Isoform-specific mutation in Dystonin-b gene causes late-onset protein aggregate myopathy and cardiomyopathy
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
The authors demonstrate that isoform-specific Dystonin-b (Dst-b) mutant mice show significant myopathy in skeletal and cardiac muscle at older ages without the peripheral neuropathy or post-natal lethality that are commonly observed by loss of function of the DST gene. The study provides novel information about the role of the Dst-b isoform in maintaining skeletal and cardiac muscle health. In addition, the study suggests that isoform-specific mutations in Dst-b gene may cause some hereditary skeletal and cardiac myopathies.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 agreed to share their name with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Dystonin ( DST ), which encodes cytoskeletal linker proteins, expresses three tissue-selective isoforms: neural DST-a, muscular DST-b, and epithelial DST-e. DST mutations cause different disorders, including hereditary sensory and autonomic neuropathy 6 (HSAN-VI) and epidermolysis bullosa simplex; however, etiology of the muscle phenotype in DST -related diseases has been unclear. Because DST-b contains all of the DST-a -encoding exons, known HSAN-VI mutations could affect both DST-a and DST-b isoforms. To investigate the specific function of DST-b in striated muscles, we generated a Dst-b -specific mutant mouse model harboring a nonsense mutation. Dst-b mutant mice exhibited late-onset protein aggregate myopathy and cardiomyopathy without neuropathy. We observed desmin aggregation, focal myofibrillar dissolution, and mitochondrial accumulation in striated muscles, which are common characteristics of myofibrillar myopathy. We also found nuclear inclusions containing p62, ubiquitin, and SUMO proteins with nuclear envelope invaginations as a unique pathological hallmark in Dst-b mutation-induced cardiomyopathy. RNA-sequencing analysis revealed changes in expression of genes responsible for cardiovascular functions. In silico analysis identified DST-b alleles with nonsense mutations in populations worldwide, suggesting that some unidentified hereditary myopathy and cardiomyopathy are caused by DST-b mutations. Here, we demonstrate that the Dst-b isoform is essential for long-term maintenance of striated muscles.
Article activity feed
-

-

Author Response
Reviewer #2 (Public Review):
In general, the study has several novel comments, the experimental design is appropriate and the manuscript is well written. While the manuscript contains a lot of data, still it is a bit descriptive. There are also some issues, which should be addressed.
- In Figure 1E, the authors demonstrate a small but significant decrease in body weight of mutant mice. The difference is not so drastic. They also mentioned that some mice showed kyphosis. Please provide data on what percentage of mutant mice showed kyphosis. Please also provide individual hind limb muscle weight normalized with body weight.
Thank you for your suggestions. The kyphosis was observed in some (more than one third of) Dst-b mutant mice as shown in the author response image 1. MRI or CT imaging of the skeleton is necessary …
Author Response
Reviewer #2 (Public Review):
In general, the study has several novel comments, the experimental design is appropriate and the manuscript is well written. While the manuscript contains a lot of data, still it is a bit descriptive. There are also some issues, which should be addressed.
- In Figure 1E, the authors demonstrate a small but significant decrease in body weight of mutant mice. The difference is not so drastic. They also mentioned that some mice showed kyphosis. Please provide data on what percentage of mutant mice showed kyphosis. Please also provide individual hind limb muscle weight normalized with body weight.
Thank you for your suggestions. The kyphosis was observed in some (more than one third of) Dst-b mutant mice as shown in the author response image 1. MRI or CT imaging of the skeleton is necessary to accurately diagnose kyphosis, however, the imaging was not performed in this paper. Therefore, we would like not to provide data on what percentage of mutant mice showed kyphosis.
We weighed the soleus of hind limb and demonstrated the data (lines 132-135).

- There is a lot of variability in the age of the mice employed for this study. For example, in Figure 3, the authors mentioned 23 months old mice (Fig. 3a) and over 20 months old and over 18 months old mice. What was the exact age of the mice? Why three different age mice were used for the same set of experiments? The authors should also comment on whether the onset of myopathy in skeletal and cardiac muscle occurs at the same or different age in mutant mice.
According to the comments, we described exact ages in each figure legends. The reason for the variability in age of mice is that we performed a lot of different kinds of experiment at different time points. We described the myopathy phenotypes occurred around 16 months of age and older (lines 128-129). As for cardiomyopathy, fibrosis was observed around 16 months of age and older (Figure 3D,E).
