Remodeling of dermal adipose tissue alleviates cutaneous toxicity induced by anti-EGFR therapy
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
This paper will be of interest to oncologists and dermatologists and has high clinical relevance. It reveals a novel mechanism of EGFR inhibitor-induced rash which be may closely related to atrophy of dermal white adipose tissue (dWAT). A series of experimental manipulations dissect the mechanism with a murine model, supporting the major claims of the paper.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. The reviewers remained anonymous to the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Anti-epidermal growth factor receptor (EGFR) therapy–associated cutaneous toxicity is a syndrome characterized by papulopustular rash, local inflammation, folliculitis, and microbial infection, resulting in a decrease in quality of life and dose interruption. However, no effective clinical intervention is available for this adverse effect. Here, we report the atrophy of dermal white adipose tissue (dWAT), a highly plastic adipose tissue with various skin-specific functions, correlates with rash occurrence and exacerbation in a murine model of EGFR inhibitor-induced rash. The reduction in dWAT is due to the inhibition of adipogenic differentiation by defects in peroxisome proliferator-activated receptor γ (PPARγ) signaling, and increased lipolysis by the induced expression of the lipolytic cytokine IL6. The activation of PPARγ by rosiglitazone maintains adipogenic differentiation and represses the transcription of IL6, eventually improving skin functions and ameliorating the severity of rash without altering the antitumor effects. Thus, activation of PPARγ represents a promising approach to ameliorate cutaneous toxicity in patients with cancer who receive anti-EGFR therapy.
Article activity feed
-

-

Author Response:
Reviewer #1:
The authors present an interesting concept for the mechanism of rash induction in EGFR inhibitor (EGFRi) treated rats. EGFRi causes production of pro-inflammatory factors in epidermal keratinocytes which may induce dedifferentiation and reduction of the dWAT compartment, presumably mediated via PPAR. Factors produced by dedifferentiated FB then recruit monocytes thereby inducing skin inflammation. This work is aiming to improve targeted cancer therapy efficiency and is therefore of potential clinical relevance.
However, most of the conclusions drawn by the authors are based on correlations, e.g. between the amount of dWAT and rash intensity. Mechanistic data have been mainly generated in vitro. The exact order of events to formulate a definitive mechanistic proof in vivo for this hypothesis is missing. …
Author Response:
Reviewer #1:
The authors present an interesting concept for the mechanism of rash induction in EGFR inhibitor (EGFRi) treated rats. EGFRi causes production of pro-inflammatory factors in epidermal keratinocytes which may induce dedifferentiation and reduction of the dWAT compartment, presumably mediated via PPAR. Factors produced by dedifferentiated FB then recruit monocytes thereby inducing skin inflammation. This work is aiming to improve targeted cancer therapy efficiency and is therefore of potential clinical relevance.
However, most of the conclusions drawn by the authors are based on correlations, e.g. between the amount of dWAT and rash intensity. Mechanistic data have been mainly generated in vitro. The exact order of events to formulate a definitive mechanistic proof in vivo for this hypothesis is missing. In particular, it is not clear which cells in the skin, apart from keratinocytes, are specifically targeted by EGFR inhibitors and/or by Rosiglitazone. The authors also do not show EGFR staining in adipocytes and its inhibition by Afa. The effects of Afa and Rosi on monocytes / macrophages are completely ignored by the authors. Additionally, some of the presented results are overinterpreted and not really supporting what is claimed.
Most importantly, the whole study is based on inhibitor treatments. Afatinib for example is not only inhibiting EGFR but all other erbB family members and as such it represents a panErbB inhibitor and it is not clear whether the observed effects are induced by inhibition of EGFR of other erbB receptors which have been shown to have also effects in the skin. For further specification of the role of EGFR, other, more specific inhibitors should be used to confirm the basic concept along with genetic proof either in genetically engineered mice or by Crispr-mediated-deletion.
To further support the hypotheses of the authors, the study needs to be further substantiated by mechanistic experiments and the clinical relevance should be strengthened by performing histologic analysis of skin samples of patients treated with EGFRi and respective analysis of rash and e.g. BMI etc.
Thanks for your positive comments on the potential impact for cancer patients suffering EGFR inhibitor induced skin rash. We have carefully considered all comments from the reviewer and revised our manuscript accordingly. In the following section, we summarize our responses to each comment of the reviewer. We believe that our responses have well addressed all concerns from the reviewer.
We agree with the reviewer’s comment that our research may need more direct mechanistic in vivo studies upon our in vitro results. In our research, we have collected evidence from previous studies and used various in vitro and ex vivo experiments to investigate our findings. However, the study was still limited by currently available technologies.
