Parallel evolution between genomic segments of seasonal human influenza viruses reveals RNA-RNA relationships
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
The influenza A virus (IAV) genome consists of eight negative-sense viral RNA (vRNA) segments that are selectively assembled into progeny virus particles through RNA-RNA interactions. To explore putative intersegmental RNA-RNA relationships, we quantified similarity between phylogenetic trees comprising each vRNA segment from seasonal human IAV. Intersegmental tree similarity differed between subtype and lineage. While intersegmental relationships were largely conserved over time in H3N2 viruses, they diverged in H1N1 strains isolated before and after the 2009 pandemic. Surprisingly, intersegmental relationships were not driven solely by protein sequence, suggesting that IAV evolution could also be driven by RNA-RNA interactions. Finally, we used confocal microscopy to determine that colocalization of highly coevolved vRNA segments is enriched over other assembly intermediates at the nuclear periphery during productive viral infection. This study illustrates how putative RNA interactions underlying selective assembly of IAV can be interrogated with phylogenetics.
Article activity feed
-

-

Author Response:
Reviewer #1:
In this paper, authors did a fine job of combining phylogenetics and molecular methods to demonstrate the parallel evolution across vRNA segments in two seasonal influenza A virus subtypes. They first estimated phylogenetic relationships between vRNA segments using Robinson-Foulds distance and identified the possibility of parallel evolution of RNA-RNA interactions driving the genomic assembly. This is indeed an interesting mechanism in addition to the traditional role for proteins for the same. Subsequently, they used molecular biology to validate such RNA-RNA driven interaction by demonstrating co-localization of vRNA segments in infected cells. They also showed that the parallel evolution between vRNA segments might vary across subtypes and virus lineages isolated from distinct host origins. Overall, …
Author Response:
Reviewer #1:
In this paper, authors did a fine job of combining phylogenetics and molecular methods to demonstrate the parallel evolution across vRNA segments in two seasonal influenza A virus subtypes. They first estimated phylogenetic relationships between vRNA segments using Robinson-Foulds distance and identified the possibility of parallel evolution of RNA-RNA interactions driving the genomic assembly. This is indeed an interesting mechanism in addition to the traditional role for proteins for the same. Subsequently, they used molecular biology to validate such RNA-RNA driven interaction by demonstrating co-localization of vRNA segments in infected cells. They also showed that the parallel evolution between vRNA segments might vary across subtypes and virus lineages isolated from distinct host origins. Overall, I find this to be excellent work with major implications for genome evolution of infectious viruses; emergence of new strains with altered genome combination.
Comments:
I am wondering if leaving out sequences (not resolving well) in the phylogenic analysis interferes with the true picture of the proposed associations. What if they reflect the evolutionary intermediates, with important implications for the pathogen evolution which is lost in the analyses?
We fully appreciate this concern and have explored this extensively. One principle assumption underlying the approach we outline in this manuscript is that the trees analyzed are robust and well- resolved. We use tree similarity as a correlate for relationships between genomic segments, so the trees must be robust enough to support our claims, as we have clarified in lines 128-131. We initially set out to examine a broader range of viral isolates in each set of trees, but larger trees containing more isolates consistently failed to be supported by bootstrapping. Bootstrapping is by far the most widely used methodology for demonstrating support for tree nodes. We provided the closest possible example to the trees presented in this manuscript for comparison. We took all 84 H3N2 strains from 2005-2014 analyzed in replicate trees 1-7 and collapsed these sequences into one tree for each vRNA segment. Figure X-A, specifically provided for the reviewers, illustrates the resultant collapsed PB2 tree, with bootstrap values of 70 or higher shown in red and individual strains coded by cluster and replicate. As expected, the majority of internal nodes on such a tree are largely unsupported by bootstrapping, indicating that relaxing our constraint of 97% sequence identity increases the uncertainty in our trees.
