revised version:
Lu et al. address my comments through responses and new experimental data. However, some of the explanations provided are inadequate.
The new experimental data using phosphomutants indeed adds to their claim that this is a PCP-independent function of Vangl2.
The addition of statistics and testing JNK pathway is appreciated by this Reviewer.
Reviewer #3 (Public Review):
Lu et al. describe Vangl2 as a negative regulator of inflammation in myeloid cells. The primary mechanism appears to be through binding p65 and promoting its degradation, albeit in an unusual autolysosome/autophagy dependent manner. Overall, these findings are novel, valuable and the crosstalk of PCP pathway protein Vangl2 with NF-kappaB is of interest. While generally solid, some concerns still remain about the rigor and conclusions drawn.
Comments on the revised version:
Lu et al. address my comments through responses and new experimental data. However, some of the explanations provided are inadequate.
The new experimental data using phosphomutants indeed adds to their claim that this is a PCP-independent function of Vangl2.
The addition of statistics and testing JNK pathway is appreciated by this Reviewer.
However, in response to my enquiry regarding directly exploring PCP effects, the authors simply assert "Our study revealed that Vangl2 recruits the E3 ubiquitin ligase PDLIM2 to facilitate K63-linked ubiquitination of p65, which is subsequently recognized by autophagy receptor NDP52 and then promotes the autophagic degradation of p65. Our findings by using autophagy inhibitors and autophagic-deficient cells indicate that Vangl2 regulates NFkB signaling through a selective autophagic pathway, rather than affecting the PCP pathway, WNT, HH/GLI, Fat-Dachsous or even mechanical tension."
I do not agree that the use of autophagy inhibitors and autophagy-deficient cells can rule out the contributions of PCP or any other pathways. Only experimentally inhibiting the pathway(s) with adequate demonstration of target inhibition/abolition of well-known effector function and documenting unaltered p65 regulation under these conditions can be considered proof. Autophagy inhibitors and autophagy-deficient cells only prove that this particular pathway is necessary. Nonetheless, I do not want to dwell on proving a negative and agree that Vangl2 is a novel regulator of p65 through its role in promoting p65 degradation. The inclusion of a statement discussing the limitations of their approach would have sufficed. The response from the authors could have been better.
I am also not satisfied with the explanation that "immune cells represent a minor fraction of the lungs and liver". There are lots of resident immune cells in the lungs and liver (alveolar macrophages in the lung and Kuppfer cells in the liver). For example, it may be so that Vangl2 is important in monocytes and not in the resident population. This might be a potential explanation. But this is not explored. The restricted tissue-specificity of the interaction between two ubiquitously present proteins is still a challenge to understand. The response from the authors is not satisfactory. There is plenty of Vangl2 in the liver in their western blot.
I had also simply pointed out PMID: 34214490 with reference to the findings described in the manuscript. There were no suggestions of contradiction. In fact, I would refer to the publication in discussion to support the findings and stress the novelty. The response from the authors could have been better.
The response to my enquiry regarding homo- or heterozygosity is unsupported by any reference or data.
The listing of 8 patients and healthy controls are also appreciated. The body temperature of #6 doesn't fall in the <36 or >38 degree C SIRS criteria. The inclusion of CRP, PCT, heart rate and respiratory rate, and other lab values would have further improved the inclusion criteria. Moreover, it is difficult to understand why there are 16 value points for healthy and sepsis cohorts in Fig 1 when there are 8 patients.
-
-
eLife assessment
This valuable work describes a novel role of Vangl2, a core planar cell polarity protein, in mechanistically linking the inflammatory NF-kB pathway to selective autophagic protein degradation. Using solid methods, the authors also establish the functional significance of the proposed mechanism in sepsis. The work may advance our understanding of NF-kB control, particularly in the context of aberrant inflammation. However, some gaps remain, and additional studies are needed to unequivocally establish the role of Vangl2 in regulating NF-kB signaling.