In the revised version, we supplemented the pEGFR and pERK staining of adipocytes in Figure 3-figure supplement 1C. The levels of phospho-EGFR and ERK in dWAT were significantly decreased after EGFRi treatment.
This study was inspired by the observations of the unusual dWAT reduction during EGFRi treatment, thus we focused on the investigation of dermal adipocytes. In addition, the roles of mastocytes, monocytes, and macrophages in EGFRi-induced cutaneous toxicity have been thought as responders to increased expressions of cytokines. Local depletion of macrophages and degranulated mastocytes just provided partial resolution, indicating a multifactorial and complicated pathology of cutaneous toxicity induced by anti-EGFR therapy(Lichtenberger et al., 2013; Mascia et al., 2013).
In terms of some inappropriate descriptions, we agree with the reviewer that they will be more convincing if there is a direct assessment from genetically engineered mice. For example, we tried to establish the relationship between S. aureus infection and EGFRi-induced rash based on a well-accepted study from Lingjuan Zhang (Zhang et al., 2015). They reported that adipose precursor cells secret antimicrobial peptide cathelicidin during differentiation to against S. aureus infection. Mice with impaired adipogenesis were more susceptible to S. aureus infection. This conclusion gave us insights into the relationship between S. aureus infection and EGFRi-induced skin inflammation. Unfortunately, the anti-CAMP antibody was made by the author’s lab and there are no mature products that can recognize CAMP in rats. To provide more mechanistic evidences, we conducted qPCR experiments to study the transcriptional level of the Camp gene both in dWAT and dFB cells isolated from rat skin (Figure 3I and 3J). dWAT in Afa group showed a lower expression level of Camp compared with control group. In addition, in different differentiation stages of dFB in vitro, transcriptional levels of Camp were decreased by Afa treatment while increased by Rosi. In summary, the data we collected could verify the causal relationship between EGFRi-induced dWAT reduction and S. aureus infection to some extent. However, the limitation of the technology is an obstacle for us to provide more evidences. Thus, in the revised manuscript, we have edited our writing to make the statement not that strong.
According to the clinical evidence, the rash can also be induced by many specific Erbb1 inhibitors. All three generations of EGFR inhibitors in the clinic have very high incidence rates of cutaneous toxicity (Supplementary file 1). In the revised version, we provided rash models induced by both first-generation EGFRi, Erlotinib, Gefitinib, and the third-generation EGFRi, Osimertinib. As shown in Figure 1-figure supplement 1D, the rash caused by Erlotinib, Gefitinib, and Osimertinib had the same phenotypes as Afatinib-induced rash.
In summary, the current form of evidences should support our findings, even more direct mechanistic studies would be better. We are now seeking the opportunity for cooperation to build a dermal adipocyte knockout mouse model platform and hope to investigate the specific roles of dermal adipocytes in the future. We also plan to have cooperation with hospitals to explore the clinical evidence of patients receiving EGFR inhibitors.
References:
Lichtenberger BM, Gerber PA, Holcmann M, Buhren BA, Amberg N, Smolle V, Schrumpf H, Boelke E, Ansari P, Mackenzie C, Wollenberg A, Kislat A, Fischer JW, Röck K, Harder J, Schröder JM, Homey B, Sibilia M. 2013. Epidermal EGFR controls cutaneous host defense and prevents inflammation. Sci Transl Med 5.
Mascia F, Lam G, Keith C, Garber C, Steinberg SM, Kohn E, Yuspa SH. 2013. Genetic ablation of epidermal EGFR reveals the dynamic origin of adverse effects of anti-EGFR therapy. Sci Transl Med 5.
Zhang L, Guerrero-juarez CF, Hata T, Bapat SP, Ramos R, Plikus M V, Gallo RL. 2015. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 347:67–72.
Reviewer #2:
Leying Chen et al. investigated the mechanism of EGFR inhibitor-induced rash. They find that atrophy of dermal white adipose tissue (dWAT), a highly plastic adipose tissue with various skin-specific functions, correlates with rash occurrence and exacerbation in a murine model. The data indicate that EGFR inhibition induces the dedifferentiation of dWAT and lipolysis , finally lead to dWAT reduction which is a hallmark of the pathophysiology of rash. Notably, they demonstrate that stimulating dermal adipocyte expansion with a high-fat diet (HFD) or the pharmacological PPARγ agonist rosiglitazone (Rosi) ameliorated the severity of rash. Therefore, PPARγ agonists may represent a promising new therapeutic strategy in the treatment of EGFRI-related skin disorders pending to be confirmed in further study.
