Regulatory T cells suppress the formation of potent KLRK1 and IL-7R expressing effector CD8 T cells by limiting IL-2
Curation statements for this article:-
Curated by eLife

eLife assessment
This manuscript is of primary interest to immunologists with a focus on the effects of interleukin-2 and T cell receptor (TCR) signaling on effector T cell differentiation and function. Extensive and well-controlled experiments support a model where TCR and interleukin-2 signals promote a specific subset of effector CD8+ T cells - termed KILR cells - with superior target cell killing properties.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Regulatory T cells (Tregs) are indispensable for maintaining self-tolerance by suppressing conventional T cells. On the other hand, Tregs promote tumor growth by inhibiting anti-cancer immunity. In this study, we identified that Tregs increase the quorum of self-reactive CD8 + T cells required for the induction of experimental autoimmune diabetes in mice. Their major suppression mechanism is limiting available IL-2, an essential T-cell cytokine. Specifically, Tregs inhibit the formation of a previously uncharacterized subset of antigen-stimulated KLRK1 + IL7R + (KILR) CD8 + effector T cells, which are distinct from conventional effector CD8 + T cells. KILR CD8 + T cells show a superior cell killing abilities in vivo. The administration of agonistic IL-2 immunocomplexes phenocopies the absence of Tregs, i.e., it induces KILR CD8 + T cells, promotes autoimmunity, and enhances anti-tumor responses in mice. Counterparts of KILR CD8 + T cells were found in the human blood, revealing them as a potential target for immunotherapy.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
By studying the effect of Treg depletion in a CD8+ T cell-dependent diabetes model the group around Ondrej Stepanek described that in the absence of Treg cells antigen-specific CD8+ OT-I T cells show an activated phenotype and accelerate the development of diabetes in mice. These cells - termed KILR cells - express CD8+ effector and NK cell gene signatures and are identified as CD49d- KLRK1+ CD127+ CD8+ T cells. The authors suggest that the generation of these cells is dependent on TCR stimulation and IL-2 signals, either provided due to the absence of Treg cells or by injection of IL-2 complexed to specific antiIL-2 mAbs. In vivo, these cells show improved target cell killing properties, while the authors report improved anti-tumor responses of combination treatments with doxorubicin …
Author Response
Reviewer #1 (Public Review):
By studying the effect of Treg depletion in a CD8+ T cell-dependent diabetes model the group around Ondrej Stepanek described that in the absence of Treg cells antigen-specific CD8+ OT-I T cells show an activated phenotype and accelerate the development of diabetes in mice. These cells - termed KILR cells - express CD8+ effector and NK cell gene signatures and are identified as CD49d- KLRK1+ CD127+ CD8+ T cells. The authors suggest that the generation of these cells is dependent on TCR stimulation and IL-2 signals, either provided due to the absence of Treg cells or by injection of IL-2 complexed to specific antiIL-2 mAbs. In vivo, these cells show improved target cell killing properties, while the authors report improved anti-tumor responses of combination treatments with doxorubicin combined with IL-2/JES6 complexes. Finally, the authors identified a similar human subset in publicly available scRNAseq datasets, supporting the translational potential of their findings.
The conclusions are mostly well supported, except for the following two considerations:
We are happy for the positive overall evaluation of our manuscript by both reviewers and we are thankful for their specific insightful comments, which helped us to improve the manuscript.
- From Fig. 4A and B it is not conclusively shown, that Tregs limit IL-2 necessary for the expansion of OT-I cells and subsequent induction of diabetes. An IL-2 depletion experiment (e.g. with combined injection of the S4B6 and JES6-1 antibodies) would further strengthen this claim. Along these lines, the authors claim "IL-2Rα expression on T cells can be induced by antigen stimulation or by IL-2 itself in a positive feedback loop [20]. Accordingly, downregulation of IL-2Rα in OT-I T cells in the presence of Tregs might be a consequence of the limited availability of IL-2.". The cited reference 20 did observe CD25 upregulation by IL-2 on T cells but the observed effect might only be caused by upregulation of CD25 on Treg cells, which increases the MFI for the whole T cell population. Did the authors observe significant upregulation of CD25 on effector CD4+ and CD8+ T cells in their experiments with IL-2/S4B6 or IL-2/JES6 treatment?
