Environmental DNA from archived leaves reveals widespread temporal turnover and biotic homogenization in forest arthropod communities
Curation statements for this article:-
Curated by eLife

eLife assessment
We admired the study by Krehenwinkel and colleagues for its novelty, depth, and ecological breadth, but have questions regarding the laboratory, bioinformatic and statistical methodologies that require clarification. It is likely to make a substantial impact in the field of plant-based arthropod metabarcoding, revealing ecological insights that can be derived from existing bio-banked material. The work, which creatively exploits herbarium material to track arthropod communities, will be interesting to a general audience in addition to ecologists, foresters, phytopathologists, and industry.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
A major limitation of current reports on insect declines is the lack of standardized, long-term, and taxonomically broad time series. Here, we demonstrate the utility of environmental DNA from archived leaf material to characterize plant-associated arthropod communities. We base our work on several multi-decadal leaf time series from tree canopies in four land use types, which were sampled as part of a long-term environmental monitoring program across Germany. Using these highly standardized and well-preserved samples, we analyze temporal changes in communities of several thousand arthropod species belonging to 23 orders using metabarcoding and quantitative PCR. Our data do not support widespread declines of α -diversity or genetic variation within sites. Instead, we find a gradual community turnover, which results in temporal and spatial biotic homogenization, across all land use types and all arthropod orders. Our results suggest that insect decline is more complex than mere α -diversity loss, but can be driven by β -diversity decay across space and time.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
In one of the most creative eDNA studies I have had the pleasure to review, the authors have taken advantage of an existing program several decades old to address whether insect declines are indeed occurring - an active area of discussion and debate within ecology. Here, they extracted arthropod environmental DNA (eDNA) from pulverized leaf samples collected from different tree species across different habitats. Their aim was to assess the arthropod community composition within the canopies of these trees during the time of collection to assess whether arthropod richness, diversity, and biomass were declining. By utilizing these leaf samples, the greatest shortcoming of assessing arthropod declines - the lack of historical data to compare to - was overcome, and strong timeseries evidence …
Author Response
Reviewer #1 (Public Review):
In one of the most creative eDNA studies I have had the pleasure to review, the authors have taken advantage of an existing program several decades old to address whether insect declines are indeed occurring - an active area of discussion and debate within ecology. Here, they extracted arthropod environmental DNA (eDNA) from pulverized leaf samples collected from different tree species across different habitats. Their aim was to assess the arthropod community composition within the canopies of these trees during the time of collection to assess whether arthropod richness, diversity, and biomass were declining. By utilizing these leaf samples, the greatest shortcoming of assessing arthropod declines - the lack of historical data to compare to - was overcome, and strong timeseries evidence can now be used to inform the discussion. Through their use of eDNA metabarcoding, they were able to determine that richness was not declining, but there was evidence of beta diversity loss due to biotic homogenization occurring across different habitats. Furthermore, their application of qPCR to assess changes in eDNA copy number temporally and associate those changes with changes to arthropod biomass provided support to the argument that arthropod biomass is indeed declining. Taken together, these data add substantial weight to the current discussion regarding how arthropods are being affected in the Anthropocene.
Thank you very much for the positive assessment of our work.
I find the conclusions of the paper to be sound and mostly defensible, though there are some issues to take note of that may undermine these findings.
Firstly, I saw no explanation of the requisite controls for such an experiment. An experiment of this scale should have detailed explanations of the field/equipment controls, extraction controls, and PCR controls to ensure there are no contamination issues that would otherwise undermine the entirety of the study. At one point in the manuscript the presence of controls is mentioned just once, so I surmise they must exist. Trusting such results needs to be taken with caution until such evidence is clearly outlined. Furthermore, the plate layout which includes these controls would help assess the extent of tag-jumping, should the plate plan proposed in Taberlet et al., 2018 be adopted.
Second, without the presence of adequate controls, filtering schemes would be unable to determine whether there were contaminants and also be unable to remove them. This would also prevent samples from being filtered out should there be excessive levels of contamination present. Without such information, it makes it difficult to fully trust the data as presented.
Finally, there is insufficient detail regarding the decontamination procedures of equipment used to prepare the samples (e.g., the cryomil). Without clear explanations of the steps the authors took to ensure samples were handled and prepared correctly, there is yet more concern that there may be unseen problems with the dataset.
