Impaired iron recycling from erythrocytes is an early hallmark of aging
Curation statements for this article:-
Curated by eLife

eLife assessment
Slusarczyk et al demonstrate that red pulp macrophages (RPM), specialized splenic cells that clear senescent red blood cells through erythrophagocytosis, show diminished function in aging mice. This impairment leads to retention of hemolytic red blood cells and formation of extracellular aggregates which further exacerbate RPM demise. Iron restriction alleviates most of these symptoms in aging RPMs. They propose RPM collapse as an early indicator of aging that could be reversed through iron limitation.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Aging affects iron homeostasis, as evidenced by tissue iron loading and anemia in the elderly. Iron needs in mammals are met primarily by iron recycling from senescent red blood cells (RBCs), a task chiefly accomplished by splenic red pulp macrophages (RPMs) via erythrophagocytosis. Given that RPMs continuously process iron, their cellular functions might be susceptible to age-dependent decline, a possibility that has been unexplored to date. Here, we found that 10- to 11-month-old female mice exhibit iron loading in RPMs, largely attributable to a drop in iron exporter ferroportin, which diminishes their erythrophagocytosis capacity and lysosomal activity. Furthermore, we identified a loss of RPMs during aging, underlain by the combination of proteotoxic stress and iron-dependent cell death resembling ferroptosis. These impairments lead to the retention of senescent hemolytic RBCs in the spleen, and the formation of undegradable iron- and heme-rich extracellular protein aggregates, likely derived from ferroptotic RPMs. We further found that feeding mice an iron-reduced diet alleviates iron accumulation in RPMs, enhances their ability to clear erythrocytes, and reduces damage. Consequently, this diet ameliorates hemolysis of splenic RBCs and reduces the burden of protein aggregates, mildly increasing serum iron availability in aging mice. Taken together, we identified RPM collapse as an early hallmark of aging and demonstrated that dietary iron reduction improves iron turnover efficacy.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
Slusarczyk et al present a very well written manuscript focused on understanding the mechanisms underlying aging of erythrophagocytic macrophages in the spleen (RPM) and its relationship to iron loading with age. The manuscript is diffuse with a broad swath of data elements. Importantly, the manuscript demonstrates that RPM erythrophagocytic capacity is diminished with age, restored in iron restricted diet fed aged mice. In addition, the mechanism for declining RPM erythrophagocytic capacity appears to be ferroptosis-mediated, insensitive to heme as it is to iron, and occur independently of ROS generation. These are compelling findings. However, some of the data relies on conjecture for conclusion and a clear causal association is not clear. The main conclusion of the manuscript points to …
Author Response
Reviewer #1 (Public Review):
Slusarczyk et al present a very well written manuscript focused on understanding the mechanisms underlying aging of erythrophagocytic macrophages in the spleen (RPM) and its relationship to iron loading with age. The manuscript is diffuse with a broad swath of data elements. Importantly, the manuscript demonstrates that RPM erythrophagocytic capacity is diminished with age, restored in iron restricted diet fed aged mice. In addition, the mechanism for declining RPM erythrophagocytic capacity appears to be ferroptosis-mediated, insensitive to heme as it is to iron, and occur independently of ROS generation. These are compelling findings. However, some of the data relies on conjecture for conclusion and a clear causal association is not clear. The main conclusion of the manuscript points to the accumulation of unavailable insoluble forms of iron as both causing and resulting from decreased RPM erythrophagocytic capacity.
We are proposing that intracellular iron accumulation progresses first and leads to global proteotoxic damage and increased lipid peroxidation. This eventually triggers the death of a fraction of aging RPMs, thus promoting the formation of extracellular iron-rich protein aggregates. More explanation can be found below. Besides, iron loading suppresses the erythrophagocytic activity of RPMs, hence further contributing to their functional impairment during aging.
In addition, the finding that IR diet leads to increased TF saturation in aged mice is surprising.
We believe that this observation implies better mobilization of splenic iron stores, and corroborates our conclusion that mice that age on an iron-reduced diet benefit from higher iron bioavailability, although these differences are relatively mild. More explanation can be found in our replies to Reviewer #2.
Furthermore, whether the finding in RPMs is intrinsic or related to RBC-related changes with aging is not addressed.
We now addressed this issue and we characterized in more detail both iron and ROS levels in RBCs.