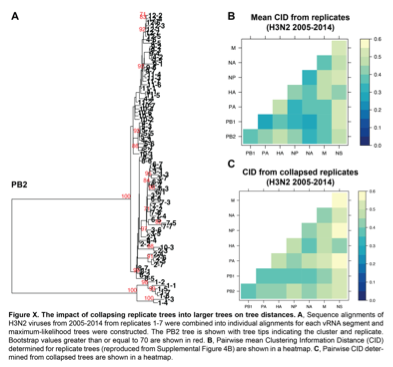
Because we agree with Reviewers #1 and #3 on the critical importance of validating our approach, we determined the distances between these new collapsed trees using a complementary approach, Clustering Information Distances (CID), that is independent of tree size (Supplemental Figure 4B and Figure X-B & X-C). Larger trees containing all sequences yielded pairwise vRNA relationships that are largely similar to those we report in the manuscript (R2 = 0.6408; P = 3.1E-07; Figure X-B vs. X-C), including higher tree similarity between PB2 and NA over NS. This observation strengthens the rationale to focus on these segments for molecular validation and correlate parallel evolution to intracellular localization in our manuscript (Figure 7). However, tree distances are generally higher in Figure X-C than in Figure X-B, which we might expect if poorly supported nodes in larger trees artificially inflate phylogenetic signal. Given the overall similarity between Figures X-B and X-C, both methods yield largely comparable results. We ultimately relied upon the more robust replicate trees with stronger bootstrap support.
Lines 50-51: Can you please elaborate? I think this might be useful for the reader to better understand the context. Also, a brief description on functional association between different known fragments might instigate curiosity among the readers from the very beginning. At present, it largely caters to people already familiar with the biology of influenza virus.
We have added additional information to reflect the complexity of intersegmental interactions and the current standing of the field (lines 49-52).
Lines 95-96 Were these strains all swine-origin? More details on these lineages will be useful for the readers.
We have clarified that all strains analyzed were isolated from humans, but were of different lineages (lines 115-120).
Lines 128-132: I think it will be nice to talk about these hypotheses well in advance, may be in the Introduction, with more functional details of viral segments.
We incorporated our hypotheses regarding tree similarity into the existing discussion of epistasis in the Introduction (lines 74-75 and 89-106).
Lines 134-136: Please rephrase this sentence to make it more direct and explain the why. E.g. "... parallel evolution between PB1 and HA is likely to be weaker than that of PB1 and PA".
The text has been modified (lines 165-168).
Lines 222-223: Please include a set of hypotheses to explain you results? Please add a perspective in the discussion on how this contribute might to the pandemic potential of H1N!?.
We have added in our interpretation of the results (lines 259-264) and expanded upon this in the Discussion (lines 418-422).
Lines 287-288: I am wondering how likely is this to be true for H1N1.
We have expanded on this in the Discussion (lines 409-410).
Reviewer #2:
The influenza A genome is made up of eight viral RNAs. Despite being segmented, many of these RNAs are known to evolve in parallel, presumably due to similar selection pressures, and influence each other's evolution. The viral protein-protein interactions have been found to be the mechanism driving the genomic evolution. Employing a range of phylogenetic and molecular methods, Jones et al. investigated the evolution of the seasonal Influenza A virus genomic segments. They found the evolutionary relationships between different RNAs varied between two subtypes, namely H1N1 and H3N2. The evolutionary relationships in case of H1N1 were also temporally more diverse than H3N2. They also reported molecular evidence that indicated the presence of RNA-RNA interaction driving the genomic coevolution, in addition to the protein interactions. These results do not only provide additional support for presence of parallel evolution and genetic interactions in Influenza A genome and but also advances the current knowledge of the field by providing novel evidence in support of RNA-RNA interactions as a driver of the genomic evolution. This work is an excellent example of hypothesis-driven scientific investigation.
The communication of the science could be improved, particularly for viral evolutionary biologists who study emergent evolutionary patterns but do not specialise in the underlying molecular mechanisms. The improvement can be easily achieved by explaining jargon (e.g., deconvolution) and methodological logics that are not immediately clear to a non-specialist.
We have clarified or eliminated jargon wherever possible throughout the text.
