The impact of local genomic properties on the evolutionary fate of genes
Curation statements for this article:-
Curated by eLife

eLife Assessment:
This study is fundamental to understanding the intrinsic driving forces of gene losses during mammalian genome evolution, linking the propensity for gene losses to the local genomic features such as mutation rate and spatially restricted expression. In general, the study is methodologically convincing because independent gene losses in at least two mammalian lineages were identified as "elusive human genes". However, additional (comparative genomics and statistical) analyses would make the current study more rigorous. This manuscript will appeal to readers interested in the evolutionary fates of genes across the phylogenetic tree.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Functionally indispensable genes are likely to be retained and otherwise to be lost during evolution. This evolutionary fate of a gene can also be affected by factors independent of gene dispensability, including the mutability of genomic positions, but such features have not been examined well. To uncover the genomic features associated with gene loss, we investigated the characteristics of genomic regions where genes have been independently lost in multiple lineages. With a comprehensive scan of gene phylogenies of vertebrates with a careful inspection of evolutionary gene losses, we identified 813 human genes whose orthologs were lost in multiple mammalian lineages: designated ‘elusive genes.’ These elusive genes were located in genomic regions with rapid nucleotide substitution, high GC content, and high gene density. A comparison of the orthologous regions of such elusive genes across vertebrates revealed that these features had been established before the radiation of the extant vertebrates approximately 500 million years ago. The association of human elusive genes with transcriptomic and epigenomic characteristics illuminated that the genomic regions containing such genes were subject to repressive transcriptional regulation. Thus, the heterogeneous genomic features driving gene fates toward loss have been in place and may sometimes have relaxed the functional indispensability of such genes. This study sheds light on the complex interplay between gene function and local genomic properties in shaping gene evolution that has persisted since the vertebrate ancestor.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
Weaknesses:
Gene expression level as a confounding factor was not well controlled throughout the study. Higher gene expression often makes genes less dispensable after gene duplication. Gene expression level is also a major determining factor of evolutionary rates (reviewed in http://www.ncbi.nlm.nih.gov/pubmed/26055156). Some proposed theories explain why gene expression level can serve as a proxy for gene importance (http://www.ncbi.nlm.nih.gov/pubmed/20884723, http://www.ncbi.nlm.nih.gov/pubmed/20485561). In that sense, many genomic/epigenomic features (such as replication timing and repressed transcriptional regulation) that were assumed "neutral" or intrinsic by the authors (or more accurately, independent of gene dispensability) cannot be easily distinguishable from the effect of …
Author Response
Reviewer #1 (Public Review):
Weaknesses:
Gene expression level as a confounding factor was not well controlled throughout the study. Higher gene expression often makes genes less dispensable after gene duplication. Gene expression level is also a major determining factor of evolutionary rates (reviewed in http://www.ncbi.nlm.nih.gov/pubmed/26055156). Some proposed theories explain why gene expression level can serve as a proxy for gene importance (http://www.ncbi.nlm.nih.gov/pubmed/20884723, http://www.ncbi.nlm.nih.gov/pubmed/20485561). In that sense, many genomic/epigenomic features (such as replication timing and repressed transcriptional regulation) that were assumed "neutral" or intrinsic by the authors (or more accurately, independent of gene dispensability) cannot be easily distinguishable from the effect of gene dispersibility.
We thank the reviewer for this important comment. We totally agree that transcriptomic and epigenomic features cannot be easily distinguished from gene dispensability and do not think that these features of the elusive genes can be explained solely by intrinsic properties of the genomes. Our motivation for investigating the expression profiles of the elusive gene is to understand how they lost their functional indispensability (original manuscript L285-286 in Results). We also discussed the possibility that sequence composition and genomic location of elusive genes may be associated with epigenetic features for expression depression, which may result in a decrease of functional constraints (original manuscript L470-474 in Discussion). Nevertheless, we think that the original manuscript may have contained misleading wordings, and thus we have edited them to better convey our view that gene expression and epigenomic features are related to gene function.
