Single-cell sequencing highlights heterogeneity and malignant progression in actinic keratosis and cutaneous squamous cell carcinoma
Curation statements for this article:-
Curated by eLife

eLife assessment
This important study delineates the molecular changes driving the progression from actinic keratosis (AK) to cutaneous squamous cell carcinoma (cSCC). Using state-of-the-art single-cell RNA profiling of 138,982 cells from 13 samples of six patients including AK, squamous cell carcinoma in situ (SCCIS), cSCC, and their matched normal tissues, thus covering comprehensive clinical courses of cSCC, the authors provide an invaluable data resource. This study identified several previously unreported and interesting candidate genes involved in different stages of the malignant progression of skin neoplasias, which have been validated in situ, and partially in vitro. Although data analysis needs improvement and comparison to other published data sets to fully support the claims and conclusions, these findings substantially advance our understanding of the molecular changes leading to skin cancer.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Cutaneous squamous cell carcinoma (cSCC) is the second most frequent of the keratinocyte-derived malignancies with actinic keratosis (AK) as a precancerous lesion. To comprehensively delineate the underlying mechanisms for the whole progression from normal skin to AK to invasive cSCC, we performed single-cell RNA sequencing (scRNA-seq) to acquire the transcriptomes of 138,982 cells from 13 samples of six patients including AK, squamous cell carcinoma in situ (SCCIS), cSCC, and their matched normal tissues, covering comprehensive clinical courses of cSCC. We identified diverse cell types, including important subtypes with different gene expression profiles and functions in major keratinocytes. In SCCIS, we discovered the malignant subtypes of basal cells with differential proliferative and migration potential. Differentially expressed genes (DEGs) analysis screened out multiple key driver genes including transcription factors along AK to cSCC progression. Immunohistochemistry (IHC)/immunofluorescence (IF) experiments and single-cell ATAC sequencing (scATAC-seq) data verified the expression changes of these genes. The functional experiments confirmed the important roles of these genes in regulating cell proliferation, apoptosis, migration, and invasion in cSCC tumor. Furthermore, we comprehensively described the tumor microenvironment (TME) landscape and potential keratinocyte-TME crosstalk in cSCC providing theoretical basis for immunotherapy. Together, our findings provide a valuable resource for deciphering the progression from AK to cSCC and identifying potential targets for anticancer treatment of cSCC.
Article activity feed
-

-

Author Response
Reviewer #2 (Public Review):
Zou et al. presented a comprehensive study where they generated single-cell RNA profiling of 138,982 cells from 13 samples of six patients including AK, squamous cell carcinoma in situ (SCCIS), cSCC, and their matched normal tissues, covering comprehensive clinical courses of cSCC. Using bioinformatics analysis, they identified keratinocytes, CAFs, immune cells, and their subpopulations. The authors further compared signatures within subpopulations of keratinocytes along with the clinical progression, especially basal cells, and identified many interesting genes. They also further validate some of the markers in an independent cohort using IHC, followed by some knockdown experiments using cSCC cell lines.
The strength of this study is the unique data set they have created, providing the …
Author Response
Reviewer #2 (Public Review):
Zou et al. presented a comprehensive study where they generated single-cell RNA profiling of 138,982 cells from 13 samples of six patients including AK, squamous cell carcinoma in situ (SCCIS), cSCC, and their matched normal tissues, covering comprehensive clinical courses of cSCC. Using bioinformatics analysis, they identified keratinocytes, CAFs, immune cells, and their subpopulations. The authors further compared signatures within subpopulations of keratinocytes along with the clinical progression, especially basal cells, and identified many interesting genes. They also further validate some of the markers in an independent cohort using IHC, followed by some knockdown experiments using cSCC cell lines.
The strength of this study is the unique data set they have created, providing the community with invaluable resources to study and validate their findings. However, a lot of analyses were not robust enough to support the claims and conclusions in the paper. More clarification and cross-comparison with polished data are needed to further strengthen the study and claims.
- Stemness markers were used. The authors used COL17A1, TP63, ITGB1, and ITGA3 to represent stemness markers. However, these were not common classic stemness markers used in cSCC. What is the source claiming these genes were stemness markers in cSCC? TP63 is a master regulator and early driver event in SCC, while COL17A1, ITGB1, and ITGA3 are all ECM genes. The authors need to use commonly well-known stem cell markers in cSCC, e.g., LGR5, to mark stem-like cells.