We are well aware of the potential issues and consequences of contamination in our work. However, we are also confident that our field and laboratory procedures adequately rule out these issues. We agree with the reviewer that we should expand more on our reasoning. Hence, we have now significantly expanded the Methods section outlining controls and sample purity, particularly under “Tree samples of the German Environmental Specimen Bank – Standardized time series samples stored at ultra-low temperatures” (lines 303-304), “Test for DNA carryover in the cryomill” (lines 448-464) and “Statistical analysis” (lines 570-575).
We ran negative control extractions as well as negative control PCRs with all samples. These controls were sequenced along with all samples and used to explore the effect of experimental contamination. With the exception of a few reads of abundant taxa, these controls were mostly clean. We report this in more detail now in the Methods under “Sequence analysis” (lines 570-575). This suggests that our data are free of experimental contamination or tag jumping issues.
We have also expanded on the avoidance of contamination in our field sampling protocols. The ESB has been set up for monitoring even the tiniest trace amounts of chemicals. Carryover between samples would render the samples useless. Hence, highly clean and standardized protocols are implemented. All samples are only collected with sterilized equipment under sterile conditions. Each piece of equipment is thoroughly decontaminated before sampling.
The cryomill is another potential source of cross-contamination. The mill is disassembled after each sample and thoroughly cleaned. Milled samples have already been tested for chemical carryover, and none was found. We have now added an additional analysis to rule out DNA carryover. We received the milling schedule of samples for the past years. Assuming samples get contaminated by carryover between milling runs, two consecutive samples should show signatures of this carryover. We tested this for singletaxon carryover as well as community-wide beta diversity, but did not find any signal of contamination. This gives us confidence that our samples are very pure. The results of this test are now reported in the manuscript (Suppl. Fig 12 & Suppl. Table 3).
Reviewer #2 (Public Review):
Krehenwinkel et al. investigated the long-term temporal dynamics of arthropod communities using environmental DNA (eDNA) remained in archived leave samples. The authors first developed a method to recover arthropod eDNA from archived leave samples and carefully tested whether the developed method could reasonably reveal the dynamics of arthropod communities where the leave samples originated. Then, using the eDNA method, the authors analyzed 30-year-long well-archived tree leaf samples in Germany and reconstructed the long-term temporal dynamics of arthropod communities associated with the tree species. The reconstructed time series includes several thousand arthropod species belonging to 23 orders, and the authors found interesting patterns in the time series. Contrary to some previous studies, the authors did not find widespread temporal α-diversity (OTU richness and haplotype diversity) declines. Instead, β-diversity among study sites gradually decreased, suggesting that the arthropod communities are more spatially homogenized in recent years. Overall, the authors suggested that the temporal dynamics of arthropod communities may be complex and involve changes in α- and β-diversity and demonstrated the usefulness of their unique eDNA-based approach.
Strengths:
The authors' idea that using eDNA remained in archived leave samples is unique and potentially applicable to other systems. For example, different types of specimens archived in museums may be utilized for reconstructing long-term community dynamics of other organisms, which would be beneficial for understanding and predicting ecosystem dynamics.
A great strength of this work is that the authors very carefully tested their method. For example, the authors tested the effects of powdered leaves input weights, sampling methods, storing methods, PCR primers, and days from last precipitation to sampling on the eDNA metabarcoding results. The results showed that the tested variables did not significantly impact the eDNA metabarcoding results, which convinced me that the proposed method reasonably recovers arthropod eDNA from the archived leaf samples. Furthermore, the authors developed a method that can separately quantify 18S DNA copy numbers of arthropods and plants, which enables the estimations of relative arthropod eDNA copy numbers. While most eDNA studies provide relative abundance only, the DNA copy numbers measured in this study provide valuable information on arthropod community dynamics.
Overall, the authors' idea is excellent, and I believe that the developed eDNA methodology reasonably reconstructed the long-term temporal dynamics of the target organisms, which are major strengths of this study.
Thank you very much for the positive assessment of our work.
Weaknesses:
Although this work has major strengths in the eDNA experimental part, there are concerns in DNA sequence processing and statistical analyses.
Statistical methods to analyze the temporal trend are too simplistic. The methods used in the study did not consider possible autocorrelation and other structures that the eDNA time series might have. It is well known that the applications of simple linear models to time series with autocorrelation structure incorrectly detect a "significant" temporal trend. For example, a linear model can often detect a significant trend even in a random walk time series.