(P.2, Introduction) This evolutionary fate of a gene can also be affected by factors independent of gene dispensability, including the mutability of genomic positions, but such features have not been examined well.
(P6, Introduction) These data assisted us to understand how intrinsic genomic features may affect gene fate, leading to gene loss by decreasing the expression level and eventually relaxing the functional importance of ʻelusiveʼ genes.
(P33, Discussion) Another factor is the spatiotemporal suppression of gene expression via epigenetic constraints. Previous studies showed that lowly expressed genes reduce their functional dispensability (Cherry, 2010; Gout et al., 2010), and so do the elusive genes.
Additionally, responding to the advices from Reviewers 1 and 2 [Rev1minor7 and Rev2-Major4], we have added a new section Elusive gene orthologs in the chicken microchromosomes in which we describe the relationship between the elusive genes and chicken microchromosomes. In this section, we also argue for the relationship between the genomic feature of the elusive genes and their transcriptomic and epigenomic characteristics. In the chicken genome, elusive genes did not show reduced pleiotropy of gene expression nor the epigenetic features relevant with the reduction, consistently with the moderation of nucleotide substitution rates. This also suggests that the relaxation of the ‘elusiveness’ is associated with the increase of functional indispensability.
(P27, Elusive gene orthologs in the chicken microchromosomes in Results) Our analyses indicates that the genomic features of the elusive genes such as high GC and high nucleotide substitutions do not always correlate with a reduction in pleiotropy of gene expression that potentially leads to an increase in functional dispensability, although these features have been well conserved across vertebrates. In addition, the avian orthologs of the elusive genes did not show higher KA and KS values than those of the non-elusive genes (Figure 3; Figure 3–figure supplement 1), likely consistent with similar expression levels between them (Figure 5–figure supplement 1) (Cherry, 2010; Zhang and Yang, 2015). With respect to the chicken genome, the sequence features of the elusive genes themselves might have been relaxed during evolution.
Ks was used by the authors to indicate mutation rates. However, synonymous mutations substantially affect gene expression levels (https://pubmed.ncbi.nlm.nih.gov/25768907/, https://pubmed.ncbi.nlm.nih.gov/35676473/). Thus, synonymous mutations cannot be simply assumed as neutral ones and may not be suitable for estimating local mutation rates. If introns can be aligned, they are better sequences for estimating the mutability of a genomic region.
We appreciate the reviewer for this meaningful suggestion. As a response, we have computed the differences in intron sequences between the human and chimpanzee genomes and compared them between the elusive and non-elusive genes. As expected, we found larger sequence differences in introns for the elusive genes than for the non-elusive genes. In Figure 2c of the revised manuscript, we have included the distribution of KI, sequence differences in introns between the human and chimpanzee genomes for the elusive and non-elusive genes. Additionally, we have added the corresponding texts to Results and the procedure to Methods as shown below.
(P11, Identification of human ‘elusive’ genes in Results) In addition, we computed nucleotide substitution rates for introns (KI) between human and chimpanzee (Pan troglodytes) orthologs and compared them between the elusive and non-elusive genes.
(P11, Identification of human ‘elusive’ genes in Results) Our analysis further illuminated larger KS and KI values for the elusive genes than in the non-elusive genes (Figure 2b, c; Figure 2–figure supplement 1). Importantly, the higher rate of synonymous and intronic nucleotide substitutions, which may not affect changes in amino acid residues, indicates that the elusive genes are also susceptible to genomic characteristics independent of selective constraints on gene functions.
(P39, Methods) To compute nucleotide sequence differences of the individual introns, we extracted 473 elusive and 4,626 non-elusive genes that harbored introns aligned with the chimpanzee genome assembly. The nucleotide differences were calculated via the whole genome alignments of hg38 and panTro6 retrieved from the UCSC genome browser.
The term "elusive gene" is not necessarily intuitive to readers.