Finally, these findings in a single strain and only female mice is intriguing but warrants tempered conclusions.
We tempered the conclusions and provided a basic characterization of the RPM aging phenotype in Balb/c female mice.
Major points:
- The main concern is that there is no clear explanation of why iron increases during aging although the authors appear to be saying that iron accumulation is both the cause of and a consequence of decreased RPM erythrophagocytic capacity. This requires more clarification of the main hypothesis on Page 4, line 17-18.
We thank the reviewer for this comment. It was previously reported that iron accumulates substantially in the spleen during aging, especially in female mice (Altamura et al., 2014). Since RPMs are those cells that process most of the iron in the spleen, we aimed to explore what is the relationship between iron accumulation and RPM functions during aging. This investigation led us to uncover that indeed iron accumulation is both the cause and the consequence of RPM dysfunction. Specifically, we propose that intracellular iron loading of RPMs precedes extracellular deposition of iron in a form of protein-rich aggregates, driven by RPMs damage. To support this, we now show that the proteome of RPMs overlaps with those proteins that are present in the age-triggered aggregates (Fig. 3F). Furthermore, corroborating our model, we now demonstrate that transient iron loading of RPMs via iron-dextran injection (new Fig. 3G) leads to the formation of protein-rich aggregates, closely resembling those present in aged spleens (new Fig. 3H). This implies that high iron content in RPMs is indeed a major driving factor that leads to aggregation of their proteome and cell damage. Importantly, we now supported this model with studies using iRPMs. We demonstrated that iron loading and blockage of ferroportin by synthetic mini-hepcidin (PR73)(Stefanova et al., 2018) cause protein aggregation in iRPMs and lead to their decreased viability only in cells that were exposed to heat shock, a well-established trigger of proteotoxicity (new Fig. 5K and L). We propose that these two factors, namely age-triggered decrease in protein homeostasis and exposure to excessive iron levels, act in concert and render RPMs particularly sensitive to damage during aging (see also Discussion, p. 16).
In parallel, our data imply that the increased iron content in aged RPMs drives their decreased erythrophagocytic activity, as we now better documented by more extensive in vitro experiments in iRPMs (new Fig 6E-H). We cannot exclude that some of the senescent splenic RBCs that are retained in the red pulp and evade erythrophagocytosis due to RPM defects in aging, may also contribute to the formation of the aggregates. This is supported by the fact that mice that lack RPMs as well exhibit iron loading in the spleen (Kohyama et al., 2009; Okreglicka et al., 2021), and that the proteome of aggregates overlaps to some extent with the proteome of erythrocytes (new Fig. 3F).
We believe that during aging intracellular iron accumulation is chiefly driven by ferroportin downregulation, as also suggested by Reviewer#3. We now show that ferroportin drops significantly already in mice aged 4 and 5 months (new Fig. 4H), preceding most of the other impairments. This drop coincides with the increase in hepcidin expression, but if this is the sole reason for ferroportin suppression during early aging would require further investigation outside the scope of the present manuscript.
In sum, to address this comment, we now modified the fragment of the introduction that refers to our hypothesis and major findings to be more clear (p. 4), we improved our manuscript by providing new data mentioned above and we added more explanation in the corresponding sections of the Results and Discussion.
- It is unclear if RPMs are in limited supply. Based on the introduction (page 4, line 13-15), they have limited self-renewal capacity and blood monocytes only partially replenished. Fig 4D suggests that there is a decrease in RPMs from aged mice. The %RPM from CD45+ compartment suggests that there may just be relatively more neutrophils or fewer monocytes recruited. There is not enough clarity on the meaning of this data point.
Thank you for this comment. We fully agree that %RPMs of CD45+ splenocytes, although well-accepted in literature (Kohyama et al., 2009; Okreglicka et al., 2021), is only a relative number. Hence, we now included additional data and explanations regarding the loss of RPMs during aging.
