Gene age shapes the transcriptional landscape of sexual morphogenesis in mushroom-forming fungi (Agaricomycetes)
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
This manuscript provides a deep characterization of transcriptional regulation and conservation across key stages of complex multicellular development during mushroom formation. The authors present evidence for extensive allele-specific expression that includes many developmentally regulated genes that appear to have evolved recently. These findings help underscore how the tuning of gene expression and gains of new genes, the function of which will need to be unraveled in future, are likely the basis for the evolution of complex morphologies in fungi. This work should be of broad interest to evolutionary biologists, and especially to those studying the evolution of gene regulation.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 and Reviewer #4 agreed to share their names with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Multicellularity has been one of the most important innovations in the history of life. The role of gene regulatory changes in driving transitions to multicellularity is being increasingly recognized; however, factors influencing gene expression patterns are poorly known in many clades. Here, we compared the developmental transcriptomes of complex multicellular fruiting bodies of eight Agaricomycetes and Cryptococcus neoformans , a closely related human pathogen with a simple morphology. In-depth analysis in Pleurotus ostreatus revealed that allele-specific expression, natural antisense transcripts, and developmental gene expression, but not RNA editing or a ‘developmental hourglass,’ act in concert to shape its transcriptome during fruiting body development. We found that transcriptional patterns of genes strongly depend on their evolutionary ages. Young genes showed more developmental and allele-specific expression variation, possibly because of weaker evolutionary constraint, suggestive of nonadaptive expression variance in fruiting bodies. These results prompted us to define a set of conserved genes specifically regulated only during complex morphogenesis by excluding young genes and accounting for deeply conserved ones shared with species showing simple sexual development. Analysis of the resulting gene set revealed evolutionary and functional associations with complex multicellularity, which allowed us to speculate they are involved in complex multicellular morphogenesis of mushroom fruiting bodies.
Article activity feed
-

-

Author Response:
Reviewer #1 (Public Review):
This study sought to systematically identify the components and driving forces of transcriptome evolution in fungi that exhibit complex multicellularity (CM). The authors examined a series of parameters or expression signatures (i.e. natural antisense transcripts, allele-specific expression, RNA-editing) concluding that the best predictor of a gene behavior in the CM transcriptome was evolutionary age.
Thus, the transcriptomes of fruiting bodies showed a distinct gene-age-related stratification, where it was possible to sort out genes related to general sexual processes from those likely linked to morphogenetic aspects of the CM fruiting bodies. Notably, their results did not support a developmental hourglass, which is the rather predominant hypothesis in metazoans, including some …
Author Response:
Reviewer #1 (Public Review):
This study sought to systematically identify the components and driving forces of transcriptome evolution in fungi that exhibit complex multicellularity (CM). The authors examined a series of parameters or expression signatures (i.e. natural antisense transcripts, allele-specific expression, RNA-editing) concluding that the best predictor of a gene behavior in the CM transcriptome was evolutionary age.
Thus, the transcriptomes of fruiting bodies showed a distinct gene-age-related stratification, where it was possible to sort out genes related to general sexual processes from those likely linked to morphogenetic aspects of the CM fruiting bodies. Notably, their results did not support a developmental hourglass, which is the rather predominant hypothesis in metazoans, including some analysis in fungi.
The studies involved analyses of new transcriptomic datasets for different developmental stages (and tissue types in some cases) of Pleurotus ostreatus and Pterula gracilis, as well as the analyses of existing datasets for other fungi.
There are diverse interesting observations such as ones regarding Allele Specific Expression (ASE), suggesting that in P. ostreatus ASE mainly occurs due to cis-regulatory allele divergence, possibly in fast evolving genes that are not under strong selection constraints, such as ones grouped in youngest gene ages categories. In addition, a large number of conserved unannotated genes among CM-specific orthogroups highlights the rather cryptic nature of CM in fungi and raises as an important area for future research.
