A human mitofusin 2 mutation can cause mitophagic cardiomyopathy
Curation statements for this article:-
Curated by eLife

eLife assessment
In this paper, the authors demonstrate an interesting link between mitofusin function (MFN2) and PARKIN recruitment and mitophagy, underlying cardiomyopathy. This is a valuable finding with broad implications for understanding the mitochondrial biology as well as mechanisms involved in heart pathologies. However, the analyses are incomplete and the main conclusions are only partially supported and need to be further evidenced.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Cardiac muscle has the highest mitochondrial density of any human tissue, but mitochondrial dysfunction is not a recognized cause of isolated cardiomyopathy. Here, we determined that the rare mitofusin (MFN) 2 R400Q mutation is 15–20× over-represented in clinical cardiomyopathy, whereas this specific mutation is not reported as a cause of MFN2 mutant-induced peripheral neuropathy, Charcot–Marie–Tooth disease type 2A (CMT2A). Accordingly, we interrogated the enzymatic, biophysical, and functional characteristics of MFN2 Q400 versus wild-type and CMT2A-causing MFN2 mutants. All MFN2 mutants had impaired mitochondrial fusion, the canonical MFN2 function. Compared to MFN2 T105M that lacked catalytic GTPase activity and exhibited normal activation-induced changes in conformation, MFN2 R400Q and M376A had normal GTPase activity with impaired conformational shifting. MFN2 R400Q did not suppress mitochondrial motility, provoke mitochondrial depolarization, or dominantly suppress mitochondrial respiration like MFN2 T105M. By contrast to MFN2 T105M and M376A, MFN2 R400Q was uniquely defective in recruiting Parkin to mitochondria. CRISPR editing of the R400Q mutation into the mouse Mfn2 gene induced perinatal cardiomyopathy with no other organ involvement; knock-in of Mfn2 T105M or M376V did not affect the heart. RNA sequencing and metabolomics of cardiomyopathic Mfn2 Q/Q400 hearts revealed signature abnormalities recapitulating experimental mitophagic cardiomyopathy. Indeed, cultured cardiomyoblasts and in vivo cardiomyocytes expressing MFN2 Q400 had mitophagy defects with increased sensitivity to doxorubicin. MFN2 R400Q is the first known natural mitophagy-defective MFN2 mutant. Its unique profile of dysfunction evokes mitophagic cardiomyopathy, suggesting a mechanism for enrichment in clinical cardiomyopathy.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
The central claim that the R400Q mutation causes cardiomyopathy in humans require(s) additional support.
We regret that the reviewer interpreted our conclusions as described. Because of the extreme rarity of the MFN2 R400Q mutation our clinical data are unavoidably limited and therefore insufficient to support a conclusion that it causes cardiomyopathy “in humans”. Importantly, this is a claim that we did not make and do not believe to be the case. Our data establish that the MFN2 R400Q mutation is sufficient to cause lethal cardiomyopathy in some mice (Q/Q400a; Figure 4) and predisposes to doxorubicin-induced cardiomyopathy in the survivors (Q/Q400n; new data, Figure 7). Based on the clinical association we propose that R400Q may act as a genetic risk modifier in human cardiomyopathy.
To …
Author Response
Reviewer #1 (Public Review):
The central claim that the R400Q mutation causes cardiomyopathy in humans require(s) additional support.
We regret that the reviewer interpreted our conclusions as described. Because of the extreme rarity of the MFN2 R400Q mutation our clinical data are unavoidably limited and therefore insufficient to support a conclusion that it causes cardiomyopathy “in humans”. Importantly, this is a claim that we did not make and do not believe to be the case. Our data establish that the MFN2 R400Q mutation is sufficient to cause lethal cardiomyopathy in some mice (Q/Q400a; Figure 4) and predisposes to doxorubicin-induced cardiomyopathy in the survivors (Q/Q400n; new data, Figure 7). Based on the clinical association we propose that R400Q may act as a genetic risk modifier in human cardiomyopathy.
To avoid further confusion we modified the manuscript title to “A human mitofusin 2 mutation can cause mitophagic cardiomyopathy” and provide a more detailed discussion of the implications and limitations of our study on page 11).
First, the claim of an association between the R400Q variant (identified in three individuals) and cardiomyopathy has some limitations based on the data presented. The initial association is suggested by comparing the frequency of the mutation in three small cohorts to that in a large database gnomAD, which aggregates whole exome and whole genome data from many other studies including those from specific disease populations. Having a matched control population is critical in these association studies.