- Authors have studied protein aggregation only in the soleus muscle of mutant mice. Do the same types of aggregates also form in cardiomyocytes? They write that desmin aggregates were observed in cardiomyocytes of mutant mice. Please show those results in a supplemental figure.
According to the suggestion, we presented the data on desmin aggregates in the cardiomyocytes of Dst-bE2610Ter/E2610Ter mice (Figure 4-figure supplement 1).
- In Figure 5, the authors suggest that mutant mice have mitochondrial abnormalities. However, this analysis is quite abstract and inconclusive. Immunohistochemical images show higher levels of CytoC and Tom20 whereas QRT-PCR demonstrates a significant decrease in mRNA levels of some of the mitochondria-related molecules. Authors should perform additional experiments to determine whether there is any difference in mitochondrial content between WT and mutant mice. In addition, they should perform some functional assays (i.e. OCR, seahorse experiment etc.) to measure mitochondria oxidative phosphorylation capacity is affected in mutant mice.
Thank you very much for the comment. Mitochondrial accumulation was a characteristic phenotype in Dst-bE2610Ter/E2610Ter muscle and also in other types of MFM. We performed quantitative analyses and added the data (Figure 5B). Mitochondrial accumulation was observed even in young stage when protein aggregates were not observed (Figure 3-figure supplement 1A). As the reviewer pointed out, it is important to demonstrate changes in mitochondrial function, but at this moment, we do not have that assay system and would like to present it as data for a future paper, including analysis on mitophagy.
- The morphology of the mitochondria in TEM images shows features that are commonly observed during oxidative damage. Is there any evidence of oxidative stress in skeletal and cardiac muscle of mutant mice?
Thank you very much for the insightful comment. Gene ontology and KEGG pathway analysis on RNA-seq data did not show alterations of oxidative stress in the heart. We performed q-PCR for genes associated with oxidative stress in soleus (Figure 1-figure supplement 3), which did not show alterations in oxidative stress. In the future, we would to investigate on this point.
Reviewer #3 (Public Review):
This manuscript by Yoshioka et al. provides an extensive analysis of cardiac and skeletal muscle in a mouse model of Dst-b mutation. The authors have generated the mutant mouse model to selectively mutate Dst-b isoform of Dystonin and show that such a mutation leads to cardiomyopathy and late-onset myofibrillar myopathy. This is a novel discovery which adds valuable information to the genetic basis and molecular mechanism of MFM mediated by Dst-b. However, the manuscript needs substantial revision and additional feasible experiments.
In Figure3A, the authors suggest that there are smaller myofibers in the mutated mice however they do not provide enough data to support that. Cross-sectional areas between the mutant and WT have to be counted and represented as bins. This can better show the presence of smaller myofibers and muscle degeneration in the mutant mice.
Thank you for the helpful comment. We quantified distribution of cross-sectional area (CSA) in the soleus and then the data was indicated in Figure 3C. It indicates that there are smaller myofibers in the mutant mice.
In Figure 3A-B, the authors show that mutant mice have significantly more myofibers with centrally located myonuclei indicating the constant degeneration and regeneration in the mutant mice. Another indicator of this is the number of activated muscle stem cells. Under homeostasis, authors can compare the number of quiescent muscle stem cells and activated muscle stem cells. If there is constant degeneration and regeneration in the mutant muscle, there will be more cycling muscle stem cells and that will further prove such phenotype in question. Alternatively, they can use EdU water and quantify the number of EdU+/Pax7+ cells between the mutant and WT.
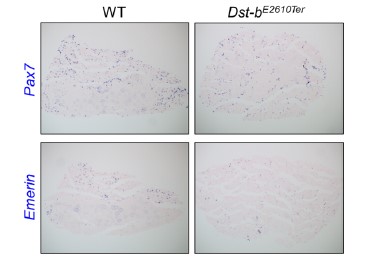
Thank you very much for the interesting comment. We agree that the subject of muscle regeneration in Dst-b mutant mice to be interesting. The authors tried to address this issue by making ISH probes for Pax7 and Emerin, which label muscle stem cells (image below). However, we were unable to reach a conclusion at this time. We intend to address this issue in the future.