We greatly appreciate the reviewer for giving the above positive comments.
The conclusions of this paper are mostly well supported by data, but some results need to be clarified and verified.
- PPAR signaling in the pathology of EGFRI-induced skin toxicity. In figure 2 , the results show Rosi reversed the dedifferentiation of dermal adipocytes induced by Afa. This may due to PPARγ upregulation but not be confirmed in the results. The relative genes expression in dWAT after treated with Afa and ROSi were not demonstrated in the results.
We thank the reviewer for reminding us for additional experiment of PPARγ. In the revised version, we collected attatched-dWAT after 5-day Afa or Rosi treatment, and performed transcriptional experiment of Pparg. The expression level of Pparg was downregulated by Afa treatment and upregulated by Rosi treatment (Figure 2-figure supplement 1D).
- the effect of PPAR signaling on PDGFRA-PI3K-AKT pathway The AKT pathway is a key downstream target of EGFR kinase, so it is reasonable to see p-AKT1 and p-AKT2 levels were decreased by Afa (figure 3C) However, addition of Rosi to Afa significantly activated both AKT1 and AKT2 . What is the underlying mechanism for the results and whether it is related to the PPAR signaling pathway.
Given the importance of the PI3K/AKT pathway in regulating AP and mature adipocyte biology(Jeffery et al., 2015), we used p-AKT to characterize the activation of dFBs. The mechanism of how modulating PPARγ affects AKT is still unknown. One study found that MAPK and PI3K are upregulated and activated by rosiglitazone that in turn might enhance adipogenesis(Fayyad et al., 2019). In skeletal muscle, PPARγ enhances insulin-stimulated PI3K and Akt activation(Marx et al., 2004). It is also reported rosiglitazone has a neuroprotection effect against oxidative stress. The PPARγ-rosiglitazone complex binds to the neurotrophic factor-α1 (NF-α1) promoter and activates the transcription of NF-α1 mRNA which is then translated to the protein. NF-α1 binds to a cognate receptor and activates the AKT and ERK pathways(Thouennon et al., 2015). Thus, further studies should be carried out to investigate the effects of rosiglitazone to PI3K/AKT pathway on adipogenesis.
- According to figure 3 F , 3G and 3H., authors draw a conclusion that " a lack of APs and mature dWAT impairs the maintenance of the host defense and hair growth in the skin" In my opinion, there are no results can directly prove this. According to figure 3H, the impairment of hair growth may be caused by EGFR inhibition of hair follicles.
We appreciate the reviewer for pointing this important point out. We tried to establish the relationship between S. aureus infection and EGFRI-induced rash based on a well-accepted study from Lingjuan Zhang (Zhang et al., 2015). They reported that adipose precursor cells secret antimicrobial peptide cathelicidin during differentiation to against S. aureus infection. Mice with impaired adipogenesis were more susceptible to S. aureus infection. This conclusion gave us insights into the relationship between S. aureus infection and EGFRI-induced skin inflammation. Unfortunately, the anti-CAMP antibody was made by the author’s lab and there are no mature products that can recognize CAMP in rats. To provide more mechanistic evidences, we conducted qPCR experiments to study the transcriptional level of the Camp gene both in dWAT and dFB cells isolated from rat skin (Figure 3I and 3J). dWAT in Afa group showed a lower expression level of Camp compared with control group. In addition, in different differentiation stages of dFB in vitro, transcriptional levels of Camp were decreased by Afa treatment while increased by Rosi. In summary, the data we collected depending on the current technology could verify the causal relationship between EGFRI-induced dWAT reduction and S. aureus infection to some extent. However, we agree with the reviewer that this conclusion needs more direct evidence. Thus, in the revised manuscript, we have edited our writing to make the statement not that strong.
Since recent reports have shown that dermal adipocytes have the capacity to support hair regeneration, we used this conclusion to characterize the function of dWAT. However, we agree with the reviewer that it needs more specific and direct experiments to verify the causality with dWAT. And we are seeking the opportunity for cooperation to build a dermal adipocyte knockout mouse model platform and hope to investigate the specific roles of dermal adipocytes in the future. In the revised manuscript, we also adjusted the statements.
- EGFRI stimulates keratinocytes (HaCaT cells) to produce lipolytic cytokines (IL-6) (Figure 4G). IL6 enhanced the lipolysis of differentiated dFB (Figure S4M) and C18 fatty acids were supposed to be released the cell matrix during lipolysis. In figure 4H, HaCaTcells supernatants and dFB supernatants were collected. IL-6 was supposed to increase in HaCaTcells supernatants and was confirmed in Figure 4SK and S4L.However, C18 fatty acids were not showed to be in the dFB supernatants in the study directly.