We have added genotyping data from the matched non-affected control population (n=861) of the Cincinnati Heart study to our analyses (page 4). The conclusions did not change.
For instance, according to gnomAD the MFN2 Q400P variant, while not observed in those of European ancestry, has a 10-fold higher frequency in the African/African American and South Asian populations (0.0004004 and 0.0003266, respectively). If the authors data in table one is compared to the gnomAD African/African American population the p-value drops to 0.029262, which would not likely survive correction for multiple comparison (e.g., Bonferroni).
Thank you for raising the important issue of racial differences in mutant allele prevalence and its association with cardiomyopathy. Sample size for this type of sub-group analysis is limited, but we are able to provide African-derived population allele frequency comparisons for both the gnomAD population and our own non-affected control group.
As now described on page 4, and just as with the gnomAD population we did not observe MFN2 R400Q in any Caucasian individuals, either cardiomyopathy or control. Its (heterozygous only) prevalence in African American cardiomyopathy is 3/674. Thus, the R400Q minor allele frequency of 3/1,345 in AA cardiomyopathy compares to 10/24,962 in African gnomAD, reflecting a statistically significant increase in this specific population group (p=0.003308; Chi2 statistic 8.6293). Moreover, all African American non-affected controls in the case-control cohort were wild-type for MFN2 (0/452 minor alleles).
(The source and characteristics of the subjects used by the authors in Table 1 is not clear from the methods.)
The details of our study cohorts were inadvertently omitted during manuscript preparation. As now reported on pages 3 and 4, the Cincinnati Heart Study is a case-control study consisting of 1,745 cardiomyopathy (1,117 Caucasian and 628 African American) subjects and 861 non-affected controls (625 Caucasian and 236 African American) (Liggett et al Nat Med 2008; Matkovich et al JCI 2010; Cappola et al PNAS 2011). The Houston hypertrophic cardiomyopathy cohort [which has been screened by linkage analysis, candidate gene sequencing or clinical genetic testing) included 286 subjects (240 Caucasians and 46 African Americans) (Osio A et al Circ Res 2007; Li L et al Circ Res 2017).
Relatedly, evaluation in a knock-in mouse model is offered as a way of bolstering the claim for an association with cardiomyopathy. Some caution should be offered here. Certain mutations have caused a cardiomyopathy in mice when knocked in have not been observed in humans with the same mutation. A recent example is the p.S59L variant in the mitochondrial protein CHCHD10, which causes cardiomyopathy in mice but not in humans (PMID: 30874923). While phenocopy is suggestive there are differences in humans and mice, which makes the correlation imperfect.
We understand that a mouse is not a man, and as noted above we view the in vitro data in multiple cell systems and the in vivo data in knock-in mice as supportive for, not proof of, the concept that MFN2 R400Q can be a genetic cardiomyopathy risk modifier. As indicated in the following responses, we have further strengthened the case by including results from 2 additional, previously undescribed human MFN2 mutation knock-in mice.
Additionally, the argument that the Mfn2 R400Q variant causes a dominant cardiomyopathy in humans would be better supported by observing of a cardiomyopathy in the heterozygous Mfn2 R400Q mice and not just in the homozygous Mfn2 R400Q mice.
We are intrigued that in the previous comment the reviewer warns that murine phenocopies are not 100% predictive of human disease, and in the next sentence he/she requests that we show that the gene dose-phenotype response is the same in mice and humans. And, we again wish to note that we never argued that MFN2 R400Q “causes a dominant cardiomyopathy in humans.” Nevertheless, we understand the underlying concerns and in the revised manuscript we present data from new doxorubicin challenge experiments comparing cardiomyopathy development and myocardial mitophagy in WT, heterozygous, and surviving (Q/Q400n) homozygous Mfn2 R400Q KI mice (new Figure 7, panels E-G). Homozygous, but not heterozygous, R400Q mice exhibited an amplified cardiomyopathic response (greater LV dilatation, reduced LV ejection performance, exaggerated LV hypertrophy) and an impaired myocardial mitophagic response to doxorubicin. These in vivo data recapitulate new in vitro results in H9c2 rat cardiomyoblasts expressing MFN2 R400Q, which exhibited enhanced cytotoxicity (cell death and TUNEL labelling) to doxorubicin associated with reduced reactive mitophagy (Parkin aggregation and mitolysosome formation) (new Figure 7, panels A-D). Thus, under the limited conditions we have explored to date we do not observe cardiomyopathy development in heterozygous Mfn2 R400Q KI mice. However, we have expanded the association between R400Q, mitophagy and cardiomyopathy thereby providing the desired additional support for our argument that it can be a cardiomyopathy risk modifier.