We have now reanalyzed our data controlling for autocorrelation and for non-linear changes of abundance and recover no change to our results. We have added this information to the manuscript under “Statistical analysis” (lines 629-644).
Also, there are some issues regarding the DNA sequence analysis and the subsequent use of the results. For example, read abundance was used in the statistical model, but the read abundance cannot be a proxy for species abundance/biomass. Because the total 18S DNA copy numbers of arthropods were quantified in the study, multiplying the sequence-based relative abundance by the total 18S DNA copy numbers may produce a better proxy of the abundance of arthropods, and the use of such a better proxy would be more appropriate here. In addition, a coverage-based rarefaction enables a more rigorous comparison of diversity (OTU diversity or haplotype diversity) than the readbased rarefaction does.
We did not use read abundance as a proxy for abundance, but used our qPCR approach to measure relative copy number of arthropods. While there are biases to this (see our explanations above), the assay proved very reliable and robust. We thus believe it should indeed provide a rough estimate of biomass. As biomass is very commonly discussed in insect decline (in fact the first study on insect decline entirely relies on biomass; Hallmann et al. 2017), we feel it is important go include a proxy for this as well. However, we also discuss the alternative option that a turnover of diversity is affecting the measured biomass. A pattern of abundance loss for common species has been described in other works on insect decline.
We liked the reviewer’s suggestion to use copy number information to perform abundance-informed rarefaction. We have done this now and added an additional analysis rarefying by copy number/biomass. A parallel analysis using this newly rarefied table was done for the total diversity as well as single species abundance change. Details can be found in the Methods and Results section of the manuscript. However, the result essentially remains the same. Even abundance-informed rarefaction does not lead to a pattern of loss of species richness over time (see “Statistical analysis”).
The overall results are supporting a scenario of no overall loss of species richness over time, but a loss of abundance for common species. And we indeed see the pattern of declining abundance for once-common species in our data, for example the loss of the Green Silver-Line moth, once a very common species in beech canopy (Suppl. Fig. 10). We have added details on this to the Discussion (lines 254-260).
These points may significantly impact the conclusions of this work.
Reviewer #3 (Public Review):
The aim of Weber and colleagues' study was to generate arthropod environmental DNA extracted from a unique 30-year time series of deep-frozen leaf material sampled at 24 German sites, that represent four different land use types. Using this dataset, they explore how the arthropod community has changed through time in these sites, using both conventional metabarcoding to reconstruct the OTUs present, and a new qPCR assay developed to estimate the overall arthropod diversity on the collected material. Overall their results show that while no clear changes in alpha diversity are found, the βdiversity dropped significantly over time in many sites, most notable in the beech forests. Overall I believe their data supports these findings, and thus their conclusion that diversity is becoming homogenized through time is valid.
Thank you for the positive assessment.
While overall I do not doubt the general findings, I have a number of comments. Firstly while I agree this is a very nice study on a unique dataset - other temporal datasets of insects that were used for eDNA studies do exist, and perhaps it would be relevant to put the findings into context (or even the study design) of other work that has been done on such datasets. One example that jumps to my mind is Thomsen et al. 2015 https://besjournals.onlinelibrary.wiley.com/doi/full/10.1111/1365-2656.12452 but I am sure there are others.
We have expanded the introduction and discussion on this citing this among other studies now (lines 71-72, 276-278).
From a technical point of view, the conclusions of course rely on several assumptions, including (1) that the biomass assay is effective and (2) that the reconstructed levels of OTU diversity are accurate,
With regards to biomass although it is stated in the manuscript that "Relative eDNA copy number should be a predictor for relative biomass ", this is in fact only true if one assumes a number of things, e.g. there is a similar copy number of 18s rDNA per species, similar numbers of mtDNA per cell, a similar number of cells per individual species etc. In this regard, on the positive side, it is gratifying to see that the authors perform a validation assay on 7 mock controls, and these seem to indicate the assay works well. Given how critical this is, I recommend discussing the details of this a bit more, and why the authors are convinced the assay is effective in the main text so that the reader is able to fully decide if they are in agreement. However perhaps on the negative side, I am concerned about the strategy taken to perform the qPCR may have not been ideal. Specifically, the assay is based on nested PCR, where the authors first perform a 15cycle amplification, this product is purified, then put into a subsequent qPCR. Given how both PCR is notorious for introducing amplification biases in general (especially when performed on low levels of DNA), and the fact that nested PCRs are notoriously contamination prone - this approach seems to be asking for trouble. This raises the question - why not just do the qPCR directly on the extracts (one can still dilute the plant DNA 100x prior to qPCR if needed). Further, given the qPCRs were run in triplicate I think the full data (Ct values) for this should be released (as opposed to just stating in the paper that the average values were used). In this way, the readers will be able to judge how replicable the assay was - something I think is critical given how noisy the patterns in Fig S10 seem to be.