We previously published a paper reporting the group of genes that we refer to as ‘elusive genes,’ lost in mammals and aves independently but retained by reptiles, in the gecko genome assembly (Hara et al., 2018, BMC Biology). We initially termed them with a more intuitive name (‘loss-prone genes’) but changed it because one of our peer-reviewers did not agree to use this name. Later on, we have continuously used this term in another paper (Hara et al., 2018, Nat. Ecol. Evol.). In addition, some other groups have used the word ‘elusive’ with a similar intention to ours (Prokop et al, 2014, PLOS ONE, doi: 10.1371/journal.pone.0092751; Ribas et al., 2011, BMC Genomics, doi: 10.1186/1471-2164-12-240). We would appreciate the reviewer’s understanding of this naming to ensure the consistency of our researches on gene loss. In the revised manuscript, we have added sentences to provide a more intuitive guide to ‘elusive genes’,
(P6, Introduction) We previously referred to the nature of genes prone to loss as ‘elusive’(Hara et al., 2018a, 2018b). In the present study, we define the elusive genes as those that are retained by modern humans but have been lost independently in multiple mammalian lineages. As a comparison of the elusive genes, we retrieved the genes that were retained by almost all of the mammalian species examined and defined them as ‘non-elusive’, representing those persistent in the genomes.
Reviewer #3 (Public Review):
Overall, the study is descriptive and adds incremental evidence to an existing body of extensive gene loss literature. The topic is specialised and will be of interest to a niche audience. The text is highly redundant, repeating the same false positive issue in the introduction, methods, and discussion sections, while no clear conclusion or interpretation of their main findings are presented.
Major comments
While some of the false discovery rate issues of gene loss detection were addressed in the presented pipeline, the authors fail to test one of the most severe cases of mis-annotating gene loss events: frameshift mutations which cause gene annotation pipelines to fail reporting these genes in the first place. Running a blastx or diamond blastx search of their elusive and non-elusive gene sets against all other genomes, should further enlighten the robustness of their gene loss detection approach
For the revised manuscript, we have refined the elusive gene set as the reviewer suggested. In the genome assemblies, we have searched for the orthologs of the elusive genes for the species in which they were missing. The search has been conducted by querying amino acid sequences of the elusive genes with tblastn as well as MMSeqs2 that performed superior to tblastn in sensitivity and computational speed. In addition, regarding another comment by Reviewer 3. we have searched for the orthologs by referring to existing ortholog annotations. We used the ortholog annotations implemented in RefSeq instead of those from the TOGA pipeline: both employ synteny conservation. We have coordinated the identified orthologs with our gene loss criteria–absence from all the species used in a particular taxon–and excluded 268 genes from the original elusive gene set. These genes contain those missing in the previous gene annotations used in the original manuscript but present in the latest ones, as well as those falsely missing due to incorrect inference of gene trees. Finally, the refined set of 813 elusive genes were subject to comparisons with the non-elusive genes. Importantly, these comparisons retained the significantly different trends of the particular genomic, transcriptomic, and epigenomic features between them except for very few cases (Table R1 included below). This indicates that both initial and revised sets of the elusive genes reflect the nature of the ‘elusiveness,’ though the initial set contained some noises. We have modified the numbers of elusive genes in the corresponding parts of the manuscript including figures and tables. Additionally, we have added the validation procedures in Methods.