We thank the reviewer for pointing this out. We conducted additional lipidomics of dFB supernatants. However, because the differentiation medium needs to be changed every two days, it is hard to accumulate enough FFAs. We collected supernatants on Day3, Day 6, and Day 9. They were all below the detection limit of mass spectrum. We agree with the reviewer that more evidences are needed to prove the correlation between C18 FFAs and lipolysis. Therefore, we performed a mass spectrometry analysis of skin tissues from Ctrl and Afa groups after 3-day treatment to confirm the releasing of C18 FFAs. The result showed an increased tendency of C18:2 and other FFAs in the Afa group (Figure 1 in response letter). However, this increase had no significant statistic difference. This might be due to the interference of sebaceous gland and dermal adipocytes. In consequence, we adjusted the descriptions in the revised manuscript to make this statement not that strong.
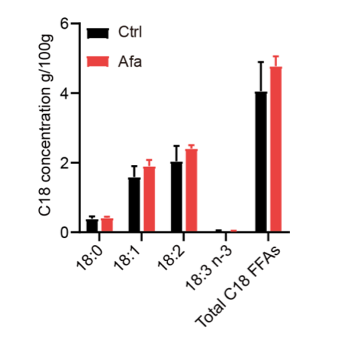
Figure 1. C18 concentrations in skin tissues from Ctrl and Afa groups after 3-day treatment. n=3.
References:
Fayyad AM, Khan AA, Abdallah SH, Alomran SS, Bajou K, Khattak MNK. 2019. Rosiglitazone Enhances Browning Adipocytes in Association with MAPK and PI3-K Pathways During the Differentiation of Telomerase-Transformed Mesenchymal Stromal Cells into Adipocytes. Int J Mol Sci 20.
Jeffery E, Church CD, Holtrup B, Colman L, Rodeheffer MS. 2015. Rapid depot-specific activation of adipocyte precursor cells at the onset of obesity. Nat Cell Biol 17:376–385.
Marx N, Duez H, Fruchart J-C, Staels B. 2004. Peroxisome proliferator-activated receptors and atherogenesis: regulators of gene expression in vascular cells. Circ Res 94:1168–1178. Thouennon E, Cheng Y, Falahatian V, Cawley NX, Loh YP. 2015. Rosiglitazone-activated PPARγ induces neurotrophic factor-α1 transcription contributing to neuroprotection. J Neurochem 134:463–470.
Zhang L, Guerrero-juarez CF, Hata T, Bapat SP, Ramos R, Plikus M V, Gallo RL. 2015. Dermal adipocytes protect against invasive Staphylococcus aureus skin infection. Science 347:67–72.
-
Evaluation Summary:
This paper will be of interest to oncologists and dermatologists and has high clinical relevance. It reveals a novel mechanism of EGFR inhibitor-induced rash which be may closely related to atrophy of dermal white adipose tissue (dWAT). A series of experimental manipulations dissect the mechanism with a murine model, supporting the major claims of the paper.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. The reviewers remained anonymous to the authors.)
-
Reviewer #1 (Public Review):
The authors present an interesting concept for the mechanism of rash induction in EGFR inhibitor (EGFRi) treated rats. EGFRi causes production of pro-inflammatory factors in epidermal keratinocytes which may induce dedifferentiation and reduction of the dWAT compartment, presumably mediated via PPAR. Factors produced by dedifferentiated FB then recruit monocytes thereby inducing skin inflammation. This work is aiming to improve targeted cancer therapy efficiency and is therefore of potential clinical relevance.
However, most of the conclusions drawn by the authors are based on correlations, e.g. between the amount of dWAT and rash intensity. Mechanistic data have been mainly generated in vitro. The exact order of events to formulate a definitive mechanistic proof in vivo for this hypothesis is missing. In …
Reviewer #1 (Public Review):
The authors present an interesting concept for the mechanism of rash induction in EGFR inhibitor (EGFRi) treated rats. EGFRi causes production of pro-inflammatory factors in epidermal keratinocytes which may induce dedifferentiation and reduction of the dWAT compartment, presumably mediated via PPAR. Factors produced by dedifferentiated FB then recruit monocytes thereby inducing skin inflammation. This work is aiming to improve targeted cancer therapy efficiency and is therefore of potential clinical relevance.