Thanks for raising this good point. We may not have provided a clear description of the markers COL17A1, TP63, ITGB1, and ITGA3 in the previous texts. We would like to clarify that these genes were used as the markers of epidermal stem cells in normal skin samples rather than tumor stem cells in cSCC. To avoid any possible misunderstanding, we revised the main text accordingly and added the references [4-11].
- Cell proportion analysis. The authors used the mean proportions to compare different clinical groups for subpopulations of keratinocytes, e.g., Figure 2B, and Figure 5B. This is not robust, as no statistics can be derived from this. For example, from Fig 2A, it is clearly shown there is a high level of heterogeneity of cellular compositions for normal samples. One cannot say which group is higher or lower simply based on mean not variance as well.
We replotted the proportion analysis with statistics and presented the new graphs in Figure 2-figure supplement 1 for Figure 2B and Figure 5-figure supplement 1 for Figure 5B.
- Basal tumour cells in SCCIS and SCC. To make the findings valid, authors need to compare these cells/populations with the keratinocyte cell populations defined by Ji et al. Cell 2020. Do basal-SCCIS-tumours cells, also in SCC samples, resemble any of the population defined in Ji et al. Ji et al. also had 10 match normal, thus the authors need to validate their findings of SCC vs normal analysis using the Ji et al. dataset.
Thanks for this valuable suggestion. We compared basal tumor cell in our study with the cell populations defined in Ji et al. Cell 2020 data using SingleCellNet [1]. The results showed that both the basal-SCCIS-tumor cells of SCCIS and basal tumor cells of cSCC in our study closely resemble the Tumor_KC_Basal subcluster defined in Ji et al’s paper (Figure 4-figure supplement 4, C and D). Tumor_KC_Basal highly expressed CCL2, CXCL14, FTH1, MT2A, which is consistent with our findings in basal tumor cells.
- Copy number analysis. Authors used inferCNV to perform copy number analysis using scRNA-seq data and identified CNVs in subpopulations of keratinocytes in SCCIS and SCC. To ensure these CNVs were not artefacts, were some of the CNVs identified by inferCNV well-known copy number changes previously reported in cSCC?
In poorly-differentiated cSCC sample, the significant gains in chromosome 7, 9 and deletion in chromosome 10 were reported in previous study, indicating the reliability of the CNV analysis results (Figure 5-figure supplement 2) [12].
- Pseudotime analysis lines 308-313. Not sure the pseudotime analysis added much as, as it is unclear two distinct subgroups were identified from this analysis. Suggest removing this to keep it neater
Thank you for this suggestion. We have deleted the result of pseudotime analysis.
- Selection of candidate genes for validation using IHC and cell line work. For example, lines 205-206, lines 352-356 and lines 437-441, authors selected several genes associated with AK and SCC to further validate using IHC and cell line knockdown work. What are the criteria for selecting those genes for validation? It is unclear to readers how these were selected. It reads like a fishing experiment, then followed by a knockdown. Clear rationale/criteria need to be elaborated.
The first consideration of candidate gene selection is the fold change of expression. We have provided the statistical results of DEGs in Supplementary file 1b, 1h, 1j-1m. Then we selected top changed genes and conducted an extensive literature search on these genes. We prioritized genes that, although not directly associated with cSCC development, have a close relationship with related pathways, as determined through functional enrichment analysis. These genes were arranged for further verification experiments. We have added more details in main text and methods section.
- TME. Compared to keratinocytes populations, the investigation of TME cells was weak. (a) can authors produce UMAP files just for T cells, DC cells, and fibroblasts separately? Figure 7B is not easy to see those subclusters. (b) similar to what was done for keratinocytes, can authors find differentially expressed clusters and genes among the different clinical groups, associated with disease progression? (c) where are the myeloid cell populations, also B cells?
Thank you for your suggestions. (a) We have added the UMAP files for T cells, DC cells and stromal cells separately in new Figure 7A. (b) We identified DEGs in TME cells among the different groups. Several key genes showed monotonically changing trends associated with disease progression. For example, with the increase of malignancy, FOS shows down-regulation while S100A8 and S100A9 monotonically increase in all three types of TME cells (Figure 7C). (c) We identified two types of myeloid cell populations, macrophage and monocyte derived DCs (MoDC). We didn’t find other myeloid cells, such as neutrophil. For B cells, there were only 28 B cells in poorly-differentiated cSCC sample, which didn’t meet the threshold for further cell-cell communication analysis.