Some of the key aspects of the analyses would need to be better exemplified such as:
– Providing a better description of the developmentally expressed TFs only in CM species
– Providing clear examples of the promoter divergence that could be the underlying mechanism behind ASE. In particular, for some cases, there may be enough information in the literature/databases to predict the appearance or disappearance of relevant cis-elements in the promoters showing the highest divergence in genes depicting the highest levels of ASE.
We appreciate the constructive comments of the Reviewer and have revised the ms in accordance with the suggestions. In particular, we link different parts of the ms better to each other, provided a more detailed discussion of developmentally expressed TFs (lines 615-621). We also provide case studies of ASE genes with cis-regulatory divergence (Figure 5 and see below), although we note that these analyses are based on inferred and not directly determined motifs, so they should be considered as preliminary.
We had considered using TF binding motifs previously, and now we gave a try to analyzing potential transcription factor binding sites in divergent promoters. We find that there are no P. ostreatus transcription factors for which motifs based on direct evidence are available; rather, all P. ostreatus motifs are based on extrapolations from experimentally determined motifs (typically in Neurospora crassa). Therefore, to avoid too general motifs, we used only those where at least 5 nucleotides show at least 80% expected frequency in the PWM-s. This left us with 158 motifs (126 excluded). High motif binding score (>=4) and self-rate (>=0.9) were also required to ignore false positive hits. Different binding ability and lack of binding in one of the parental genomes were counted for each promoter. We found that genes with allele specific expression (ASE S2 and S4) show significantly higher differences in motif binding (lacking motifs, or different binding ability) than non-ASE genes (Fig. A1). These observations show that, not only promoter divergence, but differential predicted TF binding ability is also more common among ASE genes than among non-ASE genes. This supports our conjecture that ASE arises from cis-regulatory divergence.
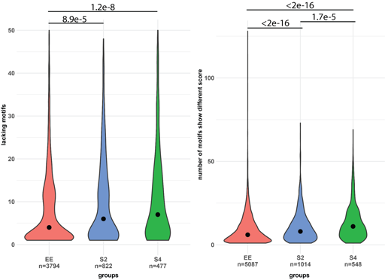
Fig A1: The left plot below shows the number of cases when the promoter of one allele of an allele pair in the two parent genomes has, but the other lacks a motif. The right plot shows the same in terms of difference in binding score.
We could find examples, such as the allele specific expression of PleosPC15_2_1031042, a Hemerythrin-like (IPR012312) protein which might be regulated by the conserved c2h2 transcription factor, containing zinc finger domain of the C2H2 type (Fig. A2). C2h2 has already been proved to be important during the initiation of primordia formation with targeted gene inactivation (Ohm et al 2011, https://pubmed.ncbi.nlm.nih.gov/21815946/). A binding site of c2h2 was detected in the upstream region of PleosPC15_2_1031042. There is a mismatch in the inferred binding motif which causes a reduced binding score in PC15 (Fig. A2/c). Indeed the PC9 nuclei contribute better to the total expression of this gene.
Despite this, and other (not shown) examples that we have found, we were not convinced about the reliability of this approach. There are many assumptions in this analysis, the positional weight matrices (PWM) that we used, are all based on indirect evidence, high number of loci these PWMs identify, uncertainty in the position of binding site from transcriptional start site, relation of difference in binding motif and expressional changes. We consider these factors to potentially contribute too much noise to the analyses for these to be robust, therefore, we are hesitant to include these results in the ms.
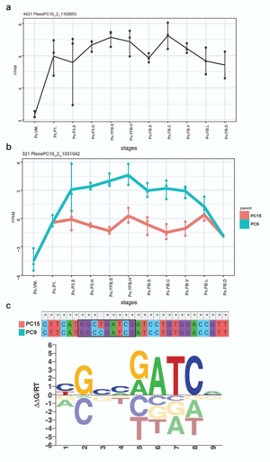
Fig A2: An example for promoter divergence a) expression of c2h2 transcription factor (TF) in P. ostreatus. b) allele-specific expression pattern of PleosPC15_2_1031042 from the two parental genomes. c) inferred binding motif of c2h2 TF and a detected potential binding site in the upstream region of PleosPC15_2_1031042 gene.