We agree with this point, and this is why we do not want to overstate the decline in copy number. This is an additional source of data next to genetic and species diversity. We have added to our discussion of turnover as another potential driver of copy number change (lines 257-260). We have also added text addressing the robustness of the mock community assay (lines 138-141).
However, we are confident of the reliability and robustness of our qPCR assay for the detection of relative arthropod copy number. We performed several validations and optimizations before using the assay. We have added additional details to the manuscript on this (see “Detection of relative arthropod DNA copy number using quantitative PCR”, lines 548-556). We got the idea for the nested qPCR from a study (Tran et al.) showing its high accuracy and reproducibility. We show that our assay has a very high replicability using triplicates of each qPCR, which we will now include in the supplementary data on Dryad. The SD of Ct values is very low (~ 0.1 on average). NTC were run with all qPCRs to rule out contamination as an issue in the experiments. We also find a very high efficiency of the assay. At dilutions far outside the observed copy number in our actual leaf data, we still find the assay to be accurate. We found very comparable abundance changes across our highly taxonomically diverse mock communities. This also suggests that abundance changes are a more likely explanation than simple turnover for the observed drop in copy number. A biomass loss for common species is well in line with recent reports on insect decline. We can also rely on several other mock community studies (Krehenwinkel et al. 2017 & 2019) where we used read abundance of 18S and found it to be a relatively good predictor of relative biomass.
The pattern in Fig. S10 is not really noisy. It just reflects typical population fluctuations for arthropods. Most arthropod taxa undergo very pronounced temporal abundance fluctuations between years.
Next, with regards to the observation that the results reveal an overall decrease in arthropod biomass over time: The authors suggest one alternate to their theory, that the dropping DNA copy number may reflect taxonomic turnover of species with different eDNA shedding rates. Could there be another potential explanation - simply be that leaves are getting denser/larger? Can this be ruled out in some way, e.g. via data on leaf mass through time for these trees? (From this dataset or indeed any other place).
This is a very good point. However, we can rule out this hypothesis, as the ESB performs intensive biometric data analysis. The average leaf weight and water content have not significantly changed in our sites. We have addressed this in the Methods section (see ”Tree samples of the German Environmental Specimen Bank – Standardized time series samples stored at ultra-low temperatures”, lines 308-311).
With regards to estimates of OTU/zOTU diversity. The authors state in the manuscript that zOTUs represent individual haplotypes, thus genetic variation within species. This is only true if they do not represent PCR and/or sequencing errors. Perhaps therefore they would be able to elaborate (for the non-computational/eDNA specialist reader) on why their sequence processing methods rule out this possibility? One very good bit of evidence would be that identical haplotypes for the individual species are found in the replicate PCRs. Or even between different extractions at single locations/timepoints.
We have repeated the analysis of genetic variation with much more stringent filtering criteria (see “Statistical analysis”, lines 611-615). Among other filtering steps, this also includes the use of only those zOTUs that occur in both technical replicates, as suggested by the reviewer. Another reason to make us believe we are dealing with true haplotypic variation here is that haplotypes show geographic variation. E.g., some haplotypes are more abundant in some sites than in others. NUMTS would consistently show a simple correlation in their abundance with the most abundant true haplotype.
With regards to the bigger picture, one thing I found very interesting from a technical point of view is that the authors explored how modifying the mass of plant material used in the extraction affects the overall results, and basically find that using more than 200mg provides no real advantage. In this regard, I draw the authors and readers attention to an excellent paper by Mata et al. (https://onlinelibrary.wiley.com/doi/full/10.1111/mec.14779) - where these authors compare the effect of increasing the amount of bat faeces used in a bat diet metabarcoding study, on the OTUs generated. Essentially Mata and colleagues report that as the amount of faeces increases, the rare taxa (e.g. those found at a low level in a single faeces) get lost - they are simply diluted out by the common taxa (e.g those in all faeces). In contrast, increasing biological replicates (in their case more individual faecal samples) increased diversity. I think these results are relevant in the context of the experiment described in this new manuscript, as they seem to show similar results - there is no benefit of considerably increasing the amount of leaf tissue used. And if so, this seems to point to a general principal of relevance to the design of metabarcoding studies, thus of likely wide interest.