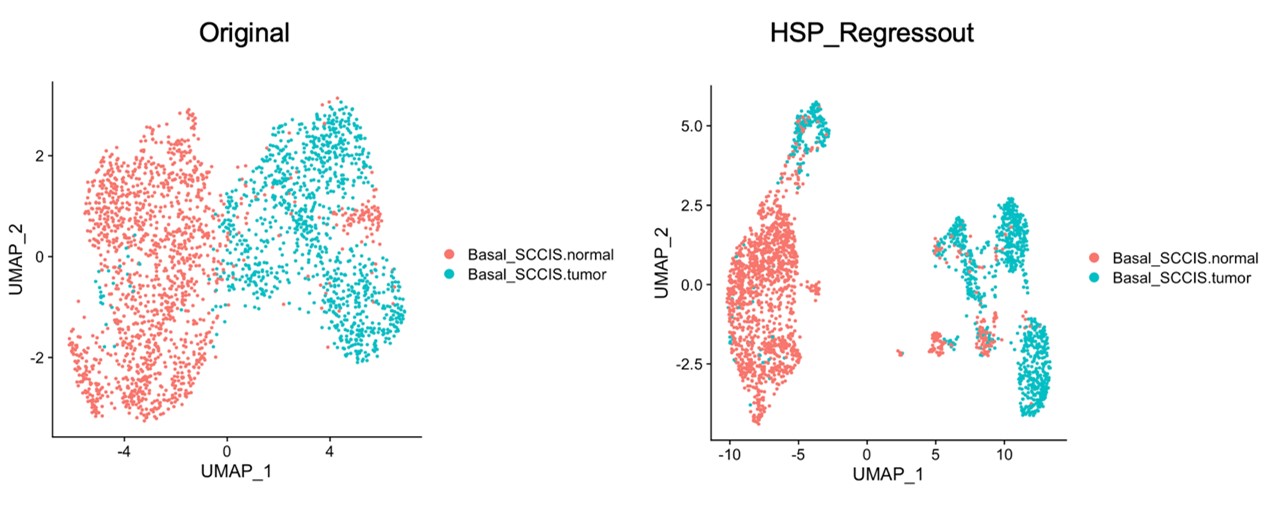
- Heat shock protein genes line 327-329. HSP signature was well-known to be induced via tissue dissociation and library prep during the scRNA experiment. How could the authors be sure these were not artefacts induced by the experiment? If authors regress their gene expression against HSP gene signatures, would this cluster still be identified?
Thank you for this valuable suggestion. It is important to note that the Basal-SCCIS-tumor cluster was identified through CNV analysis, rather than the HSP signature. To address this concern and further validate this result, “AddModuleScore” function in Seurat package was used to regress gene expression against HSP gene signatures for retrieved basal cells. Our result showed that Basal_SCCIS tumor population still can be identified after regression, even more clearly (Author response image 1).
Author response image 1.
The identity of Basal-SCCIS-tumor cluster considering regression against HSP signatures.
- Cell-cell communication analysis. The authors claimed that that cell-to-cell interaction was significantly enhanced in poorly-differentiated cSCC, and multiple interaction pathways were significantly active. How was this kind of analysis carried out? How did the authors define significance? what statistical method was used? these were all unclear. Furthermore, it is difficult to judge the robustness of the cell-cell communication analysis. Were these findings also supported by another method, such as celltalker, and cellphoneDB?
To determine the significance of the increased overall cell-to-cell interaction strength between two groups, we utilized CellChat to obtain the communication strength in different samples. We combined the communication strength based on cell type pairs, where missing values were set to 0. We performed a paired Wilcoxon test to determine whether the enhancement of cell-to-cell interaction between samples was significant.
For the comparison of outgoing or incoming interaction strength of the same cell types between two groups, we first extracted the communication strength of each signal pathway contributing to outgoing or incoming strength, and then merged the strengths of signal pathways among samples, where the strength of non-shared pathways with missing value was determined to be 0. Subsequently, we performed a paired Wilcoxon test to define the significance.
