REPOP: bacterial population quantification from plate counts
Curation statements for this article:-
Curated by eLife

eLife Assessment
This important study introduces a Bayesian method to determine bacterial counts that accounts for the experimental noise inherent to dilution and plating methods, and distinguishes it from biological uncertainty. The evidence supporting the conclusions is convincing, combining simulated data and experimental data. The method will be of interest to microbial ecologists, and potentially to the broader community interested in inference from biological data, even more so if the domain of application and the limitations are further clarified.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Abstract
Bacterial counts from native environments, such as soil or the animal gut, often show substantial variability across replicate samples. This heterogeneity is typically attributed to genetic or environmental factors. A common approach to estimating bacterial populations involves successive dilution and plating, followed by multiplying colony counts by dilution factors. This method, however, overestimates the heterogeneity in bacterial population because it conflates the inherent uncertainty in drawing a subsample from the total population with the uncertainty in the sample arising from biological origins. In other words, this approach may obscure features that may otherwise be present in the data hinting at the presence of genuine subpopulations. For example, in plate counting applied to C. elegans gut microbiota, observed multimodality is often interpreted as large host-to-host variance, while the randomness introduced by measurement is frequently ignored. To explicitly account for the uncertainty introduced by dilution and plating randomness, we introduce REPOP, a PyTorch-based library to REconstruct POpulations from Plates within a Bayesian framework. Beyond simple cases, REPOP addresses more complex scenarios, including multimodal populations and correcting the mathematically subtle, but experimentally relevant, bias introduced by excluding plates deemed too crowded to distinguish individual colonies. We demonstrate REPOP’s ability to resolve distinct population peaks otherwise obscured by standard multiplication methods. Applications to both simulated and experimental datasets, including bacterial samples of different concentrations and ones from the gut microbiota of C. elegans, show that REPOP accurately recovers the underlying multimodality by properly accounting for error propagation, where naive multiplication fails. REPOP is available on GitHub: https://github.com/PessoaP/REPOP.
Article activity feed
-

-
-

Author response:
Reviewer #1:
The only minor weakness that I found is the assumption of independence of bacterial species, which is expressed as the well-stirred approximation. One could imagine that bacterial species might cooperate, leading to non-uniform distributions that are real. How to distinguish such situations? I believe that this method can be extended to determine if this is the case or not before the application. For example, if the bacteria species are independent of each other and one can use the binomial distributions, then the Fano factor would be proportional to the overall relative fraction of bacterial species. Maybe a simple test can be added to test it before the application of REPOP. However, I believe that this is a minor issue.
This is an interesting point raised by the reviewer.
First, we need to clarify an …
Author response:
Reviewer #1:
The only minor weakness that I found is the assumption of independence of bacterial species, which is expressed as the well-stirred approximation. One could imagine that bacterial species might cooperate, leading to non-uniform distributions that are real. How to distinguish such situations? I believe that this method can be extended to determine if this is the case or not before the application. For example, if the bacteria species are independent of each other and one can use the binomial distributions, then the Fano factor would be proportional to the overall relative fraction of bacterial species. Maybe a simple test can be added to test it before the application of REPOP. However, I believe that this is a minor issue.
This is an interesting point raised by the reviewer.
First, we need to clarify an important point–we do not make a well-stirred assumption. Samples can be drawn and plated from any region of space however small and that region’s population can be quantified using our method. The stirring only occurs after we collect a sample in order to dilute the contents and pour the solution homogeneously over the plate.
As such, learning multiple independent species is possible and not impacted by the dilution (“wellstirred” assumption). In the revised manuscript we will make it clear that this assumption concerns the dilution process. Any correlation between species arises in the initial sample and should be retained in the plating. Once given the sample, the dilution itself produces independent binomial draws from that point in space from which cultures were harvested. REPOP is designed to recover the true underlying heterogeneity in species abundance (even from limited data) by leveraging a Bayesian framework that remains valid regardless of whether species are independent or correlated.
If one applies the method for multiple species as is, REPOP can recover the marginal distribution of each species in each plate if they are selectively cultured or many species at once if the colonies are sufficiently distinct. To demonstrate this, we will add a synthetic example with two species whose populations in a sample are correlated to the manuscript.