The introduction section could be better structured. The crux of this study is the parallel molecular evolution in influenza genome segments and interactions (epistasis). The authors spent the majority of the introduction section leading to those two topics and then treated them summarily. This structure, in my opinion, is diluting the story. Instead, introducing the two topics in detail at the beginning (right after introducing the system) then discussing their links to reassortments, viral emergence etc. could be a more informative, easily understandable and focused structure. The authors also failed to clearly state all the hypotheses and predictions (e.g., regarding intracellular colocalisation) near the end of the introduction.
We restructured the Introduction with more background on genomic assembly in influenza viruses, as requested by two reviewers (lines 43-52), more discussion of epistasis (lines 58-63) and provided a more thorough discussion of all hypotheses (lines 74-77, 88-92, 94-95, 97-106).
The authors used Robinson-Foulds (RF) metric to quantify topological distance between phylogenetic trees-a key variable of the study. But they did not justify using the metric despite its well-known drawbacks including lack of biological rational and lack of robustness, and particularly when more robust measures, such as generalised RF, are available.
We agree that RF has drawbacks. To address this, we performed a companion analysis using the Clustering Information Distance (CID) recently described by Smith, 2020. The mean CID can be found in Figure S4, the standard error of the mean in Figure S5, and networks depicting overall relationships between segments by CID in Figure S7E-S7H. To better assess how well RF and CID correlate with each other across influenza virus subtypes and lineages, we reanalyzed all data from both sets of distance measures by linear regression (Figure 3B, 4B-C, 5B, S6 and S9). Our results from both methods are highly comparable, which we believe strengthens our conclusions. Both analyses are included in the resubmission (lines 86-89; 162; 164; 187-188; 199-200; 207-208; 231-234; 242-244; 466-470).
Figure 1 of the paper is extremely helpful to understand the large number of methods and links between them. But it could be more useful if the authors could clearly state the goal of each step and also included the molecular methods in it. That would have connected all the hypotheses in the introduction to all the results neatly. I found a good example of such a schematic in a paper that the authors have cited (Fig. 1 of Escalera-Zamudio et al. 2020, Nature communications). Also this methodological scheme needs to be cited in the methods section.
We provided the molecular methods in a schematic in Figure 1D and the figure is cited in the Methods (lines 310; 440; 442; 456; 501).
Finally, I found the methods section to be difficult to navigate, not because it lacked any detail. The authors have been excellent in providing a considerable amount of methodological details. The difficulty arose due to the lack of a chronological structure. Ideally, the methods should be grouped under research aims (for example, Data mining and subsampling, analysis of phylogenetic concordance between genomic segments, identifying RNA-RNA interactions etc.), which will clearly link methods to specific results in one hand and the hypotheses, in the other. This structure would make the article more accessible, for a general audience in particular. The results section appeared to achieve this goal and thus often repeat or explain methodological detail, which ideally should have been restricted to the methods section.
We organized the Methods section by research aims as suggested. However, some discussion of the methods were retained in the Results section to ensure that the manuscript is accessible to audiences without formal training in phylogenetics.
Reviewer #3:
The authors sought to show how the segments of influenza viruses co-evolve in different lineages. They use phylogenetic analysis of a subset of the complete genomes of H3N2 or the two H1N1 lineages (pre and post 2009), and use a method - Robinson-Foulds distance analysis - to determine the relationships between the evolutionary patterns of each segment, and find some that are non-random.
- The phylogenetic analysis used leaves out sequences that do not resolve well in the phylogenic analysis, with the goal of achieving higher bootstrap values. It is difficult to understand how that gives the most accurate picture of the associations - those sequences represent real evolutionary intermediates, and their inclusion should not alter the relationships between the more distantly related sequences. It seems that this creates an incomplete picture that artificially emphasizes differences among the clades for each segment analyzed?
Reviewer #1 raised the same concern. Please refer to our response at the beginning of this letter where we address this issue in depth.
- It is not clear what the significance is of finding that sequences that share branching patterns in the phylogeny, and how that informs our understanding of the likelihood of genetic segments having some functional connection. What mechanism is being suggested - is this a proxy for the gene segments having been present in the same viruses - thereby revealing the favored gene segment combinations? Is there some association suggested between the RNA sequences of the different segments? The frequently evoked HA:NA associations may not be a directly relevant model as those are thought to relate to the balance of sialic acid binding and cleavage associated with mutations focused around the receptor binding site and active site, length of NA stalk, and the HA stalk - does that show up in the overall phylogeny of the HA and NA segments? Is there co-evolution of the polymerase gene segments, or has that been revealed in previous studies, as is suggested?