It was reported that the proportion of RPMs derived from bone marrow monocytes increases mildly but progressively during aging (Liu et al., 2019). This implies that due to the loss of the total RPM population, as illustrated by our data, the cells of embryonic origin are likely even more affected. We could confirm this assumption by re-analysis of the data from Liu et al. that we now included in the manuscript as Fig. 5E. These data clearly show that the representation of embryonically-derived RPMs drops more drastically than the percent of total RPMs, whereas the replenishment rate from monocytes is not affected significantly during aging. Consistent with this, we have not observed any robust change in the population of monocytes (F4/80-low, CD11b-high) or pre-RPMs (F4/80-high, CD11b-high) in the spleen at the age of 10 months (Figure 5-figure supplement 2A and B). We also have detected a mild decrease, not an increase, in the number of granulocytes (new Figure 5-figure supplement 2C). Furthermore, we measured in situ apoptosis marker and found a clear sign of apoptosis in the aged spleen (especially in the red pulp area), a phenotype that is less pronounced in mice on an IR diet (new Fig. 5O). This is consistent with the observation that apoptosis markers can be elevated in tissues upon ferroptosis induction (Friedmann Angeli et al., 2014) and that the proteotoxic stress in aged RPMs, which we now emphasized better in our manuscript, may also lead to apoptosis (Brancolini & Iuliano, 2020). Taken together, we strongly believe that the functional defect of embryonically-derived RPMs chiefly contributes to their shortage during aging.
- Anemia of aging is a complex and poorly understood mechanistically. In general, it is considered similar to anemia of chronic inflammation with increased Epo, mild drop in Hb, and erythroid expansion, similar to ineffective erythropoiesis / low Epo responsiveness. It is not surprising that IR diet did not impact this mild anemia. However, was the MCV or MCH altered in aged and IR aged mice?
We now included the data for hematocrit, RBC counts, MCV, and MCH in Figure 1-figure supplement 5. Hematocrit shows a similar tendency as hemoglobin levels, but the values for RBC counts, MCV, and MCH seem not to be altered. We also show now that the erythropoietic activity in the bone marrow is not affected in aged versus young mice. Taken together, the anemic phenotype in female C57BL/6J mice at this age is very mild, which we emphasized in the main text, and is likely affected by other factors than serum iron levels (p. 6).
- Page 6, line 23 onward: the conclusion is that KC compensate for the decreased function of RPM in the spleen, based on the expansion of KC fraction in the liver. Is there evidence that KCs are engaged in more erythrophagocytosis in aged mice? Furthermore, iron accumulation in the liver with age does not demonstrate specifically enhanced erythrophagocytosis of KC. Please clarify why liver iron accumulation would not be simply a consequence of increased parenchymal iron similar to increased splenic iron with age, independent of erythrophagocytic activity in resident macrophages in either organ.
Thanks for these questions. For the quantification of the erythrophagocytosis rate in KC, we show, as for the RPMs (Fig. 1K), the % of PKH67-positive macrophages, following transfusion of PKH67-stained stressed RBCs (Fig. 1M). The data implies a mild (not statistically significant) drop (of approx. 30%) in EP activity. We believe that it is overridden by a more pronounced (on average, 2-fold) increase in the representation of KCs (Fig. 1N). The mechanisms of iron accumulation between the spleen and the liver are very different. In the liver, we observed iron deposition in the parenchymal cells (not non-parenchymal, new Fig. 1P) that we currently characterizing in more detail in a parallel manuscript. Our data demonstrate a drop in transferrin saturation in aged mice. Hence, it is highly unlikely that aging would be hallmarked by the presence of circulating non-transferrin-bound iron that would be sequestered by hepatocytes, as shown previously (Jenkitkasemwong et al., 2015). Thus, the iron released locally by KCs is the most likely contributor to progressive hepatocytic iron loading during aging. The mechanism of iron delivery to hepatocytes from erythrophagocytosing KCs was demonstrated by Theurl et al.(Theurl et al., 2016), and we propose that it may be operational, although in a much more prolonged time scale, during aging. We now discussed this part better in our Results sections (p. 7).
- Unclear whether the effect on RPMs is intrinsic or extrinsic. Would be helpful to evaluate aged iRPMs using young RBC vs. young iRPMs using old RBCs.