Reviewer #2 (Public Review):
The evolution of complex multicellularity represents a major developmental reprogramming, and comparing related species which differ in multicellular structures may shed light on the mechanisms involved. Here, the authors compare species of Basidiomycete fungi and focus on analyzing developmental transcriptomes to identify patterns across species. Deep RNA-Seq data is generated for two species, P. ostreatus and Pt. gracilis, sampling different developmental stages. The authors report conflicting evidence for a "developmental hourglass" using a weighted transcription index vs gene age categories. There is substantial allele-specific expression in P. ostreatus, and these genes tend to have a more recent origin, have more divergent upstream regions and coding sequences, and are enriched for developmentally regulated transcripts. Antisense transcripts have low overlap with coding regions and low conservation, and a subset show a positive or negative correlation with the overlapping gene. Comparison to a species without complex multicellular development is used to further classify the developmental program.
Overall the new transcriptional data and extensive analysis provide a thorough view of the types of transcripts that appear differentially regulated, their age, and associated gene function enrichment. The gene sets identified from this analysis as well as the potential to re-analyze this data will be useful to the community studying multicellularity in fungi. The primary insights drawn in this study relate to the dating of the developmental transcriptome, however some patterns observed with young genes and noncoding transcripts are primarily reflective of expected patterns of evolutionary time.
We appreciate the Reviewer’s nice words on our ms, we think the revised version has substantial improvements in many aspects listed above.
Reviewer #3 (Public Review):
Fungi are unique in forming complex 3D multicellular reproductive structures from 2D mycelium filaments, a property used in this paper to study the genetic changes associated with the evolution of complex 3D multicellularity. The manuscript by Merenyi et al. investigates the evolution of gene expression and genome regulation during the formation of reproductive structures (fruiting bodies) in the Agaricomycetes lineage of Fungi. Transcriptome and multicellularity evolution are very exciting fundamental questions in biology that only become accessible with recent technological developments and the appropriate analysis framework. Important perspectives include understanding how genes acquire new functions and what role plays transcriptional regulation in adaptation. The study gathers a very useful dataset to this end, and relies on generally relevant hypotheses-driven analyses.
Analysis of fruiting body transcriptome in nine species revealed that prediction from the development hourglass model (that young genes are expressed in early and late but not intermediate phases of development) verified only in a few species, including Pleurotus ostreatus. An allele-specific expression (ASE) analysis in P. ostreatus showed that young genes frequently show ASE during fruiting body development. A comparative analysis with C. neoformans, which reproduces sexually without forming fruiting body, indicates that young and old (but not intermediate) genes are likely involved specifically in fruiting body morphogenesis. A number of underlying hypothesis could be presented better, and importantly the connection between the various analyses did not appear obvious to me. Some hypotheses and reasoning therefore need clarification. Some important results from the analyses are not provided and not commented, although they are required to fully meet the manuscript's objectives.
We appreciate the Reviewer’s suggestions and have revised the ms as explained below.
- I do not clearly see the connection between the developmental hourglass model studied in the first part of the ms, and the allele-specific expression patterns in the second half of the ms. Which "phase" of the hourglass is expected to contain true CM-related genes (by contrast to general sexual processes)? Was P. ostreatus chosen for the ASE analysis because evidence for a developmental hourglass pattern was detected in this species? The conclusion that "evolutionary age predicts, to a large extent, the behaviour of a gene in the CM transcriptome" was established thanks to ASE in P. ostreatus, which was also found to be rather an exception for conforming to the hourglass model of developmental evolution. To what extent is this conclusion transferable to other Agaricomycete/fungal species?