However, in order to learn the joint distribution and capture correlations between species within samples, the method would need to be extended. At present, in Eq. 5 we sum the likelihood over all values of n, using a data-driven cutoff (twice the na¨ıvely estimated count times the dilution factor). Extending this to multiple species adding up to (n1,n2), while retain the generality of the method, would require quadratically scaling memory with this cutoff in the population number. For this reason while we will comment on this in the next version of the manuscript, it will not be implemented as part of REPOP.
Reviewer #2:
A more thorough discussion of when and by how much estimated microbial population abundance distributions differ from the ground truth would be helpful in determining the best practices for applying this method. Not only would this allow researchers to understand the sampling effort necessary to achieve the results presented here, but it would also contextualize the experimental results presented in the paper. Particularly, there is a disconnect between the discussion of the large sample sizes necessary to achieve accurate multimodal distribution estimates and the small sample sizes used in both experiments.
That is a great suggestion from the reviewer. To address it, we will expand Appendix B, which currently presents the relative error between the means for the experimental results in Fig. 3, to also include a comparable evaluation for the synthetic data example in Fig. 2.
Specifically, for each example, we will report (1) the relative error in the estimated means (as already done for Fig. 3), and (2) the Kullback-Leibler (KL) divergence between the reconstructed and ground truth distributions. These metrics will be shown as a function of the size of the dataset, enabling a direct assessment of how the sampling effort affects the precision of the inference.
That said, we highlight that by explicitly modeling the dilution process within a Bayesian framework, REPOP extracts the mathematically optimal amount of information from each individual sample no matter the sample size. Our strategy therefore leads to better inference with fewer measurements, which is particularly important in applications such as plate counting, where data acquisition is laborintensive.
Reviewer #3:
While the study is promising, there are a few areas where the paper could be strengthened to increase its impact and usability. First, the extent to which dilution and plating introduce noise is not fully explored. Could this noise significantly affect experimental conclusions? And under what conditions does it matter most? Does it depend on experimental design or specific parameter values? Clarifying this would help readers appreciate when and why REPOP should be used.
We agree with the reviewer that this is an important point, and we will expand Appendix B to include a quantitative analysis using simulated data (Fig. 2), reporting both relative error and KL divergence as a function of dataset size. This complements our response to Reviewer #2 clarifying when REPOP offers the greatest benefit.
In addition, we will expand the discussion on how modeling dilution noise becomes essential when learning population dynamics. In particular, we will emphasize the role of Model 3, especially relevant when working with multiple plates and approaching the asymptotic regime—an aspect that was alluded to in Fig. 3 but not fully explored.
Second, more practical details about the tool itself would be very helpful. Simply stating that it is available on GitHub may not be enough. Readers will want to know what programming language it uses, what the input data should look like, and ideally, see a step-by-step diagram of the workflow. Packaging the tool as an easy-to-use resource, perhaps even submitting it to CRAN or including example scripts, would go a long way, especially since microbiologists tend to favor user-friendly, recipe-like solutions.
We will update the introduction to reinforce that REPOP is written in Python(PyTorch), installable via pip, and designed for ease of use. We are also expanding the tutorials to include clearer guidance on data formatting and common workflows. Author response image 1 will be added in the revised manuscript to better illustrate the full application process.
Author response image 1.
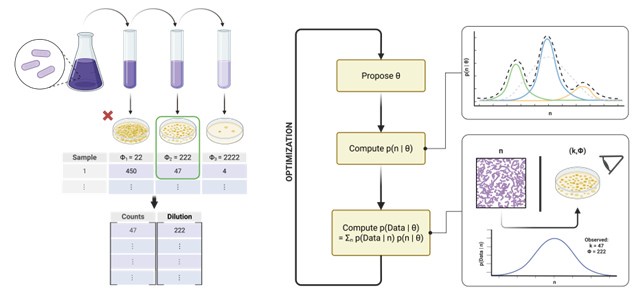
Third, it would be great to see the method tested on existing datasets, such as those from Nic Vega and Jeff Gore (2017), which explore how colonization frequency impacts abundance fluctuation distributions. Even if the general conclusions remain unchanged, showing that REPOP can better match observed patterns would strengthen the paper’s real-world relevance.