We added another reference to support our claim (Sereti, I., et al., Clin Immunol, 2000. 97(3): p. 266-76.). Along this line, we also observed that addition of IL-2 in vitro leads to IL-2Rα upregulation on CD8+ T cells (shown in Fig. 4C), which was IL-2Rα level was lower if Tregs were present. We also observed upregulation of IL-2Rα in vivo upon the stimulation of OT-I T cells with OVA and IL-2ic, which is now shown in the Fig. S6C of the revised manuscript.
To further explore if Tregs limit expansion of OT-I and diabetes progression via IL-2 limitations, we performed the proposed experiment using a combined injection of S4B6 and JES6-1 anti-IL-2 antibodies. At the beginning, we were skeptical that we could completely block the IL-2 using this approach for the following reasons. First, IL-2 is produced locally in the spleen and lymph nodes and might not be easily accessible for the antibodies for a complete block. Second, IL-2 has a relatively short turnover and is continuously produced, but the half-life of the injected antibodies is unknown, which questions the duration of such a block. Third, it is possible that some IL-2 molecules would bound only to one of the two antibodies, which will make it a hyper-stimulating immune-complex, instead of neutralizing it.
Anyway, we were curious enough to perform this experiment. We used a condition that based on our experience leads to diabetes manifestation in Tregs depleted, but not in Treg replete mice (10 k OT-I T cells, OVA + LPS immunization). One additional group of Treg-depleted mice received a single dose of S4B6 and JES6-1 anti-IL-2 (200 µg of each antibody per mouse). We observed that this IL-2 blocking delayed, but not prevented the development of diabetes in most animals (Fig. 1 below).
Overall, we believe that this experiment is rather supporting our conclusions concerning the importance of IL-2, although the effect is only partial. However, we decided not to include this experiment in the manuscript, because we do not have the evidence about how efficient the IL-2 blocking was (see above), which makes the interpretation difficult. Because the reviews and the point-by-point response is public in eLife, we believe that showing the data here is appropriate.
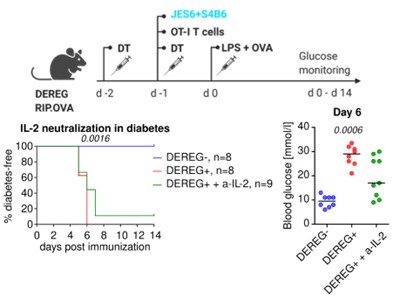
Figure 1. Role of IL-2 blocking on the development of experimental diabetes. Two independent experiments were performed. Statistical significance was calculated using Log-rank (Mantel-Cox) test for survival, and Kruskal-Wallis test for blood glucose (p-value is shown in italics).
- The anti-tumor efficacy of KILR cells is intriguing but currently, it is unclear if it is indeed mediated by KILR cells. Have KILR cells been identified by flow cytometry in the BCL1 and B16F10 models treated with doxorubicin and IL-2/JES6? Were specific KILR cell depletion studies conducted, e.g. with an anti-KLRK1 depleting antibody? Additional experiments addressing these questions would be desirable to further support the authors' claims.
We are thankful to both reviewers for their similar comments concerning the analysis of CD8+ T cells in the tumor model. Addressing these comments lead to very useful data and significantly improved our manuscript.
We performed the analysis of splenic CD8+ T cells in the BCL1 leukemia model (spleen is the major site of the leukemic cells in this model). We observed that KLRK1+ T cells represented almost half of CD8+ T cells in mice treated with DOX+IL-2, which was much higher frequency than in the control and DOX-only treated mice. Although not all KLRK1+ cells were bona fide KILR cells, the frequencies of KLRK1+ IL-7R+ and KLRK1+ CD49d- cells were also strongly elevated in the Dox+IL-2ic treated mice. Overall, the survival of DOX+IL-2ic treated mice correlated with the frequencies of KILR T cells and KLRK1+ T cells. Moreover, GZMB was almost exclusively expressed by KLRK1+ T cells. We are showing these data in Fig. 7C and Fig. S7B in the revised manuscript.