-
Reviewer #1 (Public Review):
In the manuscript titled "Vangl2 suppresses NF-κB signaling and ameliorates sepsis by targeting p65 for NDP52-mediated autophagic degradation" by Lu et al, the authors show that Vangl2, a planner cell polarity component, plays a direct role in autophagic degradation of NFkB-p65 by facilitating its ubiquitination via PDLIM2 and subsequent recognition and autophagic targeting via the autophagy adaptor protein NDP52. Conceptually it is a wonderful study with excellent execution of experiments and controls. The concerns with the manuscript are mainly on two counts - First issue is the kinetics of p65 regulation reported here, which does not fit into the kinetics of the mechanism proposed here, i.e., Vangl2-mediated ubiquitination followed by autophagic degradation of p65. The second issue is more technical- an …
Reviewer #1 (Public Review):
In the manuscript titled "Vangl2 suppresses NF-κB signaling and ameliorates sepsis by targeting p65 for NDP52-mediated autophagic degradation" by Lu et al, the authors show that Vangl2, a planner cell polarity component, plays a direct role in autophagic degradation of NFkB-p65 by facilitating its ubiquitination via PDLIM2 and subsequent recognition and autophagic targeting via the autophagy adaptor protein NDP52. Conceptually it is a wonderful study with excellent execution of experiments and controls. The concerns with the manuscript are mainly on two counts - First issue is the kinetics of p65 regulation reported here, which does not fit into the kinetics of the mechanism proposed here, i.e., Vangl2-mediated ubiquitination followed by autophagic degradation of p65. The second issue is more technical- an absolute lack of quantitative analyses. The authors rely mostly on visual qualitative interpretation to assess an increase or decrease in associations between partner molecules throughout the study. While the overall mechanism is interesting, the authors should address these concerns as highlighted below:
Major points:
- Kinetics of p65 regulation by Vangl2: As mentioned above, authors report that LPS stimulation leads to higher IKK and p65 activation in the absence of Vangl2. The mechanism of action authors subsequently work out is that- Vangl2 helps recruit E3 ligase PDLIM to p65, which causes K63 ubiquitination, which is recognised by NDP52 for autophagic targeting. Curiously, peak p65 activation is achieved within 30 minutes of LPS stimulation. The time scale of all other assays is way longer. It is not clear that in WT cells, p65 could be targeted to autophagic degradation in Vangl2 dependent manner within 30 minutes. The HA-Myc-Flag-based overexpression and Co-IP studies do confirm the interactions as proposed. However, they do not prove that this mechanism was responsible for the Vangl2-mediated modulation of p65 activation upon LPS stimulation. Moreover, the Vangl2 KO line also shows increased IKK activation. The authors do not show the cause behind increased IKK activation, which in itself can trigger increased p65 phosphorylation.
- The other major concern is regarding the lack of quantitative assessments. For Co-IP experiments, I can understand it is qualitative observation. However, when the authors infer that there is an increase or decrease in the association through co-IP immunoblots, it should also be quantified, especially since the differences are quite marginal and could be easily misinterpreted.
- Figure 4E and F: It is evident that inhibiting Autolysosome (CQ or BafA1) or autophagy (3MA) led to the recovery of p65 levels and inducing autophagy by Rapamycin led to faster decay in p65 levels. Did the authors also note/explore the possibility that Vangl2 itself may be degraded via the autophagy pathway? IB of WCL upon CQ/BAF/3MA or upon Rapa treatment does indicate the same. If true, how would that impact the dynamics of p65 activation?
- Autophagic targeting of p65 should also be shown through alternate evidence, like microscopy etc., in the LPS-stimulated WT cells.
Limitation: The mechanism behind enhanced activation of IKK in the absence of Vangl2 remains unclear. It is possible there is an autophagy-independent mechanism also involved in this regulation.
