A Novel Rapid Host Cell Entry Pathway Determines Intracellular Fate of Staphylococcus aureus
Curation statements for this article:-
Curated by eLife

eLife Assessment
This valuable study proposes a novel rapid-entry mechanism for Staphylococcus aureus, involving the rapid release of calcium from lysosomes. The paper's strength lies in its very interesting hypothesis. The methods used are solid and adequately support the conclusions.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Staphylococcus aureus is an opportunistic pathogen causing severe diseases. Recently, S. aureus was recognized as intracellular pathogen, whereby the intracellular niche promotes immune evasion and antibiotic resistance. Interaction of S. aureus with versatile host cell receptors was described previously, suggesting that internalization of the pathogen can occur via several pathways. It remains elusive whether the pathway of internalization can affect the intracellular fate of the bacteria. Here, we identified a mechanism governing cellular uptake of S. aureus which relies on lysosomal Ca2+, lysosomal exocytosis and occurs concurrently to other well-known entry pathways within the same host cell population. This internalization pathway is rapid and active within only few minutes after bacterial contact with host cells. Compared to slow bacterial internalization, the rapid pathway demonstrates altered phagosomal maturation as well as translocation of the pathogen to the host cytosol and ultimately results in different rates of intracellular bacterial replication and host cell death. We show that these alternative infection outcomes are caused by the mode of bacterial uptake.
Article activity feed
-

-
-

eLife Assessment
This valuable study proposes a novel rapid-entry mechanism for Staphylococcus aureus, involving the rapid release of calcium from lysosomes. The paper's strength lies in its very interesting hypothesis. The methods used are solid and adequately support the conclusions.
-
Reviewer #2 (Public review):
[Editors' note: This version was assessed by the editors. The authors have addressed a point raised by Reviewer #2, who thought the authors compared cells grown in low-serum and high serum conditions. This has been clarified in the latest version.]
In the manuscript Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry. Overall, this is manuscript argues for an important mechanism of a 'rapid' cellular entry pathway of S.aureus that is dependent on lysosomal exocytosis and acid sphingomyelinase and links the intracellular fate of bacterium including phagosomal dynamics, cytosolic replication and host cell death to …
Reviewer #2 (Public review):
[Editors' note: This version was assessed by the editors. The authors have addressed a point raised by Reviewer #2, who thought the authors compared cells grown in low-serum and high serum conditions. This has been clarified in the latest version.]
In the manuscript Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry. Overall, this is manuscript argues for an important mechanism of a 'rapid' cellular entry pathway of S.aureus that is dependent on lysosomal exocytosis and acid sphingomyelinase and links the intracellular fate of bacterium including phagosomal dynamics, cytosolic replication and host cell death to different modes of uptake.
A key strength is the nature of the idea proposed, while continued reliance on inhibitor treatment combined with lack of phenotype / conditional phenotype for genetic knock out is a major weakness.
In the previous version, the authors perform experiments with ASM KO cells to provide genetic evidence of the role for ASM in S. aureus entry through lysosomal modulation.
-
Author response:
The following is the authors’ response to the previous reviews.
Public Reviews:
Reviewer #2 (Public review):
In the manuscript Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry. Overall, this is manuscript argues for an important mechanism of a 'rapid' cellular entry pathway of S.aureus that is dependent on lysosomal exocytosis and acid sphingomyelinase and links the intracellular fate of bacterium including phagosomal dynamics, cytosolic replication and host cell death to different modes of uptake.
Key strength is the nature of the idea proposed, while continued reliance on inhibitor treatment combined with lack of phenotype / …
Author response:
The following is the authors’ response to the previous reviews.
Public Reviews:
Reviewer #2 (Public review):
In the manuscript Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry. Overall, this is manuscript argues for an important mechanism of a 'rapid' cellular entry pathway of S.aureus that is dependent on lysosomal exocytosis and acid sphingomyelinase and links the intracellular fate of bacterium including phagosomal dynamics, cytosolic replication and host cell death to different modes of uptake.