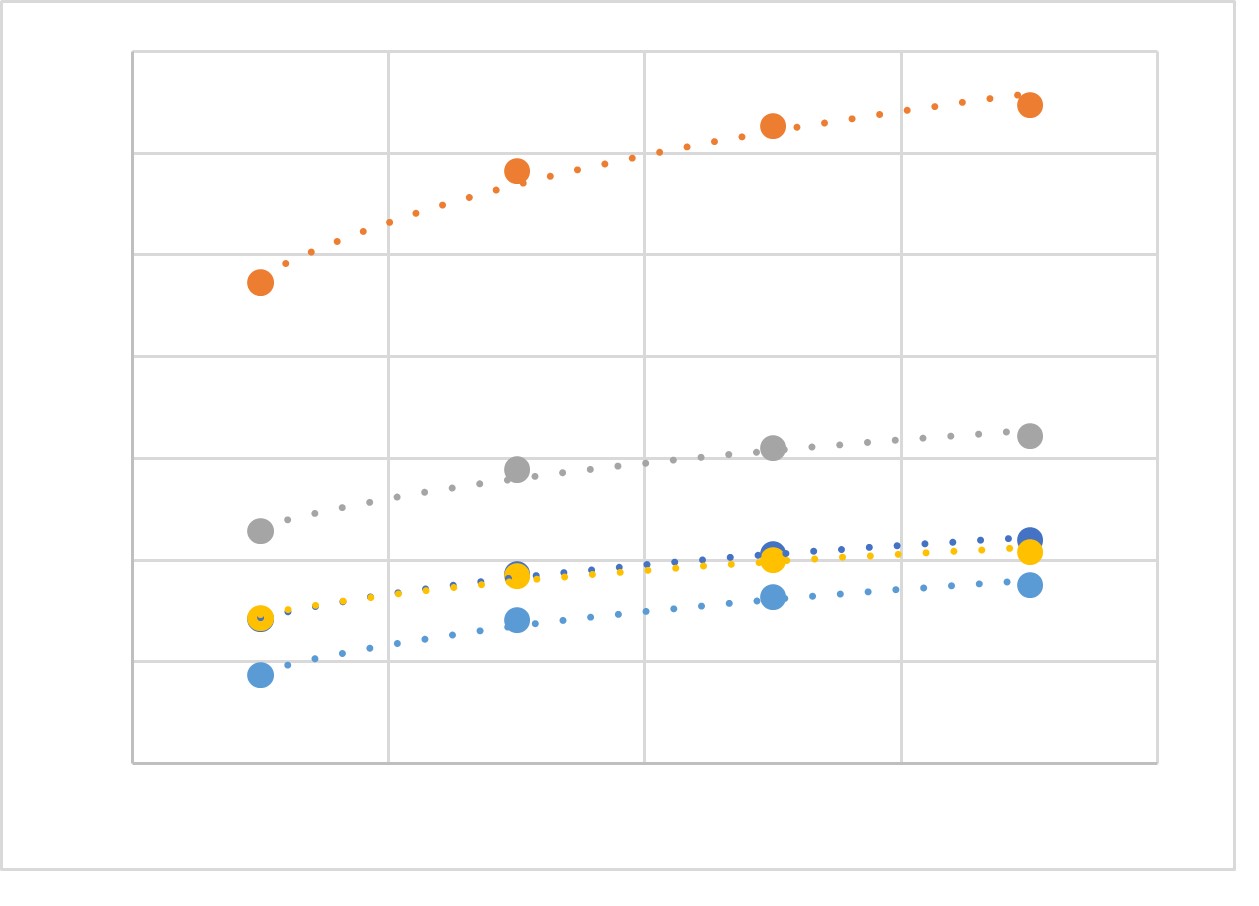
Thank you for this interesting study, which we were not aware of before. The cryomilling is an extremely efficient approach to equally disperse even traces of chemicals in a sample. This has been established for trace chemicals early during the operation of the ESB, but also seems to hold true for eDNA in the samples. We have recently done more replication experiments from different ESB samples (different terrestrial and marine samples for different taxonomic groups) and find that replication of extraction does not provide much more benefit than replication of PCR. Even after 2 replicates, diversity approaches saturation. This can be seen in the plot below, which shows recovered eDNA diversity for different ESB samples and different taxonomic groups from 1-4 replicates. A single extract of a small volume contains DNA from nearly all taxa in the community. Rare taxa can be enriched with more PCR replicates.