We clarified our working hypotheses in the Introduction (lines 89-106) and what is known about the polymerase subunits (lines 92-93). Our data do suggest that polymerase subunits share similar evolutionary trajectories that are more driven by protein than RNA (lines 291-293; Figure 2A and 6). The point about epistasis between HA and NA arising from indirect interactions is entirely fair, but these studies are nonetheless the basis for our own work. We have clarified the distinction between these prior studies and our own in the text (lines 60-63 and 74-75). Moreover, our protein trees built from HA and NA recapitulate what has been shown previously, which we highlight in the text (lines 293-296; Figure 6 and Figure S10). We also clarified our interpretation of tree similarity throughout the text (lines 165-168; 190-191; 261-264; 323-326; 419-423).
The mechanisms underlying the genomic segment associations described here are not clear. By definition they would be related to the evolution of the entire RNA segment sequence, since that is being analyzed - (1) is this because of a shared function (seems unlikely but perhaps pointing to a new activity), or is it (2) because of some RNA sequence-associated function (inter-segment hybridization, common association of RNA with some cellular or viral protein)? (3) Related to specific functions in RNA packaging - please tell us whether the current RNA packaging models inform about a possible process. Is there a known packaging assembly process based on RNA sequences, where the association leads to co-transport and packaging - in that case the co-evolution should be more strongly seen in the region involved in that function and not elsewhere? The apparent increased association in the cytoplasm of the subset of genes examined for the single virus looks mainly in the cytoplasm close to the nucleus - suggesting function (2) and/or (3)?.
It is difficult to figure out how the data found correlates with the known data on reassortment efficiency or mechanisms of systems for RNA segment selection for packaging or transport - if that is not obvious, maybe you can suggest processes that might be involved.
We provided more context on genomic packaging in the Introduction, including the current model in which direct RNA interactions are thought to drive genomic assembly (lines 43-53). Although genomic segments are bound by viral nucleoprotein (NP), accurate genomic assembly is theorized to be a result of intersegment hybridization rather than driven by viral or cellular protein. We further clarified our hypotheses regarding the colocalization data in the Results section to make the proposed mechanism clearer (lines 313-326).
-
Reviewer #3 (Public Review):
The authors sought to show how the segments of influenza viruses co-evolve in different lineages. They use phylogenetic analysis of a subset of the complete genomes of H3N2 or the two H1N1 lineages (pre and post 2009), and use a method - Robinson-Foulds distance analysis - to determine the relationships between the evolutionary patterns of each segment, and find some that are non-random.
The phylogenetic analysis used leaves out sequences that do not resolve well in the phylogenic analysis, with the goal of achieving higher bootstrap values. It is difficult to understand how that gives the most accurate picture of the associations - those sequences represent real evolutionary intermediates, and their inclusion should not alter the relationships between the more distantly related sequences. It seems that this …
Reviewer #3 (Public Review):
The authors sought to show how the segments of influenza viruses co-evolve in different lineages. They use phylogenetic analysis of a subset of the complete genomes of H3N2 or the two H1N1 lineages (pre and post 2009), and use a method - Robinson-Foulds distance analysis - to determine the relationships between the evolutionary patterns of each segment, and find some that are non-random.
The phylogenetic analysis used leaves out sequences that do not resolve well in the phylogenic analysis, with the goal of achieving higher bootstrap values. It is difficult to understand how that gives the most accurate picture of the associations - those sequences represent real evolutionary intermediates, and their inclusion should not alter the relationships between the more distantly related sequences. It seems that this creates an incomplete picture that artificially emphasizes differences among the clades for each segment analyzed?