Key strength is the nature of the idea proposed, while continued reliance on inhibitor treatment combined with lack of phenotype / conditional phenotype for genetic knock out is a major weakness.
In the revised version, the authors perform experiments with ASM KO cells to provide genetic evidence of the role for ASM in S. aureus entry through lysosomal modulation. The key additional experiment is the phenotype of reduced bacterial uptake in low serum, but not in high serum conditions. The authors suggest this could be due to the SM from serum itself affecting the entry. While this explanation is plausible, prolonged exposure of cells to low serum is well documented to alter several cellular functions, particularly in the context of this manuscript, lysosomal positioning, exocytosis and Ca2+ signaling. A better control here could be WT cells grown in low serum.
As the reviewer suggested, we did culture both, WT control cells as well as ASM knock-outs, under low serum conditions before conducting the invasion assays. Hence, the detected effects on S. aureus invasion must be caused by lack of functional ASM in the mutant.
We apologize that this did not become evident from the manuscript’s text. We thus included a change in line 259 which now reads:
”To test whether FBS confounded our invasion experiments, we cultivated WT as well as ASM K.O. cells in medium with reduced FBS concentration (1%) and determined the S. aureus invasion efficiency (Figure 2I).”
If SM in serum can interfere, why do they see such pronounced phenotype on bacterial entry in WT cells upon chemical inhibition?
We explain the differences between inhibitor-treated WT cells and ASM K.O.s by the severe accumulation of SM upon genetic ablation of ASM. We demonstrated this by HPLC-MS/MS measurements in Figure 2L. If cells were cultured in 10% FBS, an ASM K.O. resulted in approx. 4-times higher levels of cellular SM C18:0 when compared to WT cells, while amitriptyline treatment of WT cells had no effect, and ARC39 treatment increased SM C18:0 levels only by 2-fold. This likely results from different durations of SM accumulation in the cell pools which is caused either by complete absence of ASM (in case of the ASM K.O.) or only in the hour-range upon treatment with the inhibitors.
Under low serum conditions, the severe SM C18:0 accumulation in the ASM K.O. was found decreased (from 4-fold to 2-fold when compared to WT cells; Figure 2M). Here, the WT cells used as reference also were cultured in the same manner as the ASM K.O. A similar pattern was observed for other SM species (Supp. Figure 3). This correlates with the S. aureus invasion phenotype in ASM K.O.: under high serum conditions (and resulting in severe SM accumulation) we did not detect an invasion defect, while under low serum conditions (resulting in only moderate SM accumulation) S. aureus invasion was reduced in the knock-outs when compared to WT cells cultured in the same conditions, respectively.
While the authors argue a role for undetectable nano-scale Cer platforms on the cell surface caused by ASM activity, results do not rule out a SM independent role in the cellular uptake phenotype of ASM inhibitors.
Since the comments starting with the line above are identical to the previous comments by the reviewer, we assume that these points of criticism still resound with the Reviewer, although we had agreed previously with the reviewer that we do not show formation of ceramide-enriched platforms, we had changed the manuscript accordingly in the previous revision round already (see also our comment below).
The authors have attempted to address many of the points raised in the previous revision. While the new data presented provide partial evidence, the reliance on chemical inhibitors and lack of clear results directly documenting release of lysosomal Ca2+, or single bacterial tracking, or clear distinction between ASM dependent and independent processes dampen the enthusiasm.
We continue to share the reviewer’s desire to discriminate between ASM-dependent and ASMindependent processes, but the simultaneous occurrence of multiple pathways of bacterial uptake is currently the limiting factor and technological challenge in our laboratory, since these events happen rapidly. We do hope that we or others will be able to address these limitations in the future, for instance with the technologies suggested by the reviewer.