-
eLife assessment
We admired the study by Krehenwinkel and colleagues for its novelty, depth, and ecological breadth, but have questions regarding the laboratory, bioinformatic and statistical methodologies that require clarification. It is likely to make a substantial impact in the field of plant-based arthropod metabarcoding, revealing ecological insights that can be derived from existing bio-banked material. The work, which creatively exploits herbarium material to track arthropod communities, will be interesting to a general audience in addition to ecologists, foresters, phytopathologists, and industry.
-
Reviewer #1 (Public Review):
In one of the most creative eDNA studies I have had the pleasure to review, the authors have taken advantage of an existing program several decades old to address whether insect declines are indeed occurring - an active area of discussion and debate within ecology. Here, they extracted arthropod environmental DNA (eDNA) from pulverized leaf samples collected from different tree species across different habitats. Their aim was to assess the arthropod community composition within the canopies of these trees during the time of collection to assess whether arthropod richness, diversity, and biomass were declining. By utilizing these leaf samples, the greatest shortcoming of assessing arthropod declines - the lack of historical data to compare to - was overcome, and strong timeseries evidence can now be used to …
Reviewer #1 (Public Review):
In one of the most creative eDNA studies I have had the pleasure to review, the authors have taken advantage of an existing program several decades old to address whether insect declines are indeed occurring - an active area of discussion and debate within ecology. Here, they extracted arthropod environmental DNA (eDNA) from pulverized leaf samples collected from different tree species across different habitats. Their aim was to assess the arthropod community composition within the canopies of these trees during the time of collection to assess whether arthropod richness, diversity, and biomass were declining. By utilizing these leaf samples, the greatest shortcoming of assessing arthropod declines - the lack of historical data to compare to - was overcome, and strong timeseries evidence can now be used to inform the discussion. Through their use of eDNA metabarcoding, they were able to determine that richness was not declining, but there was evidence of beta diversity loss due to biotic homogenization occurring across different habitats. Furthermore, their application of qPCR to assess changes in eDNA copy number temporally and associate those changes with changes to arthropod biomass provided support to the argument that arthropod biomass is indeed declining. Taken together, these data add substantial weight to the current discussion regarding how arthropods are being affected in the Anthropocene.
I find the conclusions of the paper to be sound and mostly defensible, though there are some issues to take note of that may undermine these findings.
Firstly, I saw no explanation of the requisite controls for such an experiment. An experiment of this scale should have detailed explanations of the field/equipment controls, extraction controls, and PCR controls to ensure there are no contamination issues that would otherwise undermine the entirety of the study. At one point in the manuscript the presence of controls is mentioned just once, so I surmise they must exist. Trusting such results needs to be taken with caution until such evidence is clearly outlined. Furthermore, the plate layout which includes these controls would help assess the extent of tag-jumping, should the plate plan proposed in Taberlet et al., 2018 be adopted.
Second, without the presence of adequate controls, filtering schemes would be unable to determine whether there were contaminants and also be unable to remove them. This would also prevent samples from being filtered out should there be excessive levels of contamination present. Without such information, it makes it difficult to fully trust the data as presented.
Finally, there is insufficient detail regarding the decontamination procedures of equipment used to prepare the samples (e.g., the cryomil). Without clear explanations of the steps the authors took to ensure samples were handled and prepared correctly, there is yet more concern that there may be unseen problems with the dataset.