Summary: The study shows a new mechanism of NFkB-p65 regulation mediated by Vangl2-dependent autophagic targeting. Autophagic regulation of p65 has been reported earlier; this study brings an additional set of molecular players involved in this important regulatory event, which may have implications for chronic and acute inflammatory conditions.
-
Reviewer #2 (Public Review):
Vangl2, a core planar cell polarity protein involved in Wnt/PCP signaling, mediates cell proliferation, differentiation, homeostasis, and cell migration. Vangl2 malfunctioning has been linked to various human ailments, including autoimmune and neoplastic disorders. Interestingly, Vangl2 was shown to interact with the autophagy regulator p62, and indeed, autophagic degradation limits the activity of inflammatory mediators such as p65/NF-κB. However, if Vangl2, per se, contributes to restraining aberrant p65/NF-kB activity remains unclear.
In this manuscript, Lu et al. describe that Vangl2 expression is upregulated in human sepsis-associated PBMCs and that Vangl2 mitigates experimental sepsis in mice by negatively regulating p65/NF-κB signaling in myeloid cells. Vangl2 recruits the E3 ubiquitin ligase PDLIM2 …
Reviewer #2 (Public Review):
Vangl2, a core planar cell polarity protein involved in Wnt/PCP signaling, mediates cell proliferation, differentiation, homeostasis, and cell migration. Vangl2 malfunctioning has been linked to various human ailments, including autoimmune and neoplastic disorders. Interestingly, Vangl2 was shown to interact with the autophagy regulator p62, and indeed, autophagic degradation limits the activity of inflammatory mediators such as p65/NF-κB. However, if Vangl2, per se, contributes to restraining aberrant p65/NF-kB activity remains unclear.
In this manuscript, Lu et al. describe that Vangl2 expression is upregulated in human sepsis-associated PBMCs and that Vangl2 mitigates experimental sepsis in mice by negatively regulating p65/NF-κB signaling in myeloid cells. Vangl2 recruits the E3 ubiquitin ligase PDLIM2 to promote K63-linked poly-ubiquitination of p65. Vangl2 also facilitates the recognition of ubiquitinated p65 by the cargo receptor NDP52. These molecular processes cause selective autophagic degradation of p65. Indeed, abrogation of PDLIM2 or NDP52 functions rescued p65 from autophagic degradation, leading to extended p65/NF-κB activity.
As such, the manuscript presents a substantial body of interesting work and a novel mechanism of NF-κB control. If found true, the proposed mechanism may expand therapeutic opportunities for inflammatory diseases. However, the current draft has significant weaknesses that need to be addressed.
Specific comments
1. Vangl2 deficiency did not cause a discernible increase in the cellular level of total endogenous p65 (Fig 2A and Fig 2B) but accumulated also phosphorylated IKK.
Even Fig 4D reveals that Vangl2 exerts a rather modest effect on the total p65 level and the figure does not provide any standard error for the quantified data. Therefore, these results do not fully support the proposed model (Figure 7) - this is a significant draw back. Instead, these data provoke an alternate hypothesis that Vangl2 could be specifically mediating autophagic removal of phosphorylated IKK and phosphorylated IKK, leading to exacerbated inflammatory NF-κB response in Vangl2-deficient cells. One may need to use phosphorylation-defective mutants of p65, at least in the over-expression experiments, to dissect between these possibilities.
2. Fig 1A: The data indicates the presence of two subgroups within the sepsis cohort - one with high Vangl2 expressions and the other with relatively normal Vangle2 expression. Was there any difference with respect to NF-κB target inflammatory gene expressions between these subgroups?
3. The effect of Vangl2 deficiency was rather modest in the neutrophil. Could it be that Vangl2 mediates its effect mostly in macrophages?
4. Fig 1D and Figure 1E: Data for unstimulated Vangl2 cells should be provided. Also, the source of the IL-1β primary antibody has not been mentioned.
5. The relevance and the requirement of RNA-seq analysis are not clear in the present draft. Figure 1E already reveals upregulation of the signature NF-κB target inflammatory genes upon Vangl2 deficiency.