For multiple groups comparisons, the Kruskal-Wallis rank sum test was first performed. If the p-value is less than 0.1, the pairwise Wilcoxon test was used for subsequent pairwise comparisons. The comparison of individual signaling pathways between groups is similar to the above. We defined p-value < 0.1 as significance threshold. We have added the significance test method in figure legend for Figure 7 and Figure 8 as well as and detailed statistical data in new Supplementary file 1q-1u.
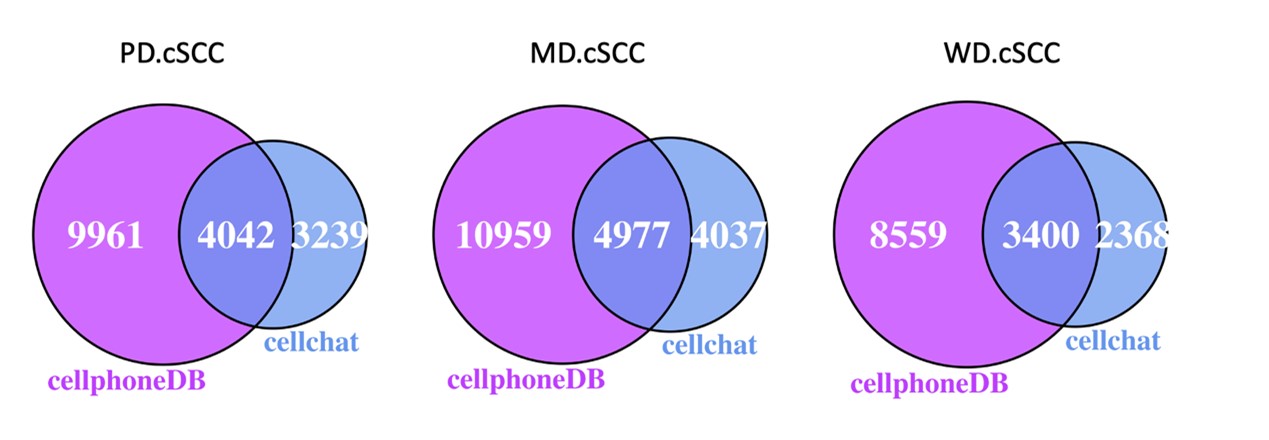
As suggested, we also used the approach of CellPhoneDB based on CellChatDB database to verify our cell-cell communication results. There are 55-58% of the ligand-receptor interactions predicted by CellChat were also predicted by CellPhoneDB (Author response image 2). The enhancement of cell interaction through MHC-II, Laminin and TNF signaling pathways in poorly-differentiated cSCC sample compare to normal sample were consistent in both CellChat and CellPhoneDB (Figure 8C and Figure 8-figure supplement 1B).
Author response image 2.
The overlap of the predicted ligand-receptor interactions between CellChat and CellPhoneDB.
- Statistics and significance. In general, the detail of statistics and significance was lacking throughout the paper. Authors need to specify what statistical tests were used, and the p-values. It is difficult to judge the correctness of the test, and robustness without seeing the stats.
We have included all statistics and significance values in the figure legend and supplemental tables, and described the statistical tests in the methods section. In this revision, we have added the necessary details of statistics and significance in the main text and figures.
- Overall, this manuscript needs a lot of re-writing. A lot of discussion was also included in the results, making it really difficult to read overall. The authors should simplify the results sections, remove the discussion bits, and further highlight and streamline with the key results of this paper.
Thanks a lot for this advice. We have revised the paper thoroughly, removed discussion in results section to make the manuscript easier to read.