We are skeptical if the generation of iRPMs cells from aged mice would be helpful – these cells are a specific type of primary macrophage culture, derived from bone marrow monocytes with MCSF1, and exposed additionally to heme and IL-33 for 4 days. We do not expect that bone marrow monocytes are heavily affected by aging, and would thus recapitulate some aspects of aged RPMs from the spleen, especially after 8-day in vitro culture. However, to address the concerns of the reviewer, we now provide additional data regarding RBC fitness. Consistent with the time life-span experiment (Fig, 2A), we show that oxidative stress in RBCs is only increased in splenic, but not circulating RBCs (new Fig. 2C, replacing the old Fig. 2B and C). In addition, we show no signs of age-triggered iron loading in RBCs, either in the spleen (new Fig. 2F) or in the circulation (new Fig. 2B). Hence, we do not envision a possibility that RPMs become iron-loaded during aging as a result of erythrophagocytosis of iron-loaded RBCs. In support of this, we also have observed that during aging first RPMs’ FPN levels drop, afterward erythrophagocytosis rate decreases, and lastly, RBCs start to exhibit significantly increased oxidative stress (presented now in new Fig. 4H, J and K).
- Discussion of aggregates in the spleen of aged mice (Fig 2G-2K and Fig 3) is very descriptive and non-specific. For example, if the iron-rich aggregates are hemosiderin, a hemosiderin-specific stain would be helpful. This data specifically is correlatory and difficult to extract value from.
Thanks for these comments. To the best of our knowledge Prussian blue Perls’ staining (Fig. 2J) is considered a hemosiderin staining. Our investigations aimed to better understand the nature and the origin of splenic iron deposits that to some extent are referred to as hemosiderin. Most importantly, as mentioned in our reply R1 Ad. 1. to assign causality to our data, we now demonstrated that iron accumulation in RPMs in response to iron-dextran (Fig. 3G) increases lipid peroxidation (Fig. 5F), tends to provoke RPMs depletion (Fig. 5G) and triggers the formation of protein-rich aggregates (new Fig. 3H). Of note, we assume that the loss of embryonically-derived RPMs in this model may be masked by simultaneous replenishment of the niche from monocytes, a phenomenon that may be addressed by future studies using Ms4a3-driven reporter mice (as shown for aged mice in our new Fig. 5E).
- The aging phenotype in RPMs appears to be initiated sometime after 2 months of age. However, there is some reversal of the phenotype with increasing age, e.g. Fig 4B with decreased lipid peroxidation in 9 month old relative to 6 month old RPMs. What does this mean? Why is there a partial spontaneous normalization?
Thanks for this comment and questions. Indeed, the degree of lipid peroxidation exhibits some kinetics, suggestive of partial normalization. Of note, such a tendency is not evident for other aging phenotypes of RPMs, hence, we did not emphasize this in the original manuscript. However, in a revised version of the manuscript, we now present the re-analysis of the published data which implies that the number of embryonically-derived RPMs drops substantially between mice at 20 weeks and 36 weeks (new Fig. 5E). We think that the higher proportion of monocyte-derived RPMs in total RPM population later in aging (9 months) might be responsible for the partial alleviation of lipid peroxidation. We now discussed this possibility in the Results sections (p. 12).
- Does the aging phenotype in RPMs respond to ferristatin? It appears that NAC, which is a glutathione generator and can reverse ferroptosis, does not reverse the decreased RPM erythrophagocytic capacity observed with age yet the authors still propose that ferroptosis is involved. A response to ferristatin is a standard and acceptable approach to evaluating ferroptosis.
We fully agree with the Reviewer that using ferristatin or Liproxstatin-1 would be very helpful to fully characterize a mechanism of RPMs depletion in mice. However, previous in vivo studies involving Liproxstatin-1 administration required daily injections of this ferroptosis inhibitor (Friedmann Angeli et al., 2014). This would be hardly feasible during aging. Regarding the experiments involving iron-dextran injection, using Liproxstatin-1 would require additional permission from the ethical committee which takes time to be processed and received. However, to address this question we now provide data from iRPMs cell cultures (new Fig.5 K-L). In essence, our results imply that both proteotoxic stress and iron overload act in concert to trigger cytotoxicity in RPM in vitro model. Interestingly, this phenomenon does not depend solely on the increased lipid peroxidation, but when we neutralize the latter with Liproxstatin-1, the cytotoxic effect is diminished (please, see also Results on p. 13 and Discussion p. 15/16).
- The possible central role for HO-1 in the pathophysiology of decreased RPM erythrophagocytic capacity with age is interesting. However, it is not clear how the authors arrived at this hypothesis and would be useful to evaluate in the least whether RBCs in young vs. aged mice have more hemoglobin as these changes may be primary drivers of how much HO-1 is needed during erythrophagocytosis.