In the B16 melanoma model, we analyzed CD8+ T cells in the spleens and also in the tumors. We observed a huge population of KLRK1+ GZMB+ CD8+ T-cell population in the spleen of DOX+IL-2ic-treated mice, but not in the untreated or DOX-only treated mice (Fig. 7F). Both KLRK1+ CD49d+ and KLRK1+ CD49d- CD8+ T cells were substantially more frequent in the DOX+IL-2ic-treated, but not in the untreated or DOX-only treated mice (Fig. S7F). In the tumor, the KLRK1+ CD49d- CD8+ T cells were found at large numbers only in the DOX+IL-2ic-treated mice (Fig. 7G). Moreover, these KLRK1+ CD49d- CD8+ T cells expressed high levels of IL-7R and GZMB only in DOX+IL-2ic-treated, but not in untreated and DOX-only treated mice (Fig. 7H).
We believe that these new data provide evidence that the combination of immunogenic chemotherapy with IL-2 treatment induced KILR cells in the spleens and in the tumors and that this correlates with the better survival.
Because the majority of non-naïve CD8+ T cells (and vast majority of GZMB+ CD8+ T cells) in the spleens and tumors of the tumor-bearing mice treated with DOX+IL-2ic were KLRK1+ and because we have shown that the protective effect of the DOX+IL-2ic therapy is largely CD8+ T cell-dependent, we did not find it essential to perform the depletion of KLRK1+ T-cells. We believe that it is almost inevitable that the depletion of KLRK1+ T cells would lead to increased tumor growth as it would probably deplete the majority of antigenspecific CD8+ T cells, mimicking the overall CD8+ T cell depletion. Moreover, we do not have this protocol established.
Reviewer #2 (Public Review):
In this study, the authors determine the superior cell killing abilities of KLRK1+ IL7R+ (KILR) CD8+ effector T cells in experimental diabetes and tumor mouse model. They also provide evidence that Tregs suppress the formation of this previously uncharacterized subset of CD8+ effector T cells by limiting IL-2.
Strength and Limitation
This study focuses on the relationship between Tregs and CD8+ T cells. They used different experimental diabetes mouse models to reveal that Tregs suppress the CD8+ effector T cells by limiting IL-2. They also found a unique subset of KLRK1+ IL7R+ (KILR) CD8+ effector T cells with superior cell killing abilities through single-cell sequencing, but killing abilities could be inhibited by Tregs. They also tested their theory in in vivo tumor model. The data, in general, support the conclusions; however, some issues need to be fully addressed, as detailed below.
We are happy for the positive overall evaluation of our manuscript by both reviewers and we are thankful for their specific insightful comments, which helped us to improve the manuscript.
- This study used the concentration of urine glucose as the standard for diabetes ({greater than or equal to} 1000 mg/dl for two consecutive days). However, multiple reasons may lead to a high level of urine glucose. As a type I diabetes mouse model, authors could use immunohistological analysis of islet to show the proportion of T cells and islet cells in islet, which can display the geographic distribution of immune cells, severity and histology structure of damaged pancreas islet directly. If possible, different subsets of immune cells, especially CD4 vs CD8+ cells should be stained for their location.
We added the histological examination of the pancreas in control, DEREG-, and DEREG+ mice using contrast H&E staining and immuno-fluorescence (Fig. 1D-E in the revised manuscript). We observed that the high glucose and blood levels are preceded by the destruction of the pancreatic islets (morphology and decreased insulin production) as well as by the infiltration of the islets with immune cells including CD4+ and CD8+ T cells.
- This article shows that KILR effector CD8+ T cells have strong cytotoxic properties. However, they do not describe the potential proliferation ability vs apoptosis of this subset from islets.
We analyzed the proliferation (KI67 expression) and apoptosis (Annexin V, cleaved Caspase 3) in T cells isolated from the pancreas of DEREG- and DEREG+ mice on day 4 after the induction of diabetes using flow cytometry (Figure 2 below). We did not observe any differences between DEREG- and DEREG+ mice or among different subsets of OT-I T cells in the DEREG+ mice. Essentially, all T cells were proliferative (KI67+) and there was a very low percentage of Annexin V or cleaved Caspase 3 positive cells.