-
eLife assessment
This important study delineates the molecular changes driving the progression from actinic keratosis (AK) to cutaneous squamous cell carcinoma (cSCC). Using state-of-the-art single-cell RNA profiling of 138,982 cells from 13 samples of six patients including AK, squamous cell carcinoma in situ (SCCIS), cSCC, and their matched normal tissues, thus covering comprehensive clinical courses of cSCC, the authors provide an invaluable data resource. This study identified several previously unreported and interesting candidate genes involved in different stages of the malignant progression of skin neoplasias, which have been validated in situ, and partially in vitro. Although data analysis needs improvement and comparison to other published data sets to fully support the claims and conclusions, these findings substantially …
eLife assessment
This important study delineates the molecular changes driving the progression from actinic keratosis (AK) to cutaneous squamous cell carcinoma (cSCC). Using state-of-the-art single-cell RNA profiling of 138,982 cells from 13 samples of six patients including AK, squamous cell carcinoma in situ (SCCIS), cSCC, and their matched normal tissues, thus covering comprehensive clinical courses of cSCC, the authors provide an invaluable data resource. This study identified several previously unreported and interesting candidate genes involved in different stages of the malignant progression of skin neoplasias, which have been validated in situ, and partially in vitro. Although data analysis needs improvement and comparison to other published data sets to fully support the claims and conclusions, these findings substantially advance our understanding of the molecular changes leading to skin cancer.
-
Reviewer #1 (Public Review):
Zou et al. employ single-cell RNA sequencing of healthy skin, actinic keratosis (AK), squamous cell carcinoma in situ (SCCIS), and cutaneous squamous cell carcinoma (cSCC) to unravel the molecular events driving the progression of AK into cSCC (n=13 samples from 6 patients), thereby filling a gap of knowledge in skin cancer research. The authors identified several previously unreported candidate genes (including ALDH3A1, IGFBP2, MAGEA4, ITGA6, and LGALS1) involved in different stages of malignant progression, the expression of which was validated in situ in a large cohort. Functional in vitro experiments confirm a possible role for these genes in the transformation from benign to malignant skin lesions.
Moreover, the authors identified epidermal cell subpopulations that may play an important role in the …
Reviewer #1 (Public Review):
Zou et al. employ single-cell RNA sequencing of healthy skin, actinic keratosis (AK), squamous cell carcinoma in situ (SCCIS), and cutaneous squamous cell carcinoma (cSCC) to unravel the molecular events driving the progression of AK into cSCC (n=13 samples from 6 patients), thereby filling a gap of knowledge in skin cancer research. The authors identified several previously unreported candidate genes (including ALDH3A1, IGFBP2, MAGEA4, ITGA6, and LGALS1) involved in different stages of malignant progression, the expression of which was validated in situ in a large cohort. Functional in vitro experiments confirm a possible role for these genes in the transformation from benign to malignant skin lesions.
Moreover, the authors identified epidermal cell subpopulations that may play an important role in the development from AK to cSCC, including an "early malignant cell" subpopulation within SCCIS basal cells with higher mutational load according to CNV analysis, which they characterized in more detail. For example, they found MAGEA4 strongly expressed in basal cells of (most) SCCIS and cSCC, as well as ITGA6. Functional assays in HaCaT and cSCC cell lines revealed that the knock-down of MAGEA4 and ITGA6 reduced proliferation, migration, and invasion but increased apoptosis in the cSCC cell lines.
Finally, they describe the tumor microenvironment of a poorly differentiated cSCC sample, and scATAC sequencing of this poorly differentiated cSCC revealed that the majority of differentially accessible chromatin regions (DARs) were located in basal epidermal cells.
Altogether, the authors provide a comprehensive transcriptional analysis of premalignant (AK, SCCIS) and malignant stages of cSCC. They suggest some key driver genes for each stage, the role of which are addressed in vitro and in situ in a large cohort. Thus, this study may provide novel biomarkers for tumor staging and diagnosis as well as potential targets for the prevention and treatment of cSCC.
-
Reviewer #2 (Public Review):
Zou et al. presented a comprehensive study where they generated single-cell RNA profiling of 138,982 cells from 13 samples of six patients including AK, squamous cell carcinoma in situ (SCCIS), cSCC, and their matched normal tissues, covering comprehensive clinical courses of cSCC. Using bioinformatics analysis, they identified keratinocytes, CAFs, immune cells, and their subpopulations. The authors further compared signatures within subpopulations of keratinocytes along with the clinical progression, especially basal cells, and identified many interesting genes. They also further validate some of the markers in an independent cohort using IHC, followed by some knockdown experiments using cSCC cell lines.