Thanks for this comment. We got interested in HO-1 levels based on the RNA sequencing data, which detected lower Hmox-1 expression in aged RPMs (Figure 3-figure supplement 1). We now show that the content of hemoglobin is not significantly altered in aged RBCs (MCH parameter, Figure 1-figure supplement 5E), hence we do not think that this is the major driver for Hmox-1 downregulation. Likewise, the levels of the Bach1 message, a gene encoding Hmox-1 transcriptional repressor, are not significantly altered according to RNAseq data. Hence, the reason for the transcriptional downregulation of Hmox-1 is not clear. Of note, HO-1 protein levels in the total spleen are higher in aged versus young mice, and we also detected a clear appearance of its nuclear truncated and enzymatically-inactive form (see a figure below, we opt not to include this in the manuscript for better clarity). The appearance of truncated HO-1 seems to be partially rescued by the IR diet. It is well established that the nuclear form of HO-1 emerges via proteolytic cleavage and migrates to the nucleus under conditions of oxidative stress (Mascaro et al., 2021). This additionally confirms that the aging spleen is hallmarked by an increased burden of ROS. Moreover, we also detected HO-1 as one of the components of the protein iron-rich aggregates. Thus, we propose that the low levels of the cytoplasmic enzymatically active form of HO-1 in RPMs (that we preferentially detect with our intracellular staining and flow cytometry) may be underlain by its nuclear translocation and sequestration in protein aggregates that evade antibody binding [this is also supported by our observation that the protein aggregates, despite the high content of ferritin (as indicated by MS analysis) are negative for L-ferritin staining. Of note, we also cannot exclude that other cell types in the aging spleen (eg. lymphocytes) express higher levels of HO-1 in response to splenic oxidative stress.
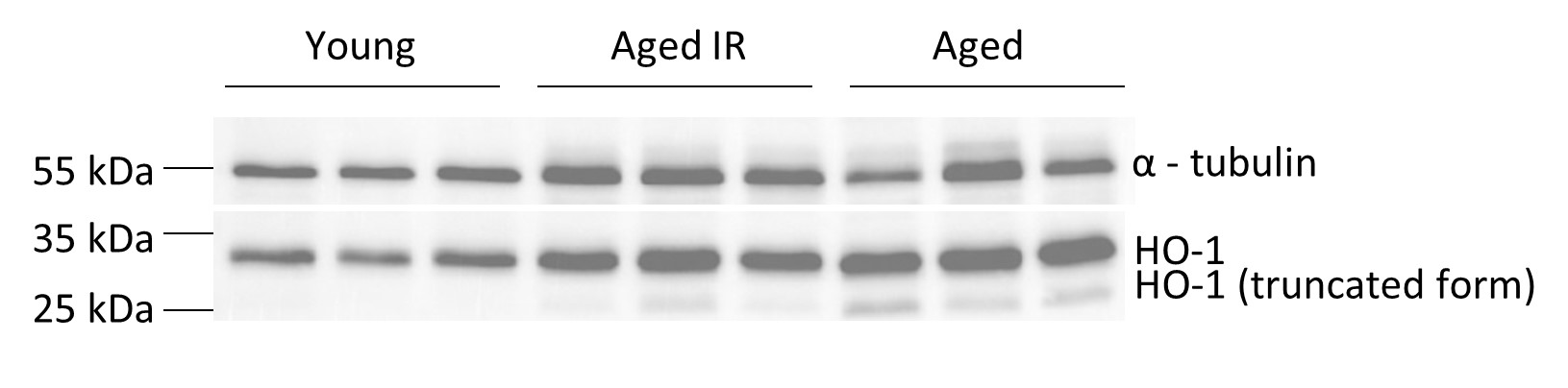
Fig. Total splenic levels of HO-1 in young, aged IR and aged mice.
Reviewer #2 (Public Review):
Slusarczyk et al. investigate the functional impairment of red pulp macrophages (RPMs) during aging. When red blood cells (RBCs) become senescent, they are recycled by RPMs via erythrophagocytosis (EP). This leads to an increase in intracellular heme and iron both of which are cytotoxic. The authors hypothesize that the continuous processing of iron by RPMs could alter their functions in an age-dependent manner. The authors used a wide variety of models: in vivo model using female mice with standard (200ppm) and restricted (25ppm) iron diet, ex vivo model using EP with splenocytes, and in vitro model with EP using iRPMs. The authors found iron accumulation in organs but markers for serum iron deficiency. They show that during aging, RPMs have a higher labile iron pool (LIP), decreased lysosomal activity with a concomitant reduction in EP. Furthermore, aging RPMs undergo ferroptosis resulting in a non-bioavailable iron deposition as intra and extracellular aggregates. Aged mice fed with an iron restricted diet restore most of the iron-recycling capacity of RPMs even though the mild-anemia remains unchanged.