Relatedly, it is not clear what the studies in the KI mouse prove over what was already known. Mfn2 function is known to be essential during the neonatal period and the authors have previously shown that the Mfn2 R400Q disrupts the ability of Mfn2 to mediate mitochondrial fusion, which is its core function. The results in the KI mouse seem consistent with those two observations, but it's not clear how they allow further conclusions to be drawn.
We strenuously disagree with the underlying proposition of this comment, which is that “mitochondrial fusion (is the) core function” of mitofusins. We also believe that our previous work, alluded to but not specified, is mischaracterized.
Our seminal study defining an essential role for Mfn2 for perinatal cardiac development (Gong et al Science 2015) reported that an engineered MFN2 mutation that was fully functional for mitochondrial fusion, but incapable of binding Parkin (MFN2 AA), caused perinatal cardiomyopathy when expressed as a transgene. By contrast, another engineered MFN2 mutant transgene that potently suppressed mitochondrial fusion, but constitutively bound Parkin (MFN2 EE) had no adverse effects on the heart.
Our initial description of MFN2 R400Q and observation that it exhibited impaired fusogenicity (Eschenbacher et al PLoS One 2012) reported results of in vitro studies and transgene overexpression in Drosophila. Importantly, a role for MFN2 in mitophagy was unknown at that time and so was not explored.
A major point both of this manuscript and our work over the last decade on mitofusin proteins has been that their biological importance extends far beyond mitochondrial fusion. As introduced/discussed throughout our manuscript, MFN2 plays important roles in mitophagy and mitochondrial motility. Because this central point seems to have been overlooked, we have gone to great lengths in the revised manuscript to unambiguously show that impaired mitochondrial fusion is not the critical functional aspect that determines disease phenotypes caused by Mfn2 mutations. To accomplish this we’ve re-structured the experiments so that R400Q is compared at every level to two other natural MFN2 mutations linked to a human disease, the peripheral neuropathy CMT2A. These comparators are MFN2 T105M in the GTPase domain and MFN2 M376A/V in the same HR1 domain as MFN2 R400Q. Each of these human MFN2 mutations is fusion-impaired, but the current studies reveal that that their spectrum of dysfunction differs in other ways as summarized in Author response table 1:
Author response table 1.
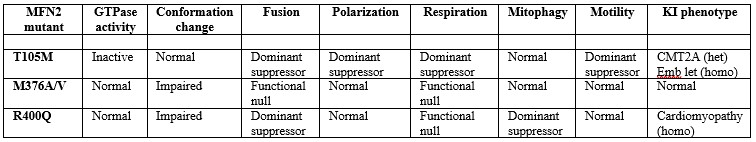
We understand that it sounds counterintuitive for a mutation in a “mitofusin” protein to evoke cardiac disease independent of its appellative function, mitochondrial fusion. But the KI mouse data clearly relate the occurrence of cardiomyopathy in R400Q mice to the unique mitophagy defect provoked in vitro and in vivo by this mutation. We hope the reviewer will agree that the KI models provide fresh scientific insight.
Additionally, the authors conclude that the effect of R400Q on the transcriptome and metabolome in a subset of animals cannot be explained by its effect on OXPHOS (based on the findings in Figure 4H). However, an alternative explanation is that the R400Q is a loss of function variant but does not act in a dominant negative fashion. According to this view, mice homozygous for R400Q (and have no wildtype copies of Mfn2) lack Mfn2 function and consequently have an OXPHOS defect giving rise to the observed transcriptomic and metabolomic changes. But in the rat heart cell line with endogenous rat Mfn2, exogenous of the MFN2 R400Q has no effect as it is loss of function and is not dominant negative.