In figure 2F, the authors show behavioral tests on the mutant mice of age 1 year. They do not show any significant difference in muscle strength. However, most of the myopathic phenotypes they observe are at 23 months of age, these behavioral tests can be repeated at that age to see if there is more muscle weakness in the mutant mice compared to the WT. Also, are these behavioral test readouts affected by the cardiomyopathy independent of skeletal muscle strength?
We have used rotarod test and wire hang test to evaluate motor coordination and have reported impairment of motor performance in dt mice (Horie et al., 2020). The purpose of these behavior tests in the present study was to evaluate motor coordination of Dst-b mutant mice compared to dt mice, not to address the skeletal muscle function. The text has been changed to clarify this point (lines 121-123).
Generally speaking, these behavioral tests, especially the rotarod test, may be affected by cardiac abnormalities. However, it is difficult to draw conclusions from the results of this study, since there were no significant differences in the behavioral experiments.
They show in Figure 3B that the number of CNF's are affected to a different extent in different muscles. These muscles have a different composition of myofibers, one consisting mostly of slow-type fibers while the other is mostly of fast-type. The question of whether Dst-b mutation effect of muscle fiber types is not clear. Is there a difference?
Thank you very much for insightful comment. We performed qPCR to evaluate whether Dst-b mutation affects the myofiber type of soleus muscle (Figure 1-figure supplement 3B). Expression levels of the genes did not change between WT and Dst-b mutant mice.
The cardiac myopathy phenotype that is clearly shown in figure 3 is shown in mice of 16 months of age whereas the skeletal muscle myopathy phenotype is shown in 23-month-old mice. The reason for the choice of the age of the mice should be discussed. Does the cardiac phenotype precede the skeletal muscle phenotype? Have they looked at the skeletal muscle phenotype at earlier ages? If so, that data should be provided as well and discussed.
Thank you for the comment. We analyzed myopathy and cardiomyopathy phenotypes in mice aged between 16-23 months and then have chosen histological photographs with the high quality. As shown in Figure 3B, CNFs increased in the soleus from all Dst-b mutant mice aged between 16-23 months. We added description that skeletal myopathy phenotypes occurred at 16-month-old mice.
The authors clearly show the formation of protein aggregates in the myofibers in the mutant mice. They further characterize the composition of these desmin aggregates by determining their co aggregates such as plectin and ab-Crystallin. Another component of the z-disk that has been shown to be involved in the aggregates in MFM is myotilin. The authors should also show the presence/ absence and co-aggregation of this protein with the desmin aggregates present in the mutant mice.
According with the suggestion, we performed immunohistochemistry of myotilin. Myotilin was abnormally accumulated in myofibers of the soleus from Dst-b mutant mice. We thank the nice comment and added the data in Figure 4-figure supplement 2.
The authors show abnormal accumulation of mitochondria through cyt c and Tom20 staining. The increased Tom20 levels in the mutant are shown in figure 5A which is from mice that are 23-month-old. However, in figure 3-figure supplement 1a they also show elevated Tom20 staining in the mutant mice that are only 1-2 months old. However, no other phenotype is observed at this age except for the disrupted mitochondria according to the data provided. This needs to be discussed and addressed.
We would like to correct that the data in figure 3-figure supplement 1a is 3-4 months old mutant mice. These data show that mitochondrial accumulation precedes CNF and desmin aggregation. We have described this point in the text (lines 206-209).
In Figure 5, the authors show changes in gene expression levels of genes involved in oxidative phosphorylation which supports the disrupted mitochondrial function. Additionally, ROS levels could be compared between the WT and mutant mice.
To address the involvement of oxidative stress, we performed q-PCR for genes associated with oxidative stress response in soleus (Figure 1-figure supplement 3C). qPCR data did not show alterations in such genes. In the future, we would like to investigate on this point.
In Figure 5 authors show disrupted oxidative phosphorylation in the mutant soleus muscle. Is this also associated with the fiber-type switch? Since mouse soleus muscle is a mix of fast and slow fiber types, they can look at differences in gene expression of key marker genes for slow and fast myofibers.
Thank you very much for the suggestion. We quantified expression levels of muscle fiber-type marker genes (Figure 1-figure supplement 3B). There is no data to suggest the fiber-type switch.