Overall, I find the manuscript to be of significant potential interest. But there are important discrepancies that need to be first resolved. The proposed model is that during aging both EP and HO-1 expression decreases in RPMs but iron and ferroportin levels are elevated. In their model, the authors show intracellular iron-rich proteinaceous aggregates. But if HO-1 levels decrease, intracellular heme levels should increase. If Fpn levels increase, intracellular iron levels should decrease. How does LIP stay high in RPMs under these conditions? I find these to be major conflicting questions in the model.
We thank the Reviewer for her/his valuable feedback. As we mentioned in our replies we can only assume that a small misunderstanding in the interpretation of the presented data underlies this comment. We show that ferroportin levels in RPMs (Fig. 1F) are modulated in a manner that fully reflects the iron status of these cells (both labile and total iron levels, Figs. 1H and I). FPN levels drop in aged RPMs and are rescued when mice are maintained on a reduced iron diet. As pointed out by Reviewer#3, and explained in our replies we believe that ferroportin levels are critical for the observed phenotypes in aging. We now described our data in a more clear way to avoid any potential misinterpretation (p.6).
Reviewer #3 (Public Review):
This is a comprehensive study of the effects of aging of the function of red pulp macrophages (RPM) involved in iron recycling from erythrocytes. The authors document that insoluble iron accumulates in the spleen, that RPM become functionally impaired, and that these effects can be ameliorated by an iron-restricted diet. The study is well written, carefully done, extensively documented, and its conclusions are well supported. It is a useful and important addition for at least three distinct fields: aging, iron and macrophage biology.
The authors do not explain why an iron-restricted diet has such a strong beneficial effect on RPM aging. This is not at all obvious. I assume that the number of erythrocytes that are recycled in the spleen, and are by far the largest source of splenic iron, is not changed much by iron restriction. Is the iron retention time in macrophages changed by the diet, i.e. the recycled iron is retained for a short time when diet is iron-restricted (making hepcidin low and ferroportin high), and long time when iron is sufficient (making hepcidin high and ferroportin low)? Longer iron retention could increase damage and account for the effect. Possibly, macrophages may not empty completely of iron before having to ingest another senescent erythrocyte, and so gradually accumulate iron.
We are very grateful to this Reviewer for emphasizing the importance of the iron export capacity of RPMs as a possible driver of the observed phenotypes. Indeed, as mentioned above, we now show in the revised version of the manuscript that ferroportin drops early during aging (revised Fig. 4). Importantly, we now also observed that iron loading and limitation of iron export from iRPMs via ferroportin aggravate the impact of heat shock (a well-accepted trigger of proteotoxicity) on both protein aggregation and cell viability (new Fig. 5K and L). Physiologically, recent findings show that aging promotes a global decrease in protein solubility [BioRxiv manuscript (Sui X. et al., 2022)], and it is very likely that the constant exposure of RPMs to high iron fluxes renders these specialized cells particularly sensitive to proteome instability. This could be further aggravated by a build-up of iron due to the drop of ferroportin early during aging, ultimately leading to the appearance of the protein aggregates as early as at 5 months of age in C57BL/6J females. Based on the new data, we emphasized this model in the revised version of the manuscript (please, see Discussion on p. 16)
-
eLife assessment
Slusarczyk et al demonstrate that red pulp macrophages (RPM), specialized splenic cells that clear senescent red blood cells through erythrophagocytosis, show diminished function in aging mice. This impairment leads to retention of hemolytic red blood cells and formation of extracellular aggregates which further exacerbate RPM demise. Iron restriction alleviates most of these symptoms in aging RPMs. They propose RPM collapse as an early indicator of aging that could be reversed through iron limitation.