6. Fig 2A reveals an increased accumulation of phosphorylated p65 and IKK in Vangl2-deficient macrophages upon LPS stimulation within 30 minutes. However, Vangl2 accumulates at around 60 minutes post-stimulation in WT cells. Similar results were obtained for neutrophils (Fig 2B). There appears to be a temporal disconnect between Vangl2 and phosphorylated p65 accumulation - this must be clarified.
7. Figure 2E and 2F do not have untreated controls. Presentations in Fig 2E may be improved to more clearly depict IL6 and TNF data, preferably with separate Y-axes.
8. Line 219: "strongly with IKKα, p65 and MyD88, and weak" - should be revised.
9. It is not clear why IKKβ was excluded from interaction studies in Fig S3G.
10. Fig 3F- In the text, authors mentioned that Vangl2 strongly associates with p65 upon LPS stimulation in BMDM. However, no controls, including input or another p65-interacting protein, were used.
11. Figure 4D - Authors claim that Vangl2-deficient BMDMs stabilized the expression of endogenous p65 after LPS treatment. However, p65 levels were particularly constitutively elevated in knockout cells, and LPS signaling did not cause any further upregulation. This again indicates the role of Vangl2 in the basal state. The authors need to explain this and revise the test accordingly. -
Reviewer #3 (Public Review):
Lu et al. describe Vangl2 as a negative regulator of inflammation in myeloid cells. The primary mechanism appears to be through binding p65 and promoting its degradation, albeit in an unusual autolysosome/autophagy dependent manner. Overall, the findings are novel and the crosstalk of PCP pathway protein Vangl2 with NF-kappaB is of interest. Whether PCP is anyway relevant or if this is a PCP-independent function of Vangl2 is not directly explored (the later appears more likely from the manuscript/discussion). PCP pathways intersect often with developmentally important pathways such as WNT, HH/GLI, Fat-Dachsous and even mechanical tension. It might be of importance to investigate whether Vangl2-dependent NF-kappaB is influenced by developmental pathways. Are Vangl2 phosphorylations (S5, S82 and S84) in anyway …
Reviewer #3 (Public Review):
Lu et al. describe Vangl2 as a negative regulator of inflammation in myeloid cells. The primary mechanism appears to be through binding p65 and promoting its degradation, albeit in an unusual autolysosome/autophagy dependent manner. Overall, the findings are novel and the crosstalk of PCP pathway protein Vangl2 with NF-kappaB is of interest. Whether PCP is anyway relevant or if this is a PCP-independent function of Vangl2 is not directly explored (the later appears more likely from the manuscript/discussion). PCP pathways intersect often with developmentally important pathways such as WNT, HH/GLI, Fat-Dachsous and even mechanical tension. It might be of importance to investigate whether Vangl2-dependent NF-kappaB is influenced by developmental pathways. Are Vangl2 phosphorylations (S5, S82 and S84) in anyway necessary for the observed effects on NF-kappaB or would a phospho-mutant (alanine substitution mutant) Vangl2 phenocopy WT Vangl2 for regulation of NF-kappaB? Another area to strengthen might be with regards to specificity of cell types where this phenomenon may be observed. LPS treatment in mice resulted in Vangl2 upregulation in spleen and lymph nodes, but not in lung and liver. What explains the specificity of organ/cell-type Vangl2 upregulation and its consequences observed here? Why is NF-kappaB signaling not more broadly or even ubiquitously affected in all cell types in a Vangl2-dependent manner, rather than being restricted to macrophages, neutrophils and peritoneal macrophages, or, for that matter, in spleen and LN and not liver and lung? After all, one may think that the PCP proteins, as well as NF-kappaB, are ubiquitous. Regardless, Vangl2 as a negative regulator of NF-kappaB is an important finding. There are, however, some concerns about methodology and statistics that need to be addressed.
-