That is a great suggestion from the reviewer. We will demonstrate the application of REPOP to datasets such as that of Vega and Gore (Ref. 27 in the manuscript), as well as other publicly available datasets, in the revised version.
Lastly, it would be helpful for the authors to briefly discuss the limitations of their method, as no approach is without its constraints. Acknowledging these would provide a more balanced and transparent perspective.
We agree with the reviewer on that. A new subsection will explicitly address the assumptions of our method, and therefore its limitations, including assumptions about species classification, computational cost of joint inference, and dependence on accurate dilution modeling. This discussion will synthesize points raised throughout our response to all reviewers.
-
eLife Assessment
This important study introduces a Bayesian method to determine bacterial counts that accounts for the experimental noise inherent to dilution and plating methods, and distinguishes it from biological uncertainty. The evidence supporting the conclusions is convincing, combining simulated data and experimental data. The method will be of interest to microbial ecologists, and potentially to the broader community interested in inference from biological data, even more so if the domain of application and the limitations are further clarified.
-
Reviewer #1 (Public review):
Summary:
The authors developed a novel theoretical/computational procedure to count bacterial populations without introducing artificial randomness effects due to dilution. Surprisingly, this very important aspect of studies of bacterial systems has been overlooked. The proposed method provides a simple and transparent approach to eliminate the randomness of bacterial accounting procedures, allowing now to fully concentrate on the intrinsic effects of the studied systems.
Strengths:
A very simple and clear procedure is introduced and explained in full detail. This elegant approach finds an excellent compromise between mathematical rigor and computational efficiency, which is important for practical applications. The provided examples are convincing beyond a doubt, clearly indicating the potential strong …
Reviewer #1 (Public review):
Summary:
The authors developed a novel theoretical/computational procedure to count bacterial populations without introducing artificial randomness effects due to dilution. Surprisingly, this very important aspect of studies of bacterial systems has been overlooked. The proposed method provides a simple and transparent approach to eliminate the randomness of bacterial accounting procedures, allowing now to fully concentrate on the intrinsic effects of the studied systems.
Strengths:
A very simple and clear procedure is introduced and explained in full detail. This elegant approach finds an excellent compromise between mathematical rigor and computational efficiency, which is important for practical applications. The provided examples are convincing beyond a doubt, clearly indicating the potential strong impact of the proposed framework. Various complications and possible issues are also discussed and analyzed. This seems to be a very powerful novel method that should significantly advance the analysis of complex biological systems.
Weaknesses:
The only minor weakness that I found is the assumption of independence of bacterial species, which is expressed as the well-stirred approximation. One could imagine that bacterial species might cooperate, leading to non-uniform distributions that are real. How to distinguish such situations?
I believe that this method can be extended to determine if this is the case or not before the application. For example, if the bacteria species are independent of each other and one can use the binomial distributions, then the Fano factor would be proportional to the overall relative fraction of bacterial species. Maybe a simple test can be added to test it before the application of REPOP. However, I believe that this is a minor issue.
-
Reviewer #2 (Public review):
Summary:
Microbial population abundances are regularly estimated by multiplying plate counts by dilution factors, with inferences made about sample heterogeneity without taking into account heterogeneity generated through dilution and plating methods. The authors have developed REPOP, a method for disentangling methodological stochasticity from ecological heterogeneity using a Bayesian framework. They present three models: a unimodal distribution, a multimodal distribution, and a multimodal distribution that incorporates a colony count cutoff. They use a combination of simulated and experimental data to show the effectiveness of the REPOP method in resolving true microbial population distributions.
Strengths:
Overall, this paper addresses a significant issue in microbial ecology and reliably demonstrates …
Reviewer #2 (Public review):
Summary:
Microbial population abundances are regularly estimated by multiplying plate counts by dilution factors, with inferences made about sample heterogeneity without taking into account heterogeneity generated through dilution and plating methods. The authors have developed REPOP, a method for disentangling methodological stochasticity from ecological heterogeneity using a Bayesian framework. They present three models: a unimodal distribution, a multimodal distribution, and a multimodal distribution that incorporates a colony count cutoff. They use a combination of simulated and experimental data to show the effectiveness of the REPOP method in resolving true microbial population distributions.