I acknowledge the author's argument of different ASM inhibitors showing similar phenotypes across different assays as pointing to a role for ASM, but the lack of phenotype in ASM KO cells is concerning. The author's argument that altered lipid composition in ASM KO cells could be overcoming the ASMmediated infection effects by other ASM-independent mechanisms is speculative, as they acknowledge, and moderates the importance of ASM-dependent pathway. The SM accumulation in ASM KO cells does not distinguish between localized alterations within the cells. If this pathway can be compensated, how central is it likely to be ?
We here want to elaborate again, since our revision experiments demonstrate the ASM-dependency of the rapid uptake under low serum conditions – see also above. We were convinced that the genetic evidence of an S. aureus invasion phenotype in ASM K.O.s under these conditions would eliminate the reviewer’s concern about the role of ASM during the bacterial invasion (see also above). Our lipidomics data of ASM K.O.s cultured in 1% and 10% FBS (Figure 2, M, Supp. Figure 3) and inhibitor-treated WT cells (Figure 2L, Supp. Figure 3) show a correlation between SM accumulation and the invasion phenotype observed by us.
We agree with the reviewer, however, that it remains elusive why changes in the sphingolipidome increase ASM-independent S. aureus internalization by host cells. One explanation is a dysfunction of the lipid raft-associated protein caveolin-1 upon strong SM accumulation, which was previously shown to appear in ASM-deficient cells (1, 2). A lack of caveolin-1 results in strongly increased host cell entry of S. aureus in certain cell types (3, 4). In other cell types, such as A549 cells, S. aureus invades in an αtoxin and caveolin-1 dependent fashion (5). It will be interesting to study, to what extent such processes as described by Goldmann and colleagues will depend on ASM. However, a characterization of the mechanism behind these observations requires further experimentation and is beyond the scope of the current manuscript.
As to the centrality of the pathway: we cannot and do not make any assumptions on the centrality of the pathway and its importance in vivo. As scientists we were intrigued by our finding of an ASM dependent uptake pathway for S. aureus – especially its speed. In different as of yet still unidentified host cell types or cell lines such a pathway may pose a major entry point for pathogens. Alternatively, we may have identified an ASM-dependent mode of receptor uptake, with which the bacteria “piggyback” into the cells.
The authors allude to lower phagosomal escape rate in ASM KO cells compared to inhibitor treatment, which appears to contradict the notion of uptake and intracellular trafficking phenotype being tightly linked. As they point out, these results might be hard to interpret.
We again want to add that we measured phagosomal escape of S. aureus in WT and ASM K.O. cells cultured in 1% FBS (low serum conditions) and compared it to escape rates obtained with host cells cultured in 10% FBS. Again, we infected cells for 10 or 30 min and determined the escape rates 3h p.i. However, the results are similar to escape rates determined with 10% FBS (see Author response image 1). This was addressed already during the manuscript’s first revision. We found that escape rates of S. aureus were significantly decreased in absence of ASM regardless of the FBS concentration in the medium.
Author response image 1.
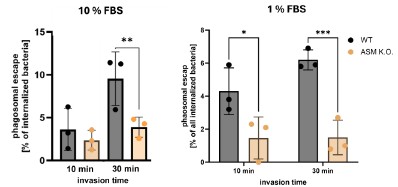
We therefore think that prolonged absence of ASM has additional side effects. For instance, certain endocytic pathways could be up- or down-regulated to adapt for the absence of ASM or could be affected by other changes in the lipidome (that can be minimized but not completely prevented by culturing cells in 1% FBS). This could, for instance, affect maturation of S. aureus-containing phagosomes and hence phagosomal escape.
As it is currently unclear in how far the prolonged absence of ASM activity affects cellular processes, we think other experiments investigating the role of ASM-dependent invasion for phagosomal escape are more reliable. Most importantly, bacteria that enter host cell early during infection (and thus, predominantly via the “rapid” ASM-dependent pathway) possess lower phagosomal escape rates than bacteria that entered host cells later during infection (Figure 5, D and E). This is confirmed by higher escapes rates upon blocking ASM-dependent invasion with Vacuolin-1 (Figure 4E) and three different ASM inhibitors (Figure 4C and D). We further demonstrate that sphingomyelin on the plasma membrane during invasion influences phagosomal escape, while sphingomyelin levels in the phagosomal membrane did not change phagosomal escape (Figure5 a and b). This is summarized in Figure 5F.