Our results in the original submission, which are retained in Figures 1D and 1E and Figure 1 Figure Supplement 1 of the revision, exclude the possibility that R400Q is a functional null mutant for, but not a dominant suppressor of, mitochondrial fusion. We have added additional data for M376A in the revision, but the original results are retained in the main figure panels and a new supplemental figure:
Figure 1D reports results of mitochondrial elongation studies (the morphological surrogate for mitochondrial fusion) performed in Mfn1/Mfn2 double knock-out (DKO) MEFs. The baseline mitochondrial aspect ratio in DKO cells infected with control (b-gal containing) virus is ~2 (white bar), and increases to ~6 (i.e. ~normal) by forced expression of WT MFN2 (black bar). By contrast, aspect ratio in DKO MEFs expressing MFN2 mutants T105M (green bar), M376A and R400Q (red bars in main figure), R94Q and K109A (green bars in the supplemental figure) is only 3-4. For these results the reviewer’s and our interpretation agree: all of the MFN2 mutants studied are non-functional as mitochondrial fusion proteins.
Importantly, Figure 1E (left panel) reports the results of parallel mitochondrial elongation studies performed in WT MEFs, i.e. in the presence of normal endogenous Mfn1 and Mfn2. Here, baseline mitochondrial aspect ratio is already normal (~6, white bar), and increases modestly to ~8 when WT MFN2 is expressed (black bar). By comparison, aspect ratio is reduced below baseline by expression of four of the five MFN2 mutants, including MFN2 R400Q (main figure and accompanying supplemental figure; green and red bars). Only MFN2 M376A failed to suppress mitochondrial fusion promoted by endogenous Mfns 1 and 2. Thus, MFN2 R400Q dominantly suppresses mitochondrial fusion. We have stressed this point in the text on page 5, first complete paragraph.
Additionally, as the authors have shown MFN2 R400Q loses its ability to promote mitochondrial fusion, and this is the central function of MFN2, it is not clear why this can't be the explanation for the mouse phenotype rather than the mitophagy mechanism the authors propose.
Please see our response #7 above beginning “We strenuously disagree...”
Finally, it is asserted that the MFN2 R400Q variant disrupts Parkin activation, by interfering with MFN2 acting a receptor for Parkin. The support for this in cell culture however is limited. Additionally, there is no assessment of mitophagy in the hearts of the KI mouse model.
The reviewer may have overlooked the studies reported in original Figure 5, in which Parkin localization to cultured cardiomyoblast mitochondria is linked both to mitochondrial autophagy (LC3-mitochondria overlay) and to formation of mito-lysosomes (MitoQC staining). These results have been retained and expanded to include MFN2 M376A in Figure 6 B-E and Figure 6 Figure Supplement 1 of the revised manuscript. Additionally, selective impairment of Parkin recruitment to mitochondria was shown in mitofusin null MEFs in current Figure 3C and Figure 3 Figure Supplement 1, panels B and C.
The in vitro and in vivo doxorubicin studies performed for the revision further strengthen the mechanistic link between cardiomyocyte toxicity, reduced parkin recruitment and impaired mitophagy in MFN2 R400Q expressing cardiac cells: MFN2 R400Q-amplified doxorubicin-induced H9c2 cell death is associated with reduced Parkin aggregation and mitolysosome formation in vitro, and the exaggerated doxorubicin-induced cardiomyopathic response in MFN2 Q/Q400 mice was associated with reduced cardiomyocyte mitophagy in vivo, measured with adenoviral Mito-QC (new Figure 7).
Reviewer #2 (Public Review):
In this manuscript, Franco et al show that the mitofusin 2 mutation MFN2 Q400 impaires mitochondrial fusion with normal GTPase activity. MFN2 Q400 fails to recruit Parkin and further disrupts Parkin-mediated mitophagy in cultured cardiac cells. They also generated MFN2 Q400 knock-in mice to show the development of lethal perinatal cardiomyopathy, which had an impairment in multiple metabolic pathways.
The major strength of this manuscript is the in vitro study that provides a thorough understanding in the characteristics of the MFN2 Q400 mutant in function of MFN2, and the effect on mitochondrial function. However, the in vivo MFN2 Q/Q400 knock-in mice are more troubling given the split phenotype of MFN2 Q/Q400a vs MFN2 Q/Q400n subtypes. Their main findings towards impaired metabolism in mutant hearts fail to distinguish between the two subtypes.