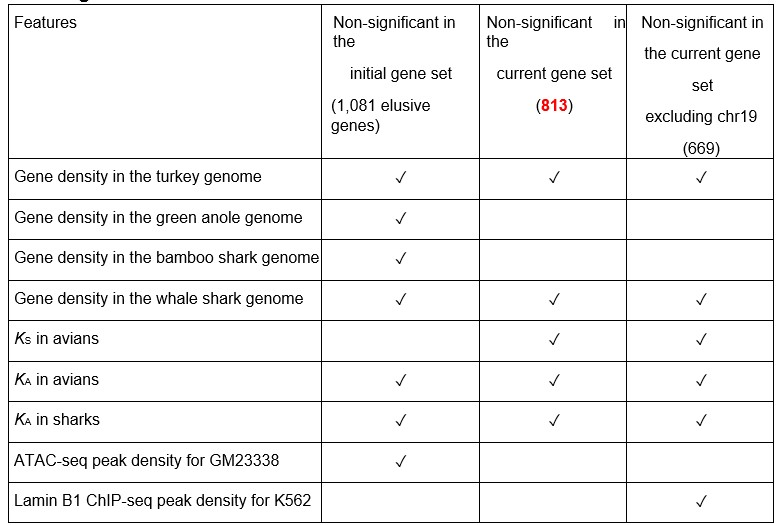
Table R1. Difference in statistical significances across different elusive gene sets *The other features showed significantly different trends between the elusive and non-elusive genes for all of the elusive gene sets and thus are not included in this table.
(P38 in Methods) The gene loss events inferred by molecular phylogeny were further assessed by synteny-based ortholog annotations implemented in RefSeq, as well as a homolog search in the genome assemblies (Table S2) with TBLASTN v2.11.0+ (Altschul et al., 1997) and MMSeqs2 (Steinegger and Söding, 2017) referring to the latest RefSeq gene annotations (last accessed on 2 Dec, 2022). This procedure resulted in the identification of 813 elusive genes that harbored three or fewer duplicates. Similarly, we extracted 8,050 human genes whose orthologs were found in all the mammalian species examined and defined them as non-elusive genes.
The reviewer also suggested us investigating falsely-missing genes due to frameshift mutations (in this case we guess that the reviewer assumed the genome assembly that falsely included frameshift mutations). This requires us to search for the orthologs by revisiting the sequencing reads because the frameshift is sometimes caused by indels of erroneous basecalling. We have selected five elusive genes and searched for the fragments of orthologs in sequencing reads for the species in which they are missing. We have retrieved sequencing reads corresponding to the genome assemblies from NCBI SRA and performed sequence similarity search using the program Diamond against the amino acid sequences of the elusive genes and could not find the frameshift that potentially causes the mis-annotation of the elusive genes.
Along this line, we noticed that when annotation files were pooled together via CD-Hit clustering, a 100% identity threshold was chosen (Methods). Since some of the pooled annotations were drawn from less high quality assemblies which yield higher likelihoods of mismatches between annotations, enforcing a 100% identity threshold will artificially remove genes due to this strict constraint. It will be paramount for this study to test the robustness of their findings when 90% and 95% identity thresholds were selected.
cd-hit clustering with 100% sequence identity only clusters those with identical (and sometimes truncated) sequences, and, in the cluster, the sequences other than the representative are discarded. This means that the sequences remain if they are not identical to the other ones. If the similarity threshold is lowered, both identical and highly similar sequences are clustered with each other, and more sequences are discarded. Therefore, our approach that employs clustering with 100% similarity may minimize false positive gene loss.
While some statistical tests were applied (although we do recommend consulting a professional statistician, since some identical distributions tend to show significantly low p-values), the authors fail to discuss the fact that their elusive gene set comprises of ~5% of all human genes (assuming 21,000 genes), while their non-elusive set represents ~40% of all genes. In other words, the authors compare their sequence and genomic features against the genomic background rather than a biological signal (nonelusiveness). An analysis whereby 1,081 genes (same number as elusive set) are randomly sampled from the 21,000 gene pool is compared against the elusive and non-elusive distributions for all presented results will reveal whether the non-elusive set follows a background distribution (noise) or not.