We chose P. ostreatus because this is the only species for which the genomes of both parental strains (PC9 and PC15) are available. Although the hourglass concept is indeed a central hypothesis in animal developmental biology (though see recent critiques some (Piasecka et al 2013), our results suggest that it simply does not generally apply to fungal development. This may be due to the unique developmental mechanisms of fungi, or the independent origin(s) of CM in fungi. Our ms might have been misleading in this respect, in the revision we clarify that the ASE and hourglass analyses are independent of each other. Our interpretation of the hourglass results is that this model is not or hardly applicable for fungal development and the fact that P. ostreatus was the only species that in fact showed an hourglass did not drive our selection of this species. We inserted a note on this in the ms.
- The authors acknowledge that fruiting body-expressed genes may relate either to CM or to more general sexual functions, and that disentangling these functions is a major challenge in their study. An overview of which gene was assigned to which function is not explicit in the ms (proposed to be described in a separate publication). Do these functional gene classes show distinct transcriptome evolution patterns (hourglass model, ASE...)?
We made accessible the complete list of CM-related genes and genes with more general sexual functions in Table S2/b-c. Due to length restrictions, we do not discuss many or each of these genes here, but provided gene ontology-based overviews (Fig 8/c-d, from lines 631). To answer the question whether CM vs shared genes show distinct transcriptomic patterns, we analyzed ASE, NATs and the hourglass model separately for CM-specific and shared genes. as follows:
-hourglass: We calculated and visualised the TAI for CM-specific and Shared gene sets of P. ostreatus separately. The average value of TAI decreased a lot in Shared genes possibly due to the overrepresentation of ancient genes here, but the patterns remained similar to the original, which imply that not simply one or the other gene set drives these patterns (Fig A3).
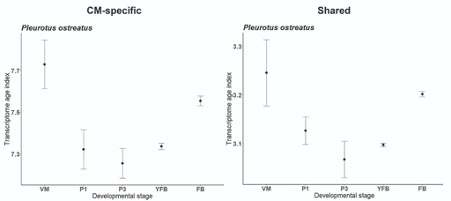
Fig A3: Transcriptome Age Index for CM-specific and Shared gene sets of P. ostreatus separately
-ASE: As we detailed in the ms, allele specific expression occurs mainly in young genes. Indeed, only 13.1% of ASE genes belong to the conserved gene sets (CMspecific: 200 and Shared: 144). Although there are more ASE genes (>2FC) among CM-specific genes, they are still underrepresented compared to young genes that are neither shared, nor CM-specific. This indicates that ASE is generally a feature of non-conserved genes and is not particularly characteristic for either conserved or CM-specific genes.
-NAT: We found that 17.3% of CM-specific (141 genes) and 18.3% of Shared genes (165 genes) overlap with antisense transcripts. Since these numbers don't differ substantially from 17.6%, which is the proportion of NATs corresponding to all protein coding genes, it implies an independent occurrence between NATs and these gene conservation groups.
3.) As far as I understand, major functions of the fruiting body transcriptome are either CM or general sexual functions. Could these genes, notably those showing ASE, play a role in general processes other than sexual development (hyphal growth, environment sensing, cell homeostasis, pathogenicity)?
Certainly, ASE might also occur in genes related to these processes. However, the processes mentioned by the Reviewer are likely associated with very conserved genes (except pathogenicity, which we can’t examine here) and our results suggest that ASE is more typical of young genes that are under weak selection. We detected ASE in 931/343 (S2/S4 genes) genes expressed in the vegetative mycelium stage of P. ostreatus. We also note that by the definition of developmentally regulated genes, we do not expect very basic fungal processes, like hyphal growth to be among the functions of the genes we identified. Genes related to such basic (housekeeping) processes usually (exceptions exist) show flat expression profiles (because they are equally important in mycelia and all fruiting body stages) and will not be picked up by our pipelines for identifying shared developmentally regulated genes.