-
Reviewer #1 (Public Review):
Slusarczyk et al present a very well written manuscript focused on understanding the mechanisms underlying aging of erythrophagocytic macrophages in the spleen (RPM) and its relationship to iron loading with age. The manuscript is diffuse with a broad swath of data elements. Importantly, the manuscript demonstrates that RPM erythrophagocytic capacity is diminished with age, restored in iron restricted diet fed aged mice. In addition, the mechanism for declining RPM erythrophagocytic capacity appears to be ferroptosis-mediated, insensitive to heme as it is to iron, and occur independently of ROS generation. These are compelling findings. However, some of the data relies on conjecture for conclusion and a clear causal association is not clear. The main conclusion of the manuscript points to the accumulation of …
Reviewer #1 (Public Review):
Slusarczyk et al present a very well written manuscript focused on understanding the mechanisms underlying aging of erythrophagocytic macrophages in the spleen (RPM) and its relationship to iron loading with age. The manuscript is diffuse with a broad swath of data elements. Importantly, the manuscript demonstrates that RPM erythrophagocytic capacity is diminished with age, restored in iron restricted diet fed aged mice. In addition, the mechanism for declining RPM erythrophagocytic capacity appears to be ferroptosis-mediated, insensitive to heme as it is to iron, and occur independently of ROS generation. These are compelling findings. However, some of the data relies on conjecture for conclusion and a clear causal association is not clear. The main conclusion of the manuscript points to the accumulation of unavailable insoluble forms of iron as both causing and resulting from decreased RPM erythrophagocytic capacity. In addition, the finding that IR diet leads to increased TF saturation in aged mice is surprising. Furthermore, whether the finding in RPMs is intrinsic or related to RBC-related changes with aging is not addressed. Finally, these findings in a single strain and only female mice is intriguing but warrants tempered conclusions.
Major points:
- The main concern is that there is no clear explanation of why iron increases during aging although the authors appear to be saying that iron accumulation is both the cause of and a consequence of decreased RPM erythrophagocytic capacity. This requires more clarification of the main hypothesis on Page 4, line 17-18.
- It is unclear if RPMs are in limited supply. Based on the introduction (page 4, line 13-15), they have limited self-renewal capacity and blood monocytes only partially replenished. Fig 4D suggests that there is a decrease in RPMs from aged mice. The %RPM from CD45+ compartment suggests that there may just be relatively more neutrophils or fewer monocytes recruited. There is not enough clarity on the meaning of this data point point.
- Anemia of aging is a complex and poorly understood mechanistically. In general, it is considered similar to anemia of chronic inflammation with increased Epo, mild drop in Hb, and erythroid expansion, similar to ineffective erythropoiesis / low Epo responsiveness. It is not surprising that IR diet did not impact this mild anemia. However, was the MCV or MCH altered in aged and IR aged mice?
- Page 6, line 23 onward: the conclusion is that KC compensate for the decreased function of RPM in the spleen, based on the expansion of KC fraction in the liver. Is there evidence that KCs are engaged in more erythrophagocytosis in aged mice? Furthermore, iron accumulation in the liver with age does not demonstrate specifically enhanced erythrophagocytosis of KC. Please clarify why liver iron accumulation would not be simply a consequence of increased parenchymal iron similar to increased splenic iron with age, independent of erythrophagocytic activity in resident macrophages in either organ.
- Unclear whether the effect on RPMs is intrinsic or extrinsic. Would be helpful to evaluate aged iRPMs using young RBC vs. young iRPMs using old RBCs.
- Discussion of aggregates in the spleen of aged mice (Fig 2G-2K and Fig 3) is very descriptive and non-specific. For example, if the iron-rich aggregates are hemosiderin, a hemosiderin-specific stain would be helpful. This data specifically is correlatory and difficult to extract value from.
- The aging phenotype in RPMs appears to be initiated sometime after 2 months of age. However, there is some reversal of the phenotype with increasing age, e.g. Fig 4B with decreased lipid peroxidation in 9 month old relative to 6 month old RPMs. What does this mean? Why is there a partial spontaneous normalization?
- Does the aging phenotype in RPMs respond to ferristatin? It appears that NAC, which is a glutathione generator and can reverse ferroptosis, does not reverse the decreased RPM erythrophagocytic capacity observed with age yet the authors still propose that ferroptosis is involved. A response to ferristatin is a standard and acceptable approach to evaluating ferroptosis.