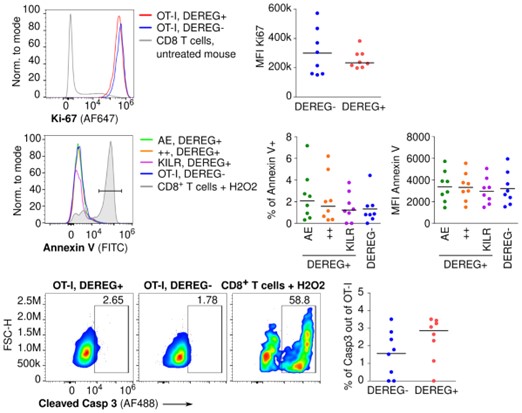
Figure 2. Lymphocytes were isolated from the pancreas of DEREG- RIP.OVA and DEREG+ RIP.OVA mice on day 4 after the induction of diabetes, and analyzed using flow cytometry. Two independent experiments were performed. Gated on OT-I T cells. Top: proliferation rate based on Ki-67 staining. Representative histogram and MFI (median is shown). Middle: Apoptosis rate based on Annexin V staining. Representative histogram shows Annexin V staining in three populations of OT-I T cells from DEREG+ mouse (“AE” - CD49d+ KLRK1-, “++” - CD49d+ KLRK1+, KILR - CD49d- KLRK1+), total OT-I T cells from DEREG-, and a positive control: WT CD8+ T cells treated with hydrogen peroxide. Middle right: Percentage of Annexin V+ cells and MFI (median is shown). Bottom: Apoptosis rate based on cleaved Caspase 3 staining. Representative dot plots show cleaved Caspase 3 staining of OT-I T cells from DEREG+, DEREG-, and a positive control: WT CD8+ T cells treated with hydrogen peroxide. Bottom right: percentage of cleaved Caspase 3+ cells (median is shown).
However, we found question concerning proliferation and apoptosis of KILR cells interesting and worth further investigation. For this reason, we assessed the proliferation, survival, and phenotypic stability of naïve, KILR, and effector T cells by their competitive transfer into CD3ε-/- mice. The phenotype of all these three subsets remained stable for 4 days (Fig. 6F), documenting that KILR cells are not just a very transient stage. Moreover, the KILR cells were ~2 fold more abundant then effector cells 3 days after their 1:1 cotransfer into CD3ε-/- mice (Fig. 6G, Fig. 6SE). This was probably caused by their slight advantages in both proliferation and survival (Fig. 6SF-G).
- Figure 7 shows that the antitumor efficacy of IL-2 depends on CD8+ T cells. But in this part, there is no data to show the change of KLRK1+ IL7R+ CD8+ effector T cells in tumor tissue. Therefore, the article needs to add more data to verify that IL-2 enhances antitumor ability via KLRK1+ IL7R+ CD8+ effector T cells.
We are thankful to both reviewers for their similar comments concerning the analysis of CD8+ T cells in the tumor model. Addressing these comments lead to very useful data and significantly improved our manuscript.
We performed the analysis of splenic CD8+ T cells in the BCL1 leukemia model (spleen is the major site of the leukemic cells in this model). We observed that KLRK1+ T cells represented almost half of CD8+ T cells in mice treated with DOX+IL-2, which was much higher frequency than in the control and DOX-only treated mice. Although not all KLRK1+ cells were bona fide KILR cells, the frequencies of KLRK1+ IL-7R+ and KLRK1+ CD49d- cells were also strongly elevated in the Dox+IL-2ic treated mice. Overall, the survival of DOX+IL-2ic treated mice correlated with the frequencies of KILR T cells and KLRK1+ T cells. Moreover, GZMB was almost exclusively expressed by KLRK1+ T cells. We are showing these data in Fig. 7C and Fig. S7B in the revised manuscript.