Could an inducible KD system recapitulate (some of) the phenotype of inhibitor treatment? If S. aureus does not escape phagosome in macrophages, could it provide a system to potentially decouple the uptake and intracellular trafficking effects by ASM (or its inhibitor treatment) ?
Knock-downs in our laboratory are based on the vector pLVTHM(6). Inducible knock-downs in the cells would require the introduction of an inducible Teton system, which the cells currently do not harbor.
However, it needs to be stated that for optimal gene knock-downs, the induction of this system has to be performed by doxycycline supplementation in the medium for 7 days thus leading to several days of growth of the cells, which will allow the cells to adapt their lipid metabolism thus reflecting a situation that we encounter for the K.O.s.
ASM-dependent uptake of S. aureus in macrophages has been demonstrated before (7). However, the course of infection in macrophages differs from non-professional phagocytes (8). E.g. in macrophages, S. aureus replicates within phagosomes, whereas in non-professional phagocytes replicates in the host cytosol. Absence of ASM therefore may influence the intracellular infection of macrophages with S. aureus in a distinct manner.
The role of ASM on cell surface remains unclear. The hypothesis proposed by the authors that the localized generation of Cer on the surface by released ASM leads to generation of Cer-enriched platforms could be plausible, but is not backed by data, technical challenges to visualize these platforms notwithstanding. These results do not rule out possible SM independent effects of ASM on the cell surface, if indeed the role of ASM is confirmed by controlled genetic depletion studies.
We agree with the reviewer that we do not show generation of ceramide-enriched platforms (see also above). We thus already had changed Figure 6F in the revised manuscript to make clear that it remains elusive whether ceramide-enriched platforms are formed. We also had added a sentence to the discussion (line 615) to emphasize that the existence of these microdomains is still debated in lipid research.
We think that the following observations support SM-dependent effects of ASM during S. aureus invasion:
(i) Reduced invasion upon removing SM from the plasma membrane (Figure 2N, Supp. Figure 2M)
(ii) Increased invasion in TPC1 and Syt7 K.O. (Figure 2, P) in presence of exogenously added SMase.
However, we agree with the reviewer that we do not directly demonstrate ASM-mediated SM cleavage during S. aureus invasion. Hence, we had added a sentence to the discussion that mentions a possible SM-independent role of ASM for invasion (line 556) that reads:
“Since it remains elusive to which extent ASM processes SM on the plasma membrane during S. aureus invasion, one may speculate that ASM could also have functions other than SM metabolization during host cell entry of the pathogen. However, we did not detect a direct interaction between S. aureus and ASM in an S. aureus-host interactome screen (9).”
The reviewer acknowledges technical challenges in directly visualizing lysosomal Ca2+ using the methods outlined. Genetically encoded lysosomal Ca2+ sensor such as Gcamp3-ML1 might provide better ways to directly visualize this during inhibitor treatment, or S. aureus infection.
We again thank the reviewer for this suggestion. We already had included the following section in our discussion (then: line 593): “Since fluorescent calcium reporters allow to monitor this process microscopically, future experiments may visualize this process in more detail and contribute to our understanding of the underlying signaling. mechanisms.”
References for the purpose of this response letter:
(1) Rappaport, J., C. Garnacho, and S. Muro, Clathrin-mediated endocytosis is impaired in type AB Niemann-Pick disease model cells and can be restored by ICAM-1-mediated enzyme replacement. Mol Pharm, 2014. 11(8): p. 2887-95.