- As stated by the authors, "the goal of this study was to systematically tease apart the components and driving forces of transcriptome evolution in CM fungi". What drives the interesting ASE pattern discovered however remains an open question at the end of the ms. The authors appropriately discuss that these patterns could be either adaptive or neutral but there is no direct evidence for any scenario in P. ostreatus. Is the expression of (some of) the young genes showing ASE required for CM? one or two case studies would allow providing support for such scenarios.
We respectfully disagree. We provide evidence that the driving force of ASE is promoter divergence (and consequently differential transcription factor binding) in genes in which it is tolerated (see conclusions, lines 708-712). Our results suggest that ASE is mostly a neutrally arising phenomenon. To get to the mechanistic bases of how promoter divergence can cause ASE (following the suggestion of Reviewer 1), we analysed putative, inferred transcription factor binding motifs in P. ostreatus and found that ASE genes had more divergence in putative TF binding sites. However, it is important to emphasise that all TF motifs we analyzed are inferred motifs and therefore these results are indicative at best.
Reviewer #4 (Public Review):
This work develops a comparative framework to test genes which support complex morphological structures and complex multicelluarity. This expands beyond simple gene sharing and phylogenomics by incorporating comparison of gene expression profiling of development of multicellular structures during sexual reproduction. This approach tests the hypothesis that genes underlying sexual reproductive structure formation are homologous and the molecular evolutionary processes that control transcriptome evolution which underlie complex multicellularity.
The approaches used are appropriate and employ modern comparative and transcriptome analyses to example allele specific expression, and evaluate an age of the evolutionary ages of genes. This work produced additional new RNAseq to examine developmental processes and combined it with existing published data to contrast fungi with either complex morphologies or yeast forms.
The strengths of work are well selected comparison organisms and efforts to have developmental stages which are appropriate comparisons.
We appreciate the Reviewer’s positive comments.
Weakness could be pointed to in how the NAT descriptions are interesting it isn't clear how they link directly to morphology variation or development. I am unclear if these are arising from new de novo promotors, are ferried by transposable elements, or if any other understanding of their genesis indicates they are more than very recent gains in a species for the most part and not part of any conserved developmental process (outside a few exemplars).
Originally, we assayed natural antisense transcripts (NAT) based on the assumption that they regulate developmental processes (e.g. Kim et al 2018 https://doi.org/10.1128/mBio.01292-18). Our analyses showed that although NATs are abundant in CM transcriptomes of fungi, they show no homology across species and so are unlikely to drive conserved developmental processes, which we are after in this ms. Rather, our data are compatible with most (but likely not all) NATs being transcriptional noise, arising from novel or random promoters. We therefore shortened this section and moved much of it to the Appendix 1.
The impact of this work will reside in how gene age intersects with variability and relative importance in CM. it will be interesting to see future work examine the functions of these genes and test how allele specific expression and specific alleles are contributing to the formation of these tissues and growth forms. I am still not sure if molecular mechanisms of how high variability in gene expression is still producing relatively uniform morphologies, or if it isn't quantification of morphological variation would be nice to link to whether ASE underlie that.
We agree that allele specific expression could influence morphologies significantly, but investigating that is beyond the scope of the current work (it would require a population genomics project). More direct evidence on allelic differences can be seen in monokaryon phenotypes, which only express one of the parental alleles. Phenotypic differences are obvious in the mycelium of the two parental monokaryons : the mycelium of PC9 is more fluffy and grows faster than that of PC15. This was reported recently by Lee et al 2021 (https://doi.org/10.1093/g3journal/jkaa008). We agree with the Reviewer that this is a very exciting future research direction.
To my read of the work, the authors achieved their goals and confirmed hypothesis about the age of genes and the variability of gene expression. I still feel there is some clarity lost in whether the findings across the large number of species compared here help inform predictions or classifications of types of genes which either have ASE or are implicated in CM. This is really work for the future as the authors have provided a detailed analysis and approach that can fuel further direction in this research area.
To address this issue we reworked the ms to make connections between ASE and CM clearer. Because ASE appears based on our results to (mostly) arise neutrally, predictions for other species are expected to be hard. On the other hand, we think we can make confident predictions on what types of genes are implicated in CM in other species, at least for conserved aspects of fruiting body development.