-
Reviewer #2 (Public Review):
Krehenwinkel et al. investigated the long-term temporal dynamics of arthropod communities using environmental DNA (eDNA) remained in archived leave samples. The authors first developed a method to recover arthropod eDNA from archived leave samples and carefully tested whether the developed method could reasonably reveal the dynamics of arthropod communities where the leave samples originated. Then, using the eDNA method, the authors analyzed 30-year-long well-archived tree leaf samples in Germany and reconstructed the long-term temporal dynamics of arthropod communities associated with the tree species. The reconstructed time series includes several thousand arthropod species belonging to 23 orders, and the authors found interesting patterns in the time series. Contrary to some previous studies, the authors …
Reviewer #2 (Public Review):
Krehenwinkel et al. investigated the long-term temporal dynamics of arthropod communities using environmental DNA (eDNA) remained in archived leave samples. The authors first developed a method to recover arthropod eDNA from archived leave samples and carefully tested whether the developed method could reasonably reveal the dynamics of arthropod communities where the leave samples originated. Then, using the eDNA method, the authors analyzed 30-year-long well-archived tree leaf samples in Germany and reconstructed the long-term temporal dynamics of arthropod communities associated with the tree species. The reconstructed time series includes several thousand arthropod species belonging to 23 orders, and the authors found interesting patterns in the time series. Contrary to some previous studies, the authors did not find widespread temporal α-diversity (OTU richness and haplotype diversity) declines. Instead, β-diversity among study sites gradually decreased, suggesting that the arthropod communities are more spatially homogenized in recent years. Overall, the authors suggested that the temporal dynamics of arthropod communities may be complex and involve changes in α- and β-diversity and demonstrated the usefulness of their unique eDNA-based approach.
Strengths:
The authors' idea that using eDNA remained in archived leave samples is unique and potentially applicable to other systems. For example, different types of specimens archived in museums may be utilized for reconstructing long-term community dynamics of other organisms, which would be beneficial for understanding and predicting ecosystem dynamics.A great strength of this work is that the authors very carefully tested their method. For example, the authors tested the effects of powdered leaves input weights, sampling methods, storing methods, PCR primers, and days from last precipitation to sampling on the eDNA metabarcoding results. The results showed that the tested variables did not significantly impact the eDNA metabarcoding results, which convinced me that the proposed method reasonably recovers arthropod eDNA from the archived leaf samples. Furthermore, the authors developed a method that can separately quantify 18S DNA copy numbers of arthropods and plants, which enables the estimations of relative arthropod eDNA copy numbers. While most eDNA studies provide relative abundance only, the DNA copy numbers measured in this study provide valuable information on arthropod community dynamics.
Overall, the authors' idea is excellent, and I believe that the developed eDNA methodology reasonably reconstructed the long-term temporal dynamics of the target organisms, which are major strengths of this study.
Weaknesses:
Although this work has major strengths in the eDNA experimental part, there are concerns in DNA sequence processing and statistical analyses.Statistical methods to analyze the temporal trend are too simplistic. The methods used in the study did not consider possible autocorrelation and other structures that the eDNA time series might have. It is well known that the applications of simple linear models to time series with autocorrelation structure incorrectly detect a "significant" temporal trend. For example, a linear model can often detect a significant trend even in a random walk time series.
Also, there are some issues regarding the DNA sequence analysis and the subsequent use of the results. For example, read abundance was used in the statistical model, but the read abundance cannot be a proxy for species abundance/biomass. Because the total 18S DNA copy numbers of arthropods were quantified in the study, multiplying the sequence-based relative abundance by the total 18S DNA copy numbers may produce a better proxy of the abundance of arthropods, and the use of such a better proxy would be more appropriate here. In addition, a coverage-based rarefaction enables a more rigorous comparison of diversity (OTU diversity or haplotype diversity) than the read-based rarefaction does.
These points may significantly impact the conclusions of this work.
-
Reviewer #3 (Public Review):
The aim of Weber and colleagues' study was to generate arthropod environmental DNA extracted from a unique 30-year time series of deep-frozen leaf material sampled at 24 German sites, that represent four different land use types. Using this dataset, they explore how the arthropod community has changed through time in these sites, using both conventional metabarcoding to reconstruct the OTUs present, and a new qPCR assay developed to estimate the overall arthropod diversity on the collected material. Overall their results show that while no clear changes in alpha diversity are found, the β-diversity dropped significantly over time in many sites, most notable in the beech forests. Overall I believe their data supports these findings, and thus their conclusion that diversity is becoming homogenized through time …
Reviewer #3 (Public Review):
The aim of Weber and colleagues' study was to generate arthropod environmental DNA extracted from a unique 30-year time series of deep-frozen leaf material sampled at 24 German sites, that represent four different land use types. Using this dataset, they explore how the arthropod community has changed through time in these sites, using both conventional metabarcoding to reconstruct the OTUs present, and a new qPCR assay developed to estimate the overall arthropod diversity on the collected material. Overall their results show that while no clear changes in alpha diversity are found, the β-diversity dropped significantly over time in many sites, most notable in the beech forests. Overall I believe their data supports these findings, and thus their conclusion that diversity is becoming homogenized through time is valid.