In figure 2, the authors show that mutant mice increase their body weight at a normal pace until 13 weeks of age after which the mutant mice become lighter than their WT counterparts. Is this suggestive of loss of muscle mass? If so, the authors show the muscle atrophy phenotype in 23-month-old mice with cross-sections. Does this mean muscle atrophy starts at an earlier age at 16 months in these mice? Please provide details on the age of the mice for each experiment. In addition, in the text (line 121) authors phrase that the mutant mice become leaner. Lean usually means a decrease in fat mass and an increase in muscle mass. Is this the case? If so, there is no data to support that and the phenotype in the mutant mice suggests there is muscle atrophy in these mice. Therefore, it would not be appropriate to suggest that these mice get lean. However, it is interesting that the bodyweight of the mutant mice gets significantly lighter after 13 weeks. EchoMRI analysis can be performed between these mice to see the total body composition to determine if there is a change in the different type of fat, lean or water composition.
Thank you for your comments. We provided exact ages in each figure legend. We described that skeletal myopathy phenotypes occur as early as 16-month-old mice, and CSA analysis showed that increased small caliber myofibers in the soleus of Dst-b mutant mice. However, muscle mass of the soleus normalized by body weight was not significantly different between control and Dst-bE2610Ter/E2610Ter mice. Therefore, muscle atrophy may be not significant enough to affect muscle weight.
Because we have not quantified the fat mass in Dst-b mutant mice, we changed the phrase from “the mutant mice become leaner” to “they become lower body weight compare with WT mice” (line 120).
Authors have performed RNA-Seq for the left ventricle from the mutant and the WT mice. Separate clustering of the WT and the mutant has to be shown at least through a PCA plot. Some IGV tracks to show the expression level changes in key genes between the mutant and WT should be shown as well. In addition, they could show how some of the genes involved in autophagy and protein degradation are affected since these are mainly the mechanism by which there is protein aggregation in MFM's.
Thank you for your helpful comment. We performed principal component analysis (PCA) and hierarchical clustering. The data showed that transcriptomic features of WT and Dst-b mutant hearts are separated (Figure 8-figure supplement 1A, B). To evaluate the change in expression level of genes, we also performed real time-PCR (Figure 8-figure supplement 1C). Our Gene ontology analysis and KEGG pathway analysis on RNA-seq data in the heart did not suggest the alterations in autophagy and protein degradation, while many genes responsible for unfolded protein response affected (Figure 8C, Figure 8-figure supplement 1C). Previous studies have reported that unfolded protein response is abnormal in several animal models for myofibrillar myopathy (Winter et al., 2014; Fang et al., J Clin Invest, 2017). We would like to investigate underlying mechanisms of protein aggregates in Dst-b mutant myofibers in the future.
-
Evaluation Summary:
The authors demonstrate that isoform-specific Dystonin-b (Dst-b) mutant mice show significant myopathy in skeletal and cardiac muscle at older ages without the peripheral neuropathy or post-natal lethality that are commonly observed by loss of function of the DST gene. The study provides novel information about the role of the Dst-b isoform in maintaining skeletal and cardiac muscle health. In addition, the study suggests that isoform-specific mutations in Dst-b gene may cause some hereditary skeletal and cardiac myopathies.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 agreed to share their name with the authors.)
-
Reviewer #1 (Public Review):
Previous work on dystonin has shown that loss of function mutations in dystonin cause hereditary sensory and autonomic neuropathy type 6. In affected patients, there is dysfunction of cardiac and skeletal muscle. In patients, the mutations disrupt both dystonin a and b. The goal of the current study was to determine whether mutations specific to dystonin b are responsible for the muscle phenotype. The identification of the distinct roles of dystonin a and b in muscle versus nerve represents an advance.
A strength of the paper is the analysis of both cardiac and skeletal muscle.
A concern is that while identification that disruption of the function of dystonin b disrupts muscle is an advance, it is unclear how important an advance it is. The paper would be more exciting if it was definitively shown that …
Reviewer #1 (Public Review):
Previous work on dystonin has shown that loss of function mutations in dystonin cause hereditary sensory and autonomic neuropathy type 6. In affected patients, there is dysfunction of cardiac and skeletal muscle. In patients, the mutations disrupt both dystonin a and b. The goal of the current study was to determine whether mutations specific to dystonin b are responsible for the muscle phenotype. The identification of the distinct roles of dystonin a and b in muscle versus nerve represents an advance.