-
Evaluation Summary:
This manuscript provides a deep characterization of transcriptional regulation and conservation across key stages of complex multicellular development during mushroom formation. The authors present evidence for extensive allele-specific expression that includes many developmentally regulated genes that appear to have evolved recently. These findings help underscore how the tuning of gene expression and gains of new genes, the function of which will need to be unraveled in future, are likely the basis for the evolution of complex morphologies in fungi. This work should be of broad interest to evolutionary biologists, and especially to those studying the evolution of gene regulation.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private …
Evaluation Summary:
This manuscript provides a deep characterization of transcriptional regulation and conservation across key stages of complex multicellular development during mushroom formation. The authors present evidence for extensive allele-specific expression that includes many developmentally regulated genes that appear to have evolved recently. These findings help underscore how the tuning of gene expression and gains of new genes, the function of which will need to be unraveled in future, are likely the basis for the evolution of complex morphologies in fungi. This work should be of broad interest to evolutionary biologists, and especially to those studying the evolution of gene regulation.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #1 and Reviewer #4 agreed to share their names with the authors.)
-
Reviewer #1 (Public Review):
This study sought to systematically identify the components and driving forces of transcriptome evolution in fungi that exhibit complex multicellularity (CM). The authors examined a series of parameters or expression signatures (i.e. natural antisense transcripts, allele-specific expression, RNA-editing) concluding that the best predictor of a gene behavior in the CM transcriptome was evolutionary age.
Thus, the transcriptomes of fruiting bodies showed a distinct gene-age-related stratification, where it was possible to sort out genes related to general sexual processes from those likely linked to morphogenetic aspects of the CM fruiting bodies. Notably, their results did not support a developmental hourglass, which is the rather predominant hypothesis in metazoans, including some analysis in fungi.The …
Reviewer #1 (Public Review):
This study sought to systematically identify the components and driving forces of transcriptome evolution in fungi that exhibit complex multicellularity (CM). The authors examined a series of parameters or expression signatures (i.e. natural antisense transcripts, allele-specific expression, RNA-editing) concluding that the best predictor of a gene behavior in the CM transcriptome was evolutionary age.
Thus, the transcriptomes of fruiting bodies showed a distinct gene-age-related stratification, where it was possible to sort out genes related to general sexual processes from those likely linked to morphogenetic aspects of the CM fruiting bodies. Notably, their results did not support a developmental hourglass, which is the rather predominant hypothesis in metazoans, including some analysis in fungi.The studies involved analyses of new transcriptomic datasets for different developmental stages (and tissue types in some cases) of Pleurotus ostreatus and Pterula gracilis, as well as the analyses of existing datasets for other fungi.
There are diverse interesting observations such as ones regarding Allele Specific Expression (ASE), suggesting that in P. ostreatus ASE mainly occurs due to cis-regulatory allele divergence, possibly in fast evolving genes that are not under strong selection constraints, such as ones grouped in youngest gene ages categories. In addition, a large number of conserved unannotated genes among CM-specific orthogroups highlights the rather cryptic nature of CM in fungi and raises as an important area for future research.Some of the key aspects of the analyses would need to be better exemplified such as:
– Providing a better description of the developmentally expressed TFs only in CM species
– Providing clear examples of the promoter divergence that could be the underlying mechanism behind ASE. In particular, for some cases, there may be enough information in the literature/databases to predict the appearance or disappearance of relevant cis-elements in the promoters showing the highest divergence in genes depicting the highest levels of ASE.