While overall I do not doubt the general findings, I have a number of comments. Firstly while I agree this is a very nice study on a unique dataset - other temporal datasets of insects that were used for eDNA studies do exist, and perhaps it would be relevant to put the findings into context (or even the study design) of other work that has been done on such datasets. One example that jumps to my mind is Thomsen et al. 2015 https://besjournals.onlinelibrary.wiley.com/doi/full/10.1111/1365-2656.12452 but I am sure there are others.
From a technical point of view, the conclusions of course rely on several assumptions, including (1) that the biomass assay is effective and (2) that the reconstructed levels of OTU diversity are accurate,
With regards to biomass although it is stated in the manuscript that "Relative eDNA copy number should be a predictor for relative biomass ", this is in fact only true if one assumes a number of things, e.g. there is a similar copy number of 18s rDNA per species, similar numbers of mtDNA per cell, a similar number of cells per individual species etc. In this regard, on the positive side, it is gratifying to see that the authors perform a validation assay on 7 mock controls, and these seem to indicate the assay works well. Given how critical this is, I recommend discussing the details of this a bit more, and why the authors are convinced the assay is effective in the main text so that the reader is able to fully decide if they are in agreement. However perhaps on the negative side, I am concerned about the strategy taken to perform the qPCR may have not been ideal. Specifically, the assay is based on nested PCR, where the authors first perform a 15cycle amplification, this product is purified, then put into a subsequent qPCR. Given how both PCR is notorious for introducing amplification biases in general (especially when performed on low levels of DNA), and the fact that nested PCRs are notoriously contamination prone - this approach seems to be asking for trouble. This raises the question - why not just do the qPCR directly on the extracts (one can still dilute the plant DNA 100x prior to qPCR if needed). Further, given the qPCRs were run in triplicate I think the full data (Ct values) for this should be released (as opposed to just stating in the paper that the average values were used). In this way, the readers will be able to judge how replicable the assay was - something I think is critical given how noisy the patterns in Fig S10 seem to be.
Next, with regards to the observation that the results reveal an overall decrease in arthropod biomass over time: The authors suggest one alternate to their theory, that the dropping DNA copy number may reflect taxonomic turnover of species with different eDNA shedding rates. Could there be another potential explanation - simply be that leaves are getting denser/larger? Can this be ruled out in some way, e.g. via data on leaf mass through time for these trees? (From this dataset or indeed any other place).
With regards to estimates of OTU/zOTU diversity. The authors state in the manuscript that zOTUs represent individual haplotypes, thus genetic variation within species. This is only true if they do not represent PCR and/or sequencing errors. Perhaps therefore they would be able to elaborate (for the non-computational/eDNA specialist reader) on why their sequence processing methods rule out this possibility? One very good bit of evidence would be that identical haplotypes for the individual species are found in the replicate PCRs. Or even between different extractions at single locations/timepoints.
With regards to the bigger picture, one thing I found very interesting from a technical point of view is that the authors explored how modifying the mass of plant material used in the extraction affects the overall results, and basically find that using more than 200mg provides no real advantage. In this regard, I draw the authors and readers attention to an excellent paper by Mata et al. (https://onlinelibrary.wiley.com/doi/full/10.1111/mec.14779) - where these authors compare the effect of increasing the amount of bat faeces used in a bat diet metabarcoding study, on the OTUs generated. Essentially Mata and colleagues report that as the amount of faeces increases, the rare taxa (e.g. those found at a low level in a single faeces) get lost - they are simply diluted out by the common taxa (e.g those in all faeces). In contrast, increasing biological replicates (in their case more individual faecal samples) increased diversity. I think these results are relevant in the context of the experiment described in this new manuscript, as they seem to show similar results - there is no benefit of considerably increasing the amount of leaf tissue used. And if so, this seems to point to a general principal of relevance to the design of metabarcoding studies, thus of likely wide interest.
-