A strength of the paper is the analysis of both cardiac and skeletal muscle.
A concern is that while identification that disruption of the function of dystonin b disrupts muscle is an advance, it is unclear how important an advance it is. The paper would be more exciting if it was definitively shown that disruption of dystonin b causes myopathy in humans.
-
Reviewer #2 (Public Review):
In general, the study has several novel comments, the experimental design is appropriate and the manuscript is well written. While the manuscript contains a lot of data, still it is a bit descriptive. There are also some issues, which should be addressed.
In Figure 1E, the authors demonstrate a small but significant decrease in body weight of mutant mice. The difference is not so drastic. They also mentioned that some mice showed kyphosis. Please provide data on what percentage of mutant mice showed kyphosis. Please also provide individual hind limb muscle weight normalized with body weight.
There is a lot of variability in the age of the mice employed for this study. For example, in Figure 3, the authors mentioned 23 months old mice (Fig. 3a) and over 20 months old and over 18 months old mice. What was the …
Reviewer #2 (Public Review):
In general, the study has several novel comments, the experimental design is appropriate and the manuscript is well written. While the manuscript contains a lot of data, still it is a bit descriptive. There are also some issues, which should be addressed.
In Figure 1E, the authors demonstrate a small but significant decrease in body weight of mutant mice. The difference is not so drastic. They also mentioned that some mice showed kyphosis. Please provide data on what percentage of mutant mice showed kyphosis. Please also provide individual hind limb muscle weight normalized with body weight.
There is a lot of variability in the age of the mice employed for this study. For example, in Figure 3, the authors mentioned 23 months old mice (Fig. 3a) and over 20 months old and over 18 months old mice. What was the exact age of the mice? Why three different age mice were used for the same set of experiments? The authors should also comment on whether the onset of myopathy in skeletal and cardiac muscle occurs at the same or different age in mutant mice.
Authors have studied protein aggregation only in the soleus muscle of mutant mice. Do the same types of aggregates also form in cardiomyocytes? They write that desmin aggregates were observed in cardiomyocytes of mutant mice. Please show those results in a supplemental figure.
In Figure 5, the authors suggest that mutant mice have mitochondrial abnormalities. However, this analysis is quite abstract and inconclusive. Immunohistochemical images show higher levels of CytoC and Tom20 whereas QRT-PCR demonstrates a significant decrease in mRNA levels of some of the mitochondria-related molecules. Authors should perform additional experiments to determine whether there is any difference in mitochondrial content between WT and mutant mice. In addition, they should perform some functional assays (i.e. OCR, seahorse experiment etc.) to measure mitochondria oxidative phosphorylation capacity is affected in mutant mice.
The morphology of the mitochondria in TEM images shows features that are commonly observed during oxidative damage. Is there any evidence of oxidative stress in skeletal and cardiac muscle of mutant mice?
-
Reviewer #3 (Public Review):
This manuscript by Yoshioka et al. provides an extensive analysis of cardiac and skeletal muscle in a mouse model of Dst-b mutation. The authors have generated the mutant mouse model to selectively mutate Dst-b isoform of Dystonin and show that such a mutation leads to cardiomyopathy and late-onset myofibrillar myopathy. This is a novel discovery which adds valuable information to the genetic basis and molecular mechanism of MFM mediated by Dst-b. However, the manuscript needs substantial revision and additional feasible experiments.
In Figure3A, the authors suggest that there are smaller myofibers in the mutated mice however they do not provide enough data to support that. Cross-sectional areas between the mutant and WT have to be counted and represented as bins. This can better show the presence of smaller …
Reviewer #3 (Public Review):
This manuscript by Yoshioka et al. provides an extensive analysis of cardiac and skeletal muscle in a mouse model of Dst-b mutation. The authors have generated the mutant mouse model to selectively mutate Dst-b isoform of Dystonin and show that such a mutation leads to cardiomyopathy and late-onset myofibrillar myopathy. This is a novel discovery which adds valuable information to the genetic basis and molecular mechanism of MFM mediated by Dst-b. However, the manuscript needs substantial revision and additional feasible experiments.
In Figure3A, the authors suggest that there are smaller myofibers in the mutated mice however they do not provide enough data to support that. Cross-sectional areas between the mutant and WT have to be counted and represented as bins. This can better show the presence of smaller myofibers and muscle degeneration in the mutant mice.