-
Reviewer #2 (Public Review):
The evolution of complex multicellularity represents a major developmental reprogramming, and comparing related species which differ in multicellular structures may shed light on the mechanisms involved. Here, the authors compare species of Basidiomycete fungi and focus on analyzing developmental transcriptomes to identify patterns across species. Deep RNA-Seq data is generated for two species, P. ostreatus and Pt. gracilis, sampling different developmental stages. The authors report conflicting evidence for a "developmental hourglass" using a weighted transcription index vs gene age categories. There is substantial allele-specific expression in P. ostreatus, and these genes tend to have a more recent origin, have more divergent upstream regions and coding sequences, and are enriched for developmentally …
Reviewer #2 (Public Review):
The evolution of complex multicellularity represents a major developmental reprogramming, and comparing related species which differ in multicellular structures may shed light on the mechanisms involved. Here, the authors compare species of Basidiomycete fungi and focus on analyzing developmental transcriptomes to identify patterns across species. Deep RNA-Seq data is generated for two species, P. ostreatus and Pt. gracilis, sampling different developmental stages. The authors report conflicting evidence for a "developmental hourglass" using a weighted transcription index vs gene age categories. There is substantial allele-specific expression in P. ostreatus, and these genes tend to have a more recent origin, have more divergent upstream regions and coding sequences, and are enriched for developmentally regulated transcripts. Antisense transcripts have low overlap with coding regions and low conservation, and a subset show a positive or negative correlation with the overlapping gene. Comparison to a species without complex multicellular development is used to further classify the developmental program.
Overall the new transcriptional data and extensive analysis provide a thorough view of the types of transcripts that appear differentially regulated, their age, and associated gene function enrichment. The gene sets identified from this analysis as well as the potential to re-analyze this data will be useful to the community studying multicellularity in fungi. The primary insights drawn in this study relate to the dating of the developmental transcriptome, however some patterns observed with young genes and noncoding transcripts are primarily reflective of expected patterns of evolutionary time.
-
Reviewer #3 (Public Review):
Fungi are unique in forming complex 3D multicellular reproductive structures from 2D mycelium filaments, a property used in this paper to study the genetic changes associated with the evolution of complex 3D multicellularity. The manuscript by Merenyi et al. investigates the evolution of gene expression and genome regulation during the formation of reproductive structures (fruiting bodies) in the Agaricomycetes lineage of Fungi. Transcriptome and multicellularity evolution are very exciting fundamental questions in biology that only become accessible with recent technological developments and the appropriate analysis framework. Important perspectives include understanding how genes acquire new functions and what role plays transcriptional regulation in adaptation. The study gathers a very useful dataset to …
Reviewer #3 (Public Review):
Fungi are unique in forming complex 3D multicellular reproductive structures from 2D mycelium filaments, a property used in this paper to study the genetic changes associated with the evolution of complex 3D multicellularity. The manuscript by Merenyi et al. investigates the evolution of gene expression and genome regulation during the formation of reproductive structures (fruiting bodies) in the Agaricomycetes lineage of Fungi. Transcriptome and multicellularity evolution are very exciting fundamental questions in biology that only become accessible with recent technological developments and the appropriate analysis framework. Important perspectives include understanding how genes acquire new functions and what role plays transcriptional regulation in adaptation. The study gathers a very useful dataset to this end, and relies on generally relevant hypotheses-driven analyses.
Analysis of fruiting body transcriptome in nine species revealed that prediction from the development hourglass model (that young genes are expressed in early and late but not intermediate phases of development) verified only in a few species, including Pleurotus ostreatus. An allele-specific expression (ASE) analysis in P. ostreatus showed that young genes frequently show ASE during fruiting body development. A comparative analysis with C. neoformans, which reproduces sexually without forming fruiting body, indicates that young and old (but not intermediate) genes are likely involved specifically in fruiting body morphogenesis. A number of underlying hypothesis could be presented better, and importantly the connection between the various analyses did not appear obvious to me. Some hypotheses and reasoning therefore need clarification. Some important results from the analyses are not provided and not commented, although they are required to fully meet the manuscript's objectives.