Our study aims to elucidate the characteristics of genes that differentiate their fates, retention or loss. To achieve this, we put this characterization into the comparison between the elusive and non-elusive genes. This comparison highlighted clearly different phylogenetic signals for gene loss between elusive and non-elusive genes, allowing us to extract the features associated with the loss-prone nature. The random sampling set suggested by Reviewer may largely consists of the remainders that were not classified by the elusive and non-elusive genes. However, these remainders may contain a considerable number of genes with distinctive phylogenetic signatures rather than the intermediates between the elusive and nonelusive genes: the genes with multiple loss events in more restricted taxa than our criterion, the ones with frequent duplication, etc. Therefore, we think that a comparison of the elusive genes with the random-sampling set does not achieve our objective: the comparison of the clearly different phylogenetic signals.
We also wondered whether the authors considered testing the links between recombination rate / LD and the genomic locations of their elusive genes (again compared against randomly sampled genes)?
We have retrieved fine-scale recombination rate data of males and females from https://www.decode.com/addendum/ (Suppl. Data of Kong, A et al., Nature, 467:1099–1103, 2010) and have compared them between the gene regions of the elusive and non-elusive genes. Both comparisons show no significant differences: average 0.829 and 0.900 recombinations/kb for the elusive and non-elusive genes, respectively, p=0.898, for males; average 0.836 and 0.846 recombinations/kb for the elusive and non-elusive genes, respectively, p=0.256, for females).
Given the evidence presented in Figure 6b, we do not agree with the statement (l.334-336): "These observations suggest that the elusive genes are unlikely to be regulated by distant regulatory elements". Here, a data population of ~1k genes is compared against a data population of ~8k genes and the presented difference between distributions could be a sample size artefact. We strongly recommend retesting this result with the ~1k randomly sampled genes from the total ~21,000 gene pool and then compare the distributions.
Analogous random sampling analysis should be performed for Fig 6a,d
As described above, our study does not intend to extract signals from background. To make the comparison objectives clear, we have revised the corresponding sentence as below.
(P22, Transcriptomic natures of elusive genes in Results) These observations suggest that the elusive genes are unlikely to be regulated by distant regulatory elements compared with the non-elusive genes (Figure 6b).
We didn't see a clear pattern in Figure 7. Please quantify enrichments with statistical tests. Even if there are enriched regions, why did the authors choose a Shannon entropy cutoff configuration of <1 (low) and >1 (high)? What was the overall entropy value range? If the maximum entropy value was 10 or 100 or even more, then denoting <1 as low and >1 as high seems rather biased.
To use Figure 7 in a new section in Results, we have added an ideogram showing the distribution of the genes that retain the chicken orthologs in microchromosomes. In response to the comment by Reviewer 2, we have performed statistical tests and found that the elusive genes were significantly more abundant in orthologs in microchromosomes than the non-elusive genes. Furthermore, the observation that the elusive genes prefer to be located in gene-rich regions was already statistically supported (Figure 2f).
As shown in Figure 5, Shannon’s H' ranged from zero to approximately 4 (exact maximum value is 3.97) and 5 (5.11) for the GTEx and Descartes gene expression datasets, respectively. Although the threshold H'=1 was an arbitrarily set, we think that it is reasonable to classify the genes with high pleiotropy from those with low pleiotropy.
-
eLife Assessment:
This study is fundamental to understanding the intrinsic driving forces of gene losses during mammalian genome evolution, linking the propensity for gene losses to the local genomic features such as mutation rate and spatially restricted expression. In general, the study is methodologically convincing because independent gene losses in at least two mammalian lineages were identified as "elusive human genes". However, additional (comparative genomics and statistical) analyses would make the current study more rigorous. This manuscript will appeal to readers interested in the evolutionary fates of genes across the phylogenetic tree.
-
Reviewer #1 (Public Review):
In this study, Hara and Kuraku identified the genes lost multiple times across the mammalian phylogenetic tree and termed them "elusive genes." They then investigated the features of these elusive genes in the species where they are well preserved. The authors identified several genomic features that drive gene fates toward loss, in addition to the long-presumed functional dispensability. This analysis explains why some genes are more likely to lose during evolution than others.