The strength of this study is the unique data set they have created, providing the community with …
Reviewer #2 (Public Review):
Zou et al. presented a comprehensive study where they generated single-cell RNA profiling of 138,982 cells from 13 samples of six patients including AK, squamous cell carcinoma in situ (SCCIS), cSCC, and their matched normal tissues, covering comprehensive clinical courses of cSCC. Using bioinformatics analysis, they identified keratinocytes, CAFs, immune cells, and their subpopulations. The authors further compared signatures within subpopulations of keratinocytes along with the clinical progression, especially basal cells, and identified many interesting genes. They also further validate some of the markers in an independent cohort using IHC, followed by some knockdown experiments using cSCC cell lines.
The strength of this study is the unique data set they have created, providing the community with invaluable resources to study and validate their findings. However, a lot of analyses were not robust enough to support the claims and conclusions in the paper. More clarification and cross-comparison with polished data are needed to further strengthen the study and claims.
Stemness markers were used. The authors used COL17A1, TP63, ITGB1, and ITGA3 to represent stemness markers. However, these were not common classic stemness markers used in cSCC. What is the source claiming these genes were stemness markers in cSCC? TP63 is a master regulator and early driver event in SCC, while COL17A1, ITGB1, and ITGA3 are all ECM genes. The authors need to use commonly well-known stem cell markers in cSCC, e.g., LGR5, to mark stem-like cells.
Cell proportion analysis. The authors used the mean proportions to compare different clinical groups for subpopulations of keratinocytes, e.g., Figure 2B, and Figure 5B. This is not robust, as no statistics can be derived from this. For example, from Fig 2A, it is clearly shown there is a high level of heterogeneity of cellular compositions for normal samples. One cannot say which group is higher or lower simply based on mean not variance as well.
Basal tumour cells in SCCIS and SCC. To make the findings valid, authors need to compare these cells/populations with the keratinocyte cell populations defined by Ji et al. Cell 2020. Do basal-SCCIS-tumours cells, also in SCC samples, resemble any of the population defined in Ji et al. Ji et al. also had 10 match normal, thus the authors need to validate their findings of SCC vs normal analysis using the Ji et al. dataset.
Copy number analysis. Authors used inferCNV to perform copy number analysis using scRNA-seq data and identified CNVs in subpopulations of keratinocytes in SCCIS and SCC. To ensure these CNVs were not artefacts, were some of the CNVs identified by inferCNV well-known copy number changes previously reported in cSCC?
Pseudotime analysis lines 308-313. Not sure the pseudotime analysis added much as, as it is unclear two distinct subgroups were identified from this analysis. Suggest removing this to keep it neater
Selection of candidate genes for validation using IHC and cell line work. For example, lines 205-206, lines 352-356 and lines 437-441, authors selected several genes associated with AK and SCC to further validate using IHC and cell line knockdown work. What are the criteria for selecting those genes for validation? It is unclear to readers how these were selected. It reads like a fishing experiment, then followed by a knockdown. Clear rationale/criteria need to be elaborated.
TME. Compared to keratinocytes populations, the investigation of TME cells was weak. (a) can authors produce UMAP files just for T cells, DC cells, and fibroblasts separately? Figure 7B is not easy to see those subclusters. (b) similar to what was done for keratinocytes, can authors find differentially expressed clusters and genes among the different clinical groups, associated with disease progression? (c) where are the myeloid cell populations, also B cells?
Heat shock protein genes line 327-329. HSP signature was well-known to be induced via tissue dissociation and library prep during the scRNA experiment. How could the authors be sure these were not artefacts induced by the experiment? If authors regress their gene expression against HSP gene signatures, would this cluster still be identified?
Cell-cell communication analysis. The authors claimed that that cell-to-cell interaction was significantly enhanced in poorly-differentiated cSCC, and multiple interaction pathways were significantly active. How was this kind of analysis carried out? How did the authors define significance? what statistical method was used? these were all unclear. Furthermore, it is difficult to judge the robustness of the cell-cell communication analysis. Were these findings also supported by another method, such as celltalker, and cellphoneDB?
Statistics and significance. In general, the detail of statistics and significance was lacking throughout the paper. Authors need to specify what statistical tests were used, and the p-values. It is difficult to judge the correctness of the test, and robustness without seeing the stats.
Overall, this manuscript needs a lot of re-writing. A lot of discussion was also included in the results, making it really difficult to read overall. The authors should simplify the results sections, remove the discussion bits, and further highlight and streamline with the key results of this paper.
-