I do not clearly see the connection between the developmental hourglass model studied in the first part of the ms, and the allele-specific expression patterns in the second half of the ms. Which "phase" of the hourglass is expected to contain true CM-related genes (by contrast to general sexual processes)? Was P. ostreatus chosen for the ASE analysis because evidence for a developmental hourglass pattern was detected in this species? The conclusion that "evolutionary age predicts, to a large extent, the behaviour of a gene in the CM transcriptome" was established thanks to ASE in P. ostreatus, which was also found to be rather an exception for conforming to the hourglass model of developmental evolution. To what extent is this conclusion transferable to other Agaricomycete/fungal species?
The authors acknowledge that fruiting body-expressed genes may relate either to CM or to more general sexual functions, and that disentangling these functions is a major challenge in their study. An overview of which gene was assigned to which function is not explicit in the ms (proposed to be described in a separate publication). Do these functional gene classes show distinct transcriptome evolution patterns (hourglass model, ASE...)?
3.) As far as I understand, major functions of the fruiting body transcriptome are either CM or general sexual functions. Could these genes, notably those showing ASE, play a role in general processes other than sexual development (hyphal growth, environment sensing, cell homeostasis, pathogenicity)?
- As stated by the authors, "the goal of this study was to systematically tease apart the components and driving forces of transcriptome evolution in CM fungi". What drives the interesting ASE pattern discovered however remains an open question at the end of the ms. The authors appropriately discuss that these patterns could be either adaptive or neutral but there is no direct evidence for any scenario in P. ostreatus. Is the expression of (some of) the young genes showing ASE required for CM? one or two case studies would allow providing support for such scenarios.
-
Reviewer #4 (Public Review):
This work develops a comparative framework to test genes which support complex morphological structures and complex multicelluarity. This expands beyond simple gene sharing and phylogenomics by incorporating comparison of gene expression profiling of development of multicellular structures during sexual reproduction. This approach tests the hypothesis that genes underlying sexual reproductive structure formation are homologous and the molecular evolutionary processes that control transcriptome evolution which underlie complex multicellularity.
The approaches used are appropriate and employ modern comparative and transcriptome analyses to example allele specific expression, and evaluate an age of the evolutionary ages of genes. This work produced additional new RNAseq to examine developmental processes and …
Reviewer #4 (Public Review):
This work develops a comparative framework to test genes which support complex morphological structures and complex multicelluarity. This expands beyond simple gene sharing and phylogenomics by incorporating comparison of gene expression profiling of development of multicellular structures during sexual reproduction. This approach tests the hypothesis that genes underlying sexual reproductive structure formation are homologous and the molecular evolutionary processes that control transcriptome evolution which underlie complex multicellularity.
The approaches used are appropriate and employ modern comparative and transcriptome analyses to example allele specific expression, and evaluate an age of the evolutionary ages of genes. This work produced additional new RNAseq to examine developmental processes and combined it with existing published data to contrast fungi with either complex morphologies or yeast forms.
The strengths of work are well selected comparison organisms and efforts to have developmental stages which are appropriate comparisons.
Weakness could be pointed to in how the NAT descriptions are interesting it isn't clear how they link directly to morphology variation or development. I am unclear if these are arising from new de novo promotors, are ferried by transposable elements, or if any other understanding of their genesis indicates they are more than very recent gains in a species for the most part and not part of any conserved developmental process (outside a few exemplars).
The impact of this work will reside in how gene age intersects with variability and relative importance in CM. it will be interesting to see future work examine the functions of these genes and test how allele specific expression and specific alleles are contributing to the formation of these tissues and growth forms. I am still not sure if molecular mechanisms of how high variability in gene expression is still producing relatively uniform morphologies, or if it isn't quantification of morphological variation would be nice to link to whether ASE underlie that.
To my read of the work, the authors achieved their goals and confirmed hypothesis about the age of genes and the variability of gene expression. I still feel there is some clarity lost in whether the findings across the large number of species compared here help inform predictions or classifications of types of genes which either have ASE or are implicated in CM. This is really work for the future as the authors have provided a detailed analysis and approach that can fuel further direction in this research area.
-
-
-
-
-
-