In the B16 melanoma model, we analyzed CD8+ T cells in the spleens and also in the tumors. We observed a huge population of KLRK1+ GZMB+ CD8+ T-cell population in the spleen of DOX+IL-2ic-treated mice, but not in the untreated or DOX-only treated mice (Fig. 7F). Both KLRK1+ CD49d+ and KLRK1+ CD49d- CD8+ T cells were substantially more frequent in the DOX+IL-2ic-treated, but not in the untreated or DOX-only treated mice (Fig. S7F). In the tumor, the KLRK1+ CD49d- CD8+ T cells were found at large numbers only in the DOX+IL-2ic-treated mice (Fig. 7G). Moreover, these KLRK1+ CD49d- CD8+ T cells expressed high levels of IL-7R and GZMB only in DOX+IL-2ic-treated, but not in untreated and DOX-only treated mice (Fig. 7H).
We believe that these new data provide evidence that the combination of immunogenic chemotherapy with IL-2 treatment induced KILR cells in the spleens and in the tumors and that this correlates with the better survival.
- It is unclear why the authors chose Dox to combine with IL-2/JES6. The authors should provide a more rational introduction to bridge such a combination. Authors should also explain the reason why there is no antitumor effect of IL-2/JES6 treatment alone.
The experiments with OT-I mice showed that the formation of KILR cells required both the antigenic stimulation and IL-2 signals. We believe that there is only very week antigenic stimulation by the tumor itself. For this reason, we combined the treatment with the chemotherapy Doxorubicin, which is known to induce immunogenic cell death of the tumor cells (e.g., Casares et al. 2005, PMID: 16365148). We believe that doxorubicin induces the death of (some) tumor cells and the release and presentation of their tumorspecific antigens. Without it, the tumor are simply too “cold” to induce sufficient T-cell response. We emphasized this in the revised version of the manuscript.
Importantly, some of us observed a similar effect of IL-2ic in a combination with check-point blockade therapy (without chemotherapy) in a different tumor model, which documents that the chemotherapy is not essential for this effect (unpublished data).
-
eLife assessment
This manuscript is of primary interest to immunologists with a focus on the effects of interleukin-2 and T cell receptor (TCR) signaling on effector T cell differentiation and function. Extensive and well-controlled experiments support a model where TCR and interleukin-2 signals promote a specific subset of effector CD8+ T cells - termed KILR cells - with superior target cell killing properties.
-
Reviewer #1 (Public Review):
By studying the effect of Treg depletion in a CD8+ T cell-dependent diabetes model the group around Ondrej Stepanek described that in the absence of Treg cells antigen-specific CD8+ OT-I T cells show an activated phenotype and accelerate the development of diabetes in mice. These cells - termed KILR cells - express CD8+ effector and NK cell gene signatures and are identified as CD49d- KLRK1+ CD127+ CD8+ T cells. The authors suggest that the generation of these cells is dependent on TCR stimulation and IL-2 signals, either provided due to the absence of Treg cells or by injection of IL-2 complexed to specific anti-IL-2 mAbs. In vivo, these cells show improved target cell killing properties, while the authors report improved anti-tumor responses of combination treatments with doxorubicin combined with …
Reviewer #1 (Public Review):
By studying the effect of Treg depletion in a CD8+ T cell-dependent diabetes model the group around Ondrej Stepanek described that in the absence of Treg cells antigen-specific CD8+ OT-I T cells show an activated phenotype and accelerate the development of diabetes in mice. These cells - termed KILR cells - express CD8+ effector and NK cell gene signatures and are identified as CD49d- KLRK1+ CD127+ CD8+ T cells. The authors suggest that the generation of these cells is dependent on TCR stimulation and IL-2 signals, either provided due to the absence of Treg cells or by injection of IL-2 complexed to specific anti-IL-2 mAbs. In vivo, these cells show improved target cell killing properties, while the authors report improved anti-tumor responses of combination treatments with doxorubicin combined with IL-2/JES6 complexes. Finally, the authors identified a similar human subset in publicly available scRNAseq datasets, supporting the translational potential of their findings.