- The possible central role for HO-1 in the pathophysiology of decreased RPM erythrophagocytic capacity with age is interesting. However, it is not clear how the authors arrived at this hypothesis and would be useful to evaluate in the least whether RBCs in young vs. aged mice have more hemoglobin as these changes may be primary drivers of how much HO-1 is needed during erythrophagocytosis.
-
Reviewer #2 (Public Review):
Slusarczyk et al. investigate the functional impairment of red pulp macrophages (RPMs) during aging. When red blood cells (RBCs) become senescent, they are recycled by RPMs via erythrophagocytosis (EP). This leads to an increase in intracellular heme and iron both of which are cytotoxic. The authors hypothesize that the continuous processing of iron by RPMs could alter their functions in an age-dependent manner. The authors used a wide variety of models: in vivo model using female mice with standard (200ppm) and restricted (25ppm) iron diet, ex vivo model using EP with splenocytes, and in vitro model with EP using iRPMs. The authors found iron accumulation in organs but markers for serum iron deficiency. They show that during aging, RPMs have a higher labile iron pool (LIP), decreased lysosomal activity with …
Reviewer #2 (Public Review):
Slusarczyk et al. investigate the functional impairment of red pulp macrophages (RPMs) during aging. When red blood cells (RBCs) become senescent, they are recycled by RPMs via erythrophagocytosis (EP). This leads to an increase in intracellular heme and iron both of which are cytotoxic. The authors hypothesize that the continuous processing of iron by RPMs could alter their functions in an age-dependent manner. The authors used a wide variety of models: in vivo model using female mice with standard (200ppm) and restricted (25ppm) iron diet, ex vivo model using EP with splenocytes, and in vitro model with EP using iRPMs. The authors found iron accumulation in organs but markers for serum iron deficiency. They show that during aging, RPMs have a higher labile iron pool (LIP), decreased lysosomal activity with a concomitant reduction in EP. Furthermore, aging RPMs undergo ferroptosis resulting in a non-bioavailable iron deposition as intra and extracellular aggregates. Aged mice fed with an iron restricted diet restore most of the iron-recycling capacity of RPMs even though the mild-anemia remains unchanged.
Overall, I find the manuscript to be of significant potential interest. But there are important discrepancies that need to be first resolved. The proposed model is that during aging both EP and HO-1 expression decreases in RPMs but iron and ferroportin levels are elevated. In their model, the authors show intracellular iron-rich proteinaceous aggregates. But if HO-1 levels decrease, intracellular heme levels should increase. If Fpn levels increase, intracellular iron levels should decrease. How does LIP stay high in RPMs under these conditions? I find these to be major conflicting questions in the model.
-
Reviewer #3 (Public Review):
This is a comprehensive study of the effects of aging of the function of red pulp macrophages (RPM) involved in iron recycling from erythrocytes. The authors document that insoluble iron accumulates in the spleen, that RPM become functionally impaired, and that these effects can be ameliorated by an iron-restricted diet. The study is well written, carefully done, extensively documented, and its conclusions are well supported. It is a useful and important addition for at least three distinct fields: aging, iron and macrophage biology.
The authors do not explain why an iron-restricted diet has such a strong beneficial effect on RPM aging. This is not at all obvious. I assume that the number of erythrocytes that are recycled in the spleen, and are by far the largest source of splenic iron, is not changed much …
Reviewer #3 (Public Review):
This is a comprehensive study of the effects of aging of the function of red pulp macrophages (RPM) involved in iron recycling from erythrocytes. The authors document that insoluble iron accumulates in the spleen, that RPM become functionally impaired, and that these effects can be ameliorated by an iron-restricted diet. The study is well written, carefully done, extensively documented, and its conclusions are well supported. It is a useful and important addition for at least three distinct fields: aging, iron and macrophage biology.
The authors do not explain why an iron-restricted diet has such a strong beneficial effect on RPM aging. This is not at all obvious. I assume that the number of erythrocytes that are recycled in the spleen, and are by far the largest source of splenic iron, is not changed much by iron restriction. Is the iron retention time in macrophages changed by the diet, i.e. the recycled iron is retained for a short time when diet is iron-restricted (making hepcidin low and ferroportin high), and long time when iron is sufficient (making hepcidin high and ferroportin low)? Longer iron retention could increase damage and account for the effect. Possibly, macrophages may not empty completely of iron before having to ingest another senescent erythrocyte, and so gradually accumulate iron.
-