It is not clear what the significance is of finding that sequences that share branching patterns in the phylogeny, and how that informs our understanding of the likelihood of genetic segments having some functional connection. What mechanism is being suggested - is this a proxy for the gene segments having been present in the same viruses - thereby revealing the favored gene segment combinations? Is there some association suggested between the RNA sequences of the different segments? The frequently evoked HA:NA associations may not be a directly relevant model as those are thought to relate to the balance of sialic acid binding and cleavage associated with mutations focused around the receptor binding site and active site, length of NA stalk, and the HA stalk - does that show up in the overall phylogeny of the HA and NA segments? Is there co-evolution of the polymerase gene segments, or has that been revealed in previous studies, as is suggested?
The mechanisms underlying the genomic segment associations described here are not clear. By definition they would be related to the evolution of the entire RNA segment sequence, since that is being analyzed - (1) is this because of a shared function (seems unlikely but perhaps pointing to a new activity), or is it (2) because of some RNA sequence-associated function (inter-segment hybridization, common association of RNA with some cellular or viral protein)? (3) Related to specific functions in RNA packaging - please tell us whether the current RNA packaging models inform about a possible process. Is there a known packaging assembly process based on RNA sequences, where the association leads to co-transport and packaging - in that case the co-evolution should be more strongly seen in the region involved in that function and not elsewhere? The apparent increased association in the cytoplasm of the subset of genes examined for the single virus looks mainly in the cytoplasm close to the nucleus - suggesting function (2) and/or (3)?.
It is difficult to figure out how the data found correlates with the known data on reassortment efficiency or mechanisms of systems for RNA segment selection for packaging or transport - if that is not obvious, maybe you can suggest processes that might be involved.
-
Reviewer #2 (Public Review):
The influenza A genome is made up of eight viral RNAs. Despite being segmented, many of these RNAs are known to evolve in parallel, presumably due to similar selection pressures, and influence each other's evolution. The viral protein-protein interactions have been found to be the mechanism driving the genomic evolution. Employing a range of phylogenetic and molecular methods, Jones et al. investigated the evolution of the seasonal Influenza A virus genomic segments. They found the evolutionary relationships between different RNAs varied between two subtypes, namely H1N1 and H3N2. The evolutionary relationships in case of H1N1 were also temporally more diverse than H3N2. They also reported molecular evidence that indicated the presence of RNA-RNA interaction driving the genomic coevolution, in addition to …
Reviewer #2 (Public Review):
The influenza A genome is made up of eight viral RNAs. Despite being segmented, many of these RNAs are known to evolve in parallel, presumably due to similar selection pressures, and influence each other's evolution. The viral protein-protein interactions have been found to be the mechanism driving the genomic evolution. Employing a range of phylogenetic and molecular methods, Jones et al. investigated the evolution of the seasonal Influenza A virus genomic segments. They found the evolutionary relationships between different RNAs varied between two subtypes, namely H1N1 and H3N2. The evolutionary relationships in case of H1N1 were also temporally more diverse than H3N2. They also reported molecular evidence that indicated the presence of RNA-RNA interaction driving the genomic coevolution, in addition to the protein interactions. These results do not only provide additional support for presence of parallel evolution and genetic interactions in Influenza A genome and but also advances the current knowledge of the field by providing novel evidence in support of RNA-RNA interactions as a driver of the genomic evolution. This work is an excellent example of hypothesis-driven scientific investigation.
The communication of the science could be improved, particularly for viral evolutionary biologists who study emergent evolutionary patterns but do not specialise in the underlying molecular mechanisms. The improvement can be easily achieved by explaining jargon (e.g., deconvolution) and methodological logics that are not immediately clear to a non-specialist.
The introduction section could be better structured. The crux of this study is the parallel molecular evolution in influenza genome segments and interactions (epistasis). The authors spent the majority of the introduction section leading to those two topics and then treated them summarily. This structure, in my opinion, is diluting the story. Instead, introducing the two topics in detail at the beginning (right after introducing the system) then discussing their links to reassortments, viral emergence etc. could be a more informative, easily understandable and focused structure. The authors also failed to clearly state all the hypotheses and predictions (e.g., regarding intracellular colocalisation) near the end of the introduction.
The authors used Robinson-Foulds (RF) metric to quantify topological distance between phylogenetic trees-a key variable of the study. But they did not justify using the metric despite its well-known drawbacks including lack of biological rational and lack of robustness, and particularly when more robust measures, such as generalised RF, are available.