This study extends the selection-mutation balance theory from nucleotide substitutions to gene losses. In the context of gene losses, functional dispensability determines the selective coefficient, and the genomic features determine the rate of gene loss mutations. While the selective force has been long presumed to be important, the …
Reviewer #1 (Public Review):
In this study, Hara and Kuraku identified the genes lost multiple times across the mammalian phylogenetic tree and termed them "elusive genes." They then investigated the features of these elusive genes in the species where they are well preserved. The authors identified several genomic features that drive gene fates toward loss, in addition to the long-presumed functional dispensability. This analysis explains why some genes are more likely to lose during evolution than others.
This study extends the selection-mutation balance theory from nucleotide substitutions to gene losses. In the context of gene losses, functional dispensability determines the selective coefficient, and the genomic features determine the rate of gene loss mutations. While the selective force has been long presumed to be important, the heterogenous genomic features that led to the mutability of gene losses were not carefully investigated in previous studies. This study fills this gap and shows that some genes are intrinsically prone to be lost (and why).
Strengths:
Identification of gene losses across the phylogenetic tree is not trivial, especially when considering the incompleteness of genomes. The authors conducted their bioinformatic analyses carefully and required two independent gene loss events, each supported by multiple species in a monophyletic group. The accuracy in the identification of elusive genes provides a solid basis for the following analyses.The authors identified genomic features associated with the gene losses in the species where the gene is preserved. This is an important strategy to avoid identifying genomic features that are formed during the gene losses but to identify the genomic features that likely formed before the gene loss. Using this strategy, the authors were able to recognize the intrinsic properties of elusive genes.
Weaknesses:
Gene expression level as a confounding factor was not well controlled throughout the study. Higher gene expression often makes genes less dispensable after gene duplication. Gene expression level is also a major determining factor of evolutionary rates (reviewed in http://www.ncbi.nlm.nih.gov/pubmed/26055156). Some proposed theories explain why gene expression level can serve as a proxy for gene importance (http://www.ncbi.nlm.nih.gov/pubmed/20884723, http://www.ncbi.nlm.nih.gov/pubmed/20485561). In that sense, many genomic/epigenomic features (such as replication timing and repressed transcriptional regulation) that were assumed "neutral" or intrinsic by the authors (or more accurately, independent of gene dispensability) cannot be easily distinguishable from the effect of gene dispersibility.
Ks was used by the authors to indicate mutation rates. However, synonymous mutations substantially affect gene expression levels (https://pubmed.ncbi.nlm.nih.gov/25768907/, https://pubmed.ncbi.nlm.nih.gov/35676473/). Thus, synonymous mutations cannot be simply assumed as neutral ones and may not be suitable for estimating local mutation rates. If introns can be aligned, they are better sequences for estimating the mutability of a genomic region.
The term "elusive gene" is not necessarily intuitive to readers.
-
Reviewer #2 (Public Review):
By analyzing hundreds of genomes, authors studied the so-called elusive genes, i.e., genes present in human genome but their orthologs deleted in some other mammals. Authors showed their bioinformatic pipeline of identifying these genes (Fig. 1), the genomic or evolutionary features of these genes (e.g. high GC content, Fig. 2), conservation of these features in other vertebrates including remotely related gar or shark (Fig. 3) together with polymorphism level, transcriptional features, epigenetic features of these genes (Fig. 4-6). Finally, in the Discussion section, the authors showed the chromosomal contributions of elusive genes and argued that these genes could be derived from ancient microchromosomes (Fig. 7).