Thanks for the comments. We do not fully understand the statement that “impaired metabolism in mutant hearts fails to distinguish between the two (in vivo) subtypes.” The data in current Figure 5 and its accompanying figure supplements show that impaired metabolism measured both as metabolomic and transcriptomic changes in the subtypes (orange Q400n vs red Q400a in Figure 5 panels A and D) are reflected in the histopathological analyses. Moreover, newly presented data on ROS-modifying pathways (Figure 5C) suggest that a central difference between Mfn2 Q/Q400 hearts that can compensate for the underlying impairment in mitophagic quality control (Q400n) vs those that cannot (Q400a) is the capacity to manage downstream ROS effects of metabolic derangements and mitochondrial uncoupling. Additional support for this idea is provided in the newly performed doxorubicin challenge experiments (Figure 7), demonstrating that mitochondrial ROS levels are in fact increased at baseline in adult Q400n mice.
While the data support the conclusion that MFN2 Q400 causes cardiomyopathy, several experiments are needed to further understand mechanism.
We thank the reviewer for agreeing with our conclusion that MFN2 Q400 can cause cardiomyopathy, which was the major issue raised by R1. As detailed below we have performed a great deal of additional experimentation, including on two completely novel MFN2 mutant knock-in mouse models, to validate the underlying mechanism.
This manuscript will likely impact the field of MFN2 mutation-related diseases and show how MFN2 mutation leads to perinatal cardiomyopathy in support of previous literature.
Thank you again. We think our findings have relevance beyond the field of MFN2 mutant-related disease as they provide the first evidence (to our knowledge) that a naturally occurring primary defect in mitophagy can manifest as myocardial disease.
-
eLife assessment
In this paper, the authors demonstrate an interesting link between mitofusin function (MFN2) and PARKIN recruitment and mitophagy, underlying cardiomyopathy. This is a valuable finding with broad implications for understanding the mitochondrial biology as well as mechanisms involved in heart pathologies. However, the analyses are incomplete and the main conclusions are only partially supported and need to be further evidenced.
-
Reviewer #1 (Public Review):
In their manuscript titled "A human mitofusion 2 mutation causes mitophagic cardiomyopathy", Franco et al suggest that a rare mutation in MFN2 (R400Q) is over-represented in patients with cardiomyopathy, causes loss of conformational malleability, leading to mitochondrial fusion defects, impaired Parkin recruitment to mitochondria, and suppressed MFN2-Parkin mediated mitophagy. This work is an extension of previous work from the same group that found the MFN2 R400Q mutation is loss of function in a Drosophila model. Unlike MFN2 R94Q and T105M that cause Charcot-Marie-Tooth disease type 2 A, the MFN2 R400Q mutant has normal GTPase activity and mitochondrial electrochemical integrity, motility, and respiration. MFN2 R400Q knock-in mice exhibit cardiac-specific phenotypes.
Strengths include detailed …
Reviewer #1 (Public Review):
In their manuscript titled "A human mitofusion 2 mutation causes mitophagic cardiomyopathy", Franco et al suggest that a rare mutation in MFN2 (R400Q) is over-represented in patients with cardiomyopathy, causes loss of conformational malleability, leading to mitochondrial fusion defects, impaired Parkin recruitment to mitochondria, and suppressed MFN2-Parkin mediated mitophagy. This work is an extension of previous work from the same group that found the MFN2 R400Q mutation is loss of function in a Drosophila model. Unlike MFN2 R94Q and T105M that cause Charcot-Marie-Tooth disease type 2 A, the MFN2 R400Q mutant has normal GTPase activity and mitochondrial electrochemical integrity, motility, and respiration. MFN2 R400Q knock-in mice exhibit cardiac-specific phenotypes.
Strengths include detailed characterization of the MFN2 R400Q variant in variety of models, including cell models and novel knock-in mouse model.
However, there are some weaknesses. The central claim that the R400Q mutation causes cardiomyopathy in humans and the claim that the pathogenetic mechanism is decreased mitophagy require additional support.First, the claim of an association between the R400Q variant (identified in three individuals) and cardiomyopathy has some limitations based on the data presented. The initial association is suggested by comparing the frequency of the mutation in three small cohorts to that in a large database gnomAD, which aggregates whole exome and whole genome data from many other studies including those from specific disease populations. Having a matched control population is critical in these association studies. For instance, according to gnomAD the MFN2 Q400P variant, while not observed in those of European ancestry, has a 10-fold higher frequency in the African/African American and South Asian populations (0.0004004 and 0.0003266, respectively). If the authors data in table one is compared to the gnomAD African/African American population the p-value drops to 0.029262, which would not likely survive correction for multiple comparison (e.g., Bonferroni). (The source and characteristics of the subjects used by the authors in Table 1 is not clear from the methods.)