Strengths:
Overall, this paper addresses a significant issue in microbial ecology and reliably demonstrates that the REPOP method improves upon current methods of estimating microbial population heterogeneity, particularly with simulation data. The three models presented build upon each other and are discussed in a way that is fairly accessible to a broad audience. The authors also show that leveraging the information provided by non-countable plates is important. Additionally, the authors address the potential for extending this method to other sources of methodological stochasticity that may occur in microbial plating. However, it does seem that they could extend this further by discussing ways that this method could be applied to non-microbial systems, allowing this work to appeal to a broader audience.
Weaknesses:
A more thorough discussion of when and by how much estimated microbial population abundance distributions differ from the ground truth would be helpful in determining the best practices for applying this method. Not only would this allow researchers to understand the sampling effort necessary to achieve the results presented here, but it would also contextualize the experimental results presented in the paper. Particularly, there is a disconnect between the discussion of the large sample sizes necessary to achieve accurate multimodal distribution estimates and the small sample sizes used in both experiments.
-
Reviewer #3 (Public review):
Summary:
In microbiology, accurately characterizing microbial populations and communities is essential. One widely used approach is to measure the absolute or relative abundance of microbial species. Recent research in microbial ecology, for instance, has shown that even genetically identical hosts exposed to the same microbial pool can develop very different gut microbiota, largely due to random colonization events. This study builds on that idea but adds a valuable layer: it suggests that some of the observed variability might actually result from experimental noise, specifically the randomness introduced by dilution and plate counting techniques. To address this, the authors introduce REPOP, a new tool designed to improve the quantification of microbial populations by explicitly accounting for the …
Reviewer #3 (Public review):
Summary:
In microbiology, accurately characterizing microbial populations and communities is essential. One widely used approach is to measure the absolute or relative abundance of microbial species. Recent research in microbial ecology, for instance, has shown that even genetically identical hosts exposed to the same microbial pool can develop very different gut microbiota, largely due to random colonization events. This study builds on that idea but adds a valuable layer: it suggests that some of the observed variability might actually result from experimental noise, specifically the randomness introduced by dilution and plate counting techniques. To address this, the authors introduce REPOP, a new tool designed to improve the quantification of microbial populations by explicitly accounting for the inherent stochasticity in these methods. They test REPOP using both simulated and experimental datasets, showing how it can help recover meaningful trends.
Strengths:
Overall, this paper is a good contribution to the field. The motivation is clear: improving our ability to quantify microbial populations is crucial for many research areas. The authors make a strong case that ignoring experimental noise is no longer acceptable, and they offer a well-argued solution. The manuscript is well-written and easy to follow, and the logic behind REPOP is convincingly laid out. The use of simulated data is especially valuable, as it allows the authors to test whether the method can recover known inputs, an important validation step. Even with experimental data, where true values are unknown, the method seems to behave in a reasonable and expected way, which is reassuring. All in all, this is an important step forward in how we quantify microbial populations.
Weaknesses:
While the study is promising, there are a few areas where the paper could be strengthened to increase its impact and usability. First, the extent to which dilution and plating introduce noise is not fully explored. Could this noise significantly affect experimental conclusions? And under what conditions does it matter most? Does it depend on experimental design or specific parameter values? Clarifying this would help readers appreciate when and why REPOP should be used. Second, more practical details about the tool itself would be very helpful. Simply stating that it is available on GitHub may not be enough. Readers will want to know what programming language it uses, what the input data should look like, and ideally, see a step-by-step diagram of the workflow. Packaging the tool as an easy-to-use resource, perhaps even submitting it to CRAN or including example scripts, would go a long way, especially since microbiologists tend to favor user-friendly, recipe-like solutions. Third, it would be great to see the method tested on existing datasets, such as those from Nic Vega and Jeff Gore (2017), which explore how colonization frequency impacts abundance fluctuation distributions. Even if the general conclusions remain unchanged, showing that REPOP can better match observed patterns would strengthen the paper's real-world relevance. Lastly, it would be helpful for the authors to briefly discuss the limitations of their method, as no approach is without its constraints. Acknowledging these would provide a more balanced and transparent perspective.
-