The conclusions are mostly well supported, except for the following two considerations:
From Fig. 4A and B it is not conclusively shown, that Tregs limit IL-2 necessary for the expansion of OT-I cells and subsequent induction of diabetes. An IL-2 depletion experiment (e.g. with combined injection of the S4B6 and JES6-1 antibodies) would further strengthen this claim. Along these lines, the authors claim "IL-2Rα expression on T cells can be induced by antigen stimulation or by IL-2 itself in a positive feedback loop [20]. Accordingly, downregulation of IL-2Rα in OT-I T cells in the presence of Tregs might be a consequence of the limited availability of IL-2.". The cited reference 20 did observe CD25 upregulation by IL-2 on T cells but the observed effect might only be caused by upregulation of CD25 on Treg cells, which increases the MFI for the whole T cell population. Did the authors observe significant upregulation of CD25 on effector CD4+ and CD8+ T cells in their experiments with IL-2/S4B6 or IL-2/JES6 treatment?
The anti-tumor efficacy of KILR cells is intriguing but currently, it is unclear if it is indeed mediated by KILR cells. Have KILR cells been identified by flow cytometry in the BCL1 and B16F10 models treated with doxorubicin and IL-2/JES6? Were specific KILR cell depletion studies conducted, e.g. with an anti-KLRK1 depleting antibody? Additional experiments addressing these questions would be desirable to further support the authors' claims.
-
Reviewer #2 (Public Review):
In this study, the authors determine the superior cell killing abilities of KLRK1+ IL7R+ (KILR) CD8+ effector T cells in experimental diabetes and tumor mouse model. They also provide evidence that Tregs suppress the formation of this previously uncharacterized subset of CD8+ effector T cells by limiting IL-2.
Strength and Limitation
This study focuses on the relationship between Tregs and CD8+ T cells. They used different experimental diabetes mouse models to reveal that Tregs suppress the CD8+ effector T cells by limiting IL-2. They also found a unique subset of KLRK1+ IL7R+ (KILR) CD8+ effector T cells with superior cell killing abilities through single-cell sequencing, but killing abilities could be inhibited by Tregs. They also tested their theory in in vivo tumor model. The data, in general, support …
Reviewer #2 (Public Review):
In this study, the authors determine the superior cell killing abilities of KLRK1+ IL7R+ (KILR) CD8+ effector T cells in experimental diabetes and tumor mouse model. They also provide evidence that Tregs suppress the formation of this previously uncharacterized subset of CD8+ effector T cells by limiting IL-2.
Strength and Limitation
This study focuses on the relationship between Tregs and CD8+ T cells. They used different experimental diabetes mouse models to reveal that Tregs suppress the CD8+ effector T cells by limiting IL-2. They also found a unique subset of KLRK1+ IL7R+ (KILR) CD8+ effector T cells with superior cell killing abilities through single-cell sequencing, but killing abilities could be inhibited by Tregs. They also tested their theory in in vivo tumor model. The data, in general, support the conclusions; however, some issues need to be fully addressed, as detailed below.
1. This study used the concentration of urine glucose as the standard for diabetes ({greater than or equal to} 1000 mg/dl for two consecutive days). However, multiple reasons may lead to a high level of urine glucose. As a type I diabetes mouse model, authors could use immunohistological analysis of islet to show the proportion of T cells and islet cells in islet, which can display the geographic distribution of immune cells, severity and histology structure of damaged pancreas islet directly. If possible, different subsets of immune cells, especially CD4 vs CD8+ cells should be stained for their location.
2. This article shows that KILR effector CD8+ T cells have strong cytotoxic properties. However, they do not describe the potential proliferation ability vs apoptosis of this subset from islets.
3. Figure 7 shows that the antitumor efficacy of IL-2 depends on CD8+ T cells. But in this part, there is no data to show the change of KLRK1+ IL7R+ CD8+ effector T cells in tumor tissue. Therefore, the article needs to add more data to verify that IL-2 enhances antitumor ability via KLRK1+ IL7R+ CD8+ effector T cells.
4. It is unclear why the authors chose Dox to combine with IL-2/JES6. The authors should provide a more rational introduction to bridge such a combination. Authors should also explain the reason why there is no antitumor effect of IL-2/JES6 treatment alone.
-