-
Reviewer #3 (Public Review):
The manuscript by Hara and Kuraku addresses the question of whether some genes have a diverging gene fate (gene loss) due to underlying sequence or genomic properties. To approach this task, the authors introduce a gene loss detection pipeline that takes some previously raised technical concerns of overestimating gene loss (e.g. variations in assembly quality) into account. When applying their pipeline to >100 species, the authors report ~1,000 human genes whose orthologues were lost in multiple mammalian lineages (which they refer to as elusive genes). The study then focuses on integrating all functional evidence that can be obtained from large-scale databases for these elusive genes and test whether their genomic and evolutionary properties in the genomes of human and various other vertebrates (chimpanzee, …
Reviewer #3 (Public Review):
The manuscript by Hara and Kuraku addresses the question of whether some genes have a diverging gene fate (gene loss) due to underlying sequence or genomic properties. To approach this task, the authors introduce a gene loss detection pipeline that takes some previously raised technical concerns of overestimating gene loss (e.g. variations in assembly quality) into account. When applying their pipeline to >100 species, the authors report ~1,000 human genes whose orthologues were lost in multiple mammalian lineages (which they refer to as elusive genes). The study then focuses on integrating all functional evidence that can be obtained from large-scale databases for these elusive genes and test whether their genomic and evolutionary properties in the genomes of human and various other vertebrates (chimpanzee, mouse, chicken, turkey, green anole, central bearded dragon, western clawed frog, coelacanth, spotted gar, bamboo shark, whale shark) differs from the properties of the ~8,000 non-elusive genes (genes stably conserved across the compared species). In addition, the authors further analyse the human genome for the population-level variations, expression profiles and epigenetic features of elusive genes.
Overall, the study is descriptive and adds incremental evidence to an existing body of extensive gene loss literature. The topic is specialised and will be of interest to a niche audience. The text is highly redundant, repeating the same false positive issue in the introduction, methods, and discussion sections, while no clear conclusion or interpretation of their main findings are presented.
Major comments
- While some of the false discovery rate issues of gene loss detection were addressed in the presented pipeline, the authors fail to test one of the most severe cases of mis-annotating gene loss events: frameshift mutations which cause gene annotation pipelines to fail reporting these genes in the first place. Running a blastx or diamond blastx search of their elusive and non-elusive gene sets against all other genomes, should further enlighten the robustness of their gene loss detection approach
- Along this line, we noticed that when annotation files were pooled together via CD-Hit clustering, a 100% identity threshold was chosen (Methods). Since some of the pooled annotations were drawn from less high quality assemblies which yield higher likelihoods of mismatches between annotations, enforcing a 100% identity threshold will artificially remove genes due to this strict constraint. It will be paramount for this study to test the robustness of their findings when 90% and 95% identity thresholds were selected.
- While some statistical tests were applied (although we do recommend consulting a professional statistician, since some identical distributions tend to show significantly low p-values), the authors fail to discuss the fact that their elusive gene set comprises of ~5% of all human genes (assuming 21,000 genes), while their non-elusive set represents ~40% of all genes. In other words, the authors compare their sequence and genomic features against the genomic background rather than a biological signal (non-elusiveness). An analysis whereby 1,081 genes (same number as elusive set) are randomly sampled from the 21,000 gene pool is compared against the elusive and non-elusive distributions for all presented results will reveal whether the non-elusive set follows a background distribution (noise) or not.
- We also wondered whether the authors considered testing the links between recombination rate / LD and the genomic locations of their elusive genes (again compared against randomly sampled genes)?
- Given the evidence presented in Figure 6b, we do not agree with the statement (l.334-336): "These observations suggest that the elusive genes are unlikely to be regulated by distant regulatory elements". Here, a data population of ~1k genes is compared against a data population of ~8k genes and the presented difference between distributions could be a sample size artefact. We strongly recommend retesting this result with the ~1k randomly sampled genes from the total ~21,000 gene pool and then compare the distributions.
- Analogous random sampling analysis should be performed for Fig 6a,d
- We didn't see a clear pattern in Figure 7. Please quantify enrichments with statistical tests. Even if there are enriched regions, why did the authors choose a Shannon entropy cutoff configuration of <1 (low) and >1 (high)? What was the overall entropy value range? If the maximum entropy value was 10 or 100 or even more, then denoting <1 as low and >1 as high seems rather biased.
-