Figure 1 of the paper is extremely helpful to understand the large number of methods and links between them. But it could be more useful if the authors could clearly state the goal of each step and also included the molecular methods in it. That would have connected all the hypotheses in the introduction to all the results neatly. I found a good example of such a schematic in a paper that the authors have cited (Fig. 1 of Escalera-Zamudio et al. 2020, Nature communications). Also this methodological scheme needs to be cited in the methods section.
Finally, I found the methods section to be difficult to navigate, not because it lacked any detail. The authors have been excellent in providing a considerable amount of methodological details. The difficulty arose due to the lack of a chronological structure. Ideally, the methods should be grouped under research aims (for example, Data mining and subsampling, analysis of phylogenetic concordance between genomic segments, identifying RNA-RNA interactions etc.), which will clearly link methods to specific results in one hand and the hypotheses, in the other. This structure would make the article more accessible, for a general audience in particular. The results section appeared to achieve this goal and thus often repeat or explain methodological detail, which ideally should have been restricted to the methods section.
-
Reviewer #1 (Public Review):
In this paper, authors did a fine job of combining phylogenetics and molecular methods to demonstrate the parallel evolution across vRNA segments in two seasonal influenza A virus subtypes. They first estimated phylogenetic relationships between vRNA segments using Robinson-Foulds distance and identified the possibility of parallel evolution of RNA-RNA interactions driving the genomic assembly. This is indeed an interesting mechanism in addition to the traditional role for proteins for the same. Subsequently, they used molecular biology to validate such RNA-RNA driven interaction by demonstrating co-localization of vRNA segments in infected cells. They also showed that the parallel evolution between vRNA segments might vary across subtypes and virus lineages isolated from distinct host origins. Overall, I …
Reviewer #1 (Public Review):
In this paper, authors did a fine job of combining phylogenetics and molecular methods to demonstrate the parallel evolution across vRNA segments in two seasonal influenza A virus subtypes. They first estimated phylogenetic relationships between vRNA segments using Robinson-Foulds distance and identified the possibility of parallel evolution of RNA-RNA interactions driving the genomic assembly. This is indeed an interesting mechanism in addition to the traditional role for proteins for the same. Subsequently, they used molecular biology to validate such RNA-RNA driven interaction by demonstrating co-localization of vRNA segments in infected cells. They also showed that the parallel evolution between vRNA segments might vary across subtypes and virus lineages isolated from distinct host origins. Overall, I find this to be excellent work with major implications for genome evolution of infectious viruses; emergence of new strains with altered genome combination.
Comments:
I am wondering if leaving out sequences (not resolving well) in the phylogenic analysis interferes with the true picture of the proposed associations. What if they reflect the evolutionary intermediates, with important implications for the pathogen evolution which is lost in the analyses?
Lines 50-51: Can you please elaborate? I think this might be useful for the reader to better understand the context. Also, a brief description on functional association between different known fragments might instigate curiosity among the readers from the very beginning. At present, it largely caters to people already familiar with the biology of influenza virus.
Lines 95-96 Were these strains all swine-origin? More details on these lineages will be useful for the readers.
Lines 128-132: I think it will be nice to talk about these hypotheses well in advance, may be in the Introduction, with more functional details of viral segments.
Lines 134-136: Please rephrase this sentence to make it more direct and explain the why. E.g. "... parallel evolution between PB1 and HA is likely to be weaker than that of PB1 and PA" .
Lines 222-223: Please include a set of hypotheses to explain you results? Please add a perspective in the discussion on how this contribute might to the pandemic potential of H1N!?.
Lines 287-288: I am wondering how likely is this to be true for H1N1.
-
Evaluation Summary:
The manuscript reports phylogenetic and molecular evidence of novel RNA-RNA interactions driving the genomic coevolution of Influenza virus subtypes, in addition to protein interactions. With a few minor changes, this study could reveal how the likelihood of certain genetic combinations might lead to new viral variants emerging with the possibility of new antigenic properties and implications in disease spread.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #2 agreed to share their name with the authors.)
-