However, most of the conclusions drawn by the authors are based on correlations, e.g. between the amount of dWAT and rash intensity. Mechanistic data have been mainly generated in vitro. The exact order of events to formulate a definitive mechanistic proof in vivo for this hypothesis is missing. In particular, it is not clear which cells in the skin, apart from keratinocytes, are specifically targeted by EGFR inhibitors and/or by Rosiglitazone. The authors also do not show EGFR staining in adipocytes and its inhibition by Afa. The effects of Afa and Rosi on monocytes / macrophages are completely ignored by the authors. Additionally, some of the presented results are overinterpreted and not really supporting what is claimed.
Most importantly, the whole study is based on inhibitor treatments. Afatinib for example is not only inhibiting EGFR but all other erbB family members and as such it represents a panErbB inhibitor and it is not clear whether the observed effects are induced by inhibition of EGFR of other erbB receptors which have been shown to have also effects in the skin. For further specification of the role of EGFR, other, more specific inhibitors should be used to confirm the basic concept along with genetic proof either in genetically engineered mice or by Crispr-mediated-deletion.
To further support the hypotheses of the authors, the study needs to be further substantiated by mechanistic experiments and the clinical relevance should be strengthened by performing histologic analysis of skin samples of patients treated with EGFRi and respective analysis of rash and e.g. BMI etc.
-
Reviewer #2 (Public Review):
Leying Chen et al. investigated the mechanism of EGFR inhibitor-induced rash. They find that atrophy of dermal white adipose tissue (dWAT), a highly plastic adipose tissue with various skin-specific functions, correlates with rash occurrence and exacerbation in a murine model. The data indicate that EGFR inhibition induces the dedifferentiation of dWAT and lipolysis , finally lead to dWAT reduction which is a hallmark of the pathophysiology of rash. Notably, they demonstrate that stimulating dermal adipocyte expansion with a high-fat diet (HFD) or the pharmacological PPARγ agonist rosiglitazone (Rosi) ameliorated the severity of rash. Therefore, PPARγ agonists may represent a promising new therapeutic strategy in the treatment of EGFRI-related skin disorders pending to be confirmed in further study.
The …
Reviewer #2 (Public Review):
Leying Chen et al. investigated the mechanism of EGFR inhibitor-induced rash. They find that atrophy of dermal white adipose tissue (dWAT), a highly plastic adipose tissue with various skin-specific functions, correlates with rash occurrence and exacerbation in a murine model. The data indicate that EGFR inhibition induces the dedifferentiation of dWAT and lipolysis , finally lead to dWAT reduction which is a hallmark of the pathophysiology of rash. Notably, they demonstrate that stimulating dermal adipocyte expansion with a high-fat diet (HFD) or the pharmacological PPARγ agonist rosiglitazone (Rosi) ameliorated the severity of rash. Therefore, PPARγ agonists may represent a promising new therapeutic strategy in the treatment of EGFRI-related skin disorders pending to be confirmed in further study.
The conclusions of this paper are mostly well supported by data, but some results need to be clarified and verified.
PPAR signaling in the pathology of EGFRI-induced skin toxicity.
In figure 2 , the results show Rosi reversed the dedifferentiation of dermal adipocytes induced by Afa. This may due to PPARγ upregulation but not be confirmed in the results. The relative genes expression in dWAT after treated with Afa and ROSi were not demonstrated in the results.the effect of PPAR signaling on PDGFRA-PI3K-AKT pathway
The AKT pathway is a key downstream target of EGFR kinase, so it is reasonable to see p-AKT1 and p-AKT2 levels were decreased by Afa (figure 3C) However, addition of Rosi to Afa significantly activated both AKT1 and AKT2 . What is the underlying mechanism for the results and whether it is related to the PPAR signaling pathway.According to figure 3 F , 3G and 3H., authors draw a conclusion that " a lack of APs and mature dWAT impairs the maintenance of the host defense and hair growth in the skin" In my opinion, there are no results can directly prove this. According to figure 3H, the impairment of hair growth may be caused by EGFR inhibition of hair follicles.
EGFRI stimulates keratinocytes (HaCaT cells) to produce lipolytic cytokines (IL-6) (Figure 4G). IL6 enhanced the lipolysis of differentiated dFB (Figure S4M) and C18 fatty acids were supposed to be released the cell matrix during lipolysis.
In figure 4H, HaCaTcells supernatants and dFB supernatants were collected. IL-6 was supposed to increase in HaCaTcells supernatants and was confirmed in Figure 4SK and S4L.However, C18 fatty acids were not showed to be in the dFB supernatants in the study directly.
-