Relatedly, evaluation in a knock-in mouse model is offered as a way of bolstering the claim for an association with cardiomyopathy. Some caution should be offered here. Certain mutations have caused a cardiomyopathy in mice when knocked in have not been observed in humans with the same mutation. A recent example is the p.S59L variant in the mitochondrial protein CHCHD10, which causes cardiomyopathy in mice but not in humans (PMID: 30874923). While phenocopy is suggestive there are differences in humans and mice, which makes the correlation imperfect.
Additionally, the argument that the Mfn2 R400Q variant causes a dominant cardiomyopathy in humans would be better supported by observing of a cardiomyopathy in the heterozygous Mfn2 R400Q mice and not just in the homozygous Mfn2 R400Q mice. Relatedly, it is not clear what the studies in the KI mouse prove over what was already known. Mfn2 function is known to be essential during the neonatal period and the authors have previously shown that the Mfn2 R400Q disrupts the ability of Mfn2 to mediate mitochondrial fusion, which is its core function. The results in the KI mouse seem consistent with those two observations, but it's not clear how they allow further conclusions to be drawn.
Additionally, the authors conclude that the effect of R400Q on the transcriptome and metabolome in a subset of animals cannot be explained by its effect on OXPHOS (based on the findings in Figure 4H). However, an alternative explanation is that the R400Q is a loss of function variant but does not act in a dominant negative fashion. According to this view, mice homozygous for R400Q (and have no wildtype copies of Mfn2) lack Mfn2 function and consequently have an OXPHOS defect giving rise to the observed transcriptomic and metabolomic changes. But in the rat heart cell line with endogenous rat Mfn2, exogenous of the MFN2 R400Q has no effect as it is loss of function and is not dominant negative. Additionally, as the authors have shown MFN2 R400Q loses its ability to promote mitochondrial fusion, and this is the central function of MFN2, it is not clear why this can't be the explanation for the mouse phenotype rather than the mitophagy mechanism the authors propose.
Finally, it is asserted that the MFN2 R400Q variant disrupts Parkin activation, by interfering with MFN2 acting a receptor for Parkin. The support for this in cell culture however is limited. Additionally, there is no assessment of mitophagy in the hearts of the KI mouse model.
-
Reviewer #2 (Public Review):
In this manuscript, Franco et al show that the mitofusin 2 mutation MFN2 Q400 impaires mitochondrial fusion with normal GTPase activity. MFN2 Q400 fails to recruit Parkin and further disrupts Parkin-mediated mitophagy in cultured cardiac cells. They also generated MFN2 Q400 knock-in mice to show the development of lethal perinatal cardiomyopathy, which had an impairment in multiple metabolic pathways.
The major strength of this manuscript is the in vitro study that provides a thorough understanding in the characteristics of the MFN2 Q400 mutant in function of MFN2, and the effect on mitochondrial function. However, the in vivo MFN2 Q/Q400 knock-in mice are more troubling given the split phenotype of MFN2 Q/Q400a vs MFN2 Q/Q400n subtypes. Their main findings towards impaired metabolism in mutant hearts fail …
Reviewer #2 (Public Review):
In this manuscript, Franco et al show that the mitofusin 2 mutation MFN2 Q400 impaires mitochondrial fusion with normal GTPase activity. MFN2 Q400 fails to recruit Parkin and further disrupts Parkin-mediated mitophagy in cultured cardiac cells. They also generated MFN2 Q400 knock-in mice to show the development of lethal perinatal cardiomyopathy, which had an impairment in multiple metabolic pathways.
The major strength of this manuscript is the in vitro study that provides a thorough understanding in the characteristics of the MFN2 Q400 mutant in function of MFN2, and the effect on mitochondrial function. However, the in vivo MFN2 Q/Q400 knock-in mice are more troubling given the split phenotype of MFN2 Q/Q400a vs MFN2 Q/Q400n subtypes. Their main findings towards impaired metabolism in mutant hearts fail to distinguish between the two subtypes.
While the data support the conclusion that MFN2 Q400 causes cardiomyopathy, several experiments are needed to further understand mechanism. This manuscript will likely impact the field of MFN2 mutation-related diseases and show how MFN2 mutation leads to perinatal cardiomyopathy in support of previous literature.
-