In Figure 3A-B, the authors show that mutant mice have significantly more myofibers with centrally located myonuclei indicating the constant degeneration and regeneration in the mutant mice. Another indicator of this is the number of activated muscle stem cells. Under homeostasis, authors can compare the number of quiescent muscle stem cells and activated muscle stem cells. If there is constant degeneration and regeneration in the mutant muscle, there will be more cycling muscle stem cells and that will further prove such phenotype in question. Alternatively, they can use EdU water and quantify the number of EdU+/Pax7+ cells between the mutant and WT.
In figure 2F, the authors show behavioral tests on the mutant mice of age 1 year. They do not show any significant difference in muscle strength. However, most of the myopathic phenotypes they observe are at 23 months of age, these behavioral tests can be repeated at that age to see if there is more muscle weakness in the mutant mice compared to the WT. Also, are these behavioral test readouts affected by the cardiomyopathy independent of skeletal muscle strength?
They show in Figure 3B that the number of CNF's are affected to a different extent in different muscles. These muscles have a different composition of myofibers, one consisting mostly of slow-type fibers while the other is mostly of fast-type. The question of whether Dst-b mutation effect of muscle fiber types is not clear. Is there a difference?
The cardiac myopathy phenotype that is clearly shown in figure 3 is shown in mice of 16 months of age whereas the skeletal muscle myopathy phenotype is shown in 23-month-old mice. The reason for the choice of the age of the mice should be discussed. Does the cardiac phenotype precede the skeletal muscle phenotype? Have they looked at the skeletal muscle phenotype at earlier ages? If so, that data should be provided as well and discussed.
The authors clearly show the formation of protein aggregates in the myofibers in the mutant mice. They further characterize the composition of these desmin aggregates by determining their co aggregates such as plectin and ab-Crystallin. Another component of the z-disk that has been shown to be involved in the aggregates in MFM is myotilin. The authors should also show the presence/ absence and co-aggregation of this protein with the desmin aggregates present in the mutant mice.
The authors show abnormal accumulation of mitochondria through cyt c and Tom20 staining. The increased Tom20 levels in the mutant are shown in figure 5A which is from mice that are 23-month-old. However, in figure 3-figure supplement 1a they also show elevated Tom20 staining in the mutant mice that are only 1-2 months old. However, no other phenotype is observed at this age except for the disrupted mitochondria according to the data provided. This needs to be discussed and addressed.
In Figure 5, the authors show changes in gene expression levels of genes involved in oxidative phosphorylation which supports the disrupted mitochondrial function. Additionally, ROS levels could be compared between the WT and mutant mice.
In Figure 5 authors show disrupted oxidative phosphorylation in the mutant soleus muscle. Is this also associated with the fiber-type switch? Since mouse soleus muscle is a mix of fast and slow fiber types, they can look at differences in gene expression of key marker genes for slow and fast myofibers.
In figure 2, the authors show that mutant mice increase their body weight at a normal pace until 13 weeks of age after which the mutant mice become lighter than their WT counterparts. Is this suggestive of loss of muscle mass? If so, the authors show the muscle atrophy phenotype in 23-month-old mice with cross-sections. Does this mean muscle atrophy starts at an earlier age at 16 months in these mice? Please provide details on the age of the mice for each experiment. In addition, in the text authors phrase that the mutant mice become leaner. Lean usually means a decrease in fat mass and an increase in muscle mass. Is this the case? If so, there is no data to support that and the phenotype in the mutant mice suggests there is muscle atrophy in these mice. Therefore, it would not be appropriate to suggest that these mice get lean. However, it is interesting that the bodyweight of the mutant mice gets significantly lighter after 13 weeks. EchoMRI analysis can be performed between these mice to see the total body composition to determine if there is a change in the different type of fat, lean or water composition.
Authors have performed RNA-Seq for the left ventricle from the mutant and the WT mice. Separate clustering of the WT and the mutant has to be shown at least through a PCA plot. Some IGV tracks to show the expression level changes in key genes between the mutant and WT should be shown as well. In addition, they could show how some of the genes involved in autophagy and protein degradation are affected since these are mainly the mechanism by which there is protein aggregation in MFM's.
-