(2) Rappaport, J., et al., Altered Clathrin-Independent Endocytosis in Type A Niemann-Pick Disease Cells and Rescue by ICAM-1-Targeted Enzyme Delivery. Mol Pharm, 2015. 12(5): p. 1366-76.
(3) Hoffmann, C., et al., Caveolin limits membrane microdomain mobility and integrin-mediated uptake of fibronectin-binding pathogens. J Cell Sci, 2010. 123(Pt 24): p. 4280-91.
(4) Tricou, L.-P., et al., Staphylococcus aureus can use an alternative pathway to be internalized by osteoblasts in absence of β1 integrins. Scientific Reports, 2024. 14(1): p. 28643.
(5) Goldmann, O., et al., Alpha-hemolysin promotes internalization of Staphylococcus aureus into human lung epithelial cells via caveolin-1- and cholesterol-rich lipid rafts. Cell Mol Life Sci, 2024. 81(1): p. 435.
(6) Wiznerowicz, M. and D. Trono, Conditional suppression of cellular genes: lentivirus vectormediated drug-inducible RNA interference. J Virol, 2003. 77(16): p. 8957-61.
(7) Li, C., et al., Regulation of Staphylococcus aureus Infection of Macrophages by CD44, Reactive Oxygen Species, and Acid Sphingomyelinase. Antioxid Redox Signal, 2018. 28(10): p. 916-934.
(8) Moldovan, A. and M.J. Fraunholz, In or out: Phagosomal escape of Staphylococcus aureus. Cell Microbiol, 2019. 21(3): p. e12997.
(9) Rühling, M., et al., Identification of the Staphylococcus aureus endothelial cell surface interactome by proximity labeling. mBio, 2025. 0(0): p. e03654-24.
-
-
eLife Assessment
This valuable study proposes a novel rapid-entry mechanism for S. aureus, involving the rapid release of calcium from lysosomes. The paper's strength lies in its very interesting hypothesis. The methods used are solid and adequately support the conclusions.
-
Reviewer #2 (Public review):
In the manuscript Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry. Overall, this is manuscript argues for an important mechanism of a 'rapid' cellular entry pathway of S.aureus that is dependent on lysosomal exocytosis and acid sphingomyelinase and links the intracellular fate of bacterium including phagosomal dynamics, cytosolic replication and host cell death to different modes of uptake.
Key strength is the nature of the idea proposed, while continued reliance on inhibitor treatment combined with lack of phenotype / conditional phenotype for genetic knock out is a major weakness.
In the revised version, the …
Reviewer #2 (Public review):
In the manuscript Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry. Overall, this is manuscript argues for an important mechanism of a 'rapid' cellular entry pathway of S.aureus that is dependent on lysosomal exocytosis and acid sphingomyelinase and links the intracellular fate of bacterium including phagosomal dynamics, cytosolic replication and host cell death to different modes of uptake.
Key strength is the nature of the idea proposed, while continued reliance on inhibitor treatment combined with lack of phenotype / conditional phenotype for genetic knock out is a major weakness.
In the revised version, the authors perform experiments with ASM KO cells to provide genetic evidence of the role for ASM in S. aureus entry through lysosomal modulation. The key additional experiment is the phenotype of reduced bacterial uptake in low serum, but not in high serum conditions. The authors suggest this could be due to the SM from serum itself affecting the entry. While this explanation is plausible, prolonged exposure of cells to low serum is well documented to alter several cellular functions, particularly in the context of this manuscript, lysosomal positioning, exocytosis and Ca2+ signaling. A better control here could be WT cells grown in low serum. If SM in serum can interfere, why do they see such pronounced phenotype on bacterial entry in WT cells upon chemical inhibition?
While the authors argue a role for undetectable nano-scale Cer platforms on the cell surface caused by ASM activity, results do not rule out a SM independent role in the cellular uptake phenotype of ASM inhibitors.
The authors have attempted to address many of the points raised in the previous revision. While the new data presented provide partial evidence, the reliance on chemical inhibitors and lack of clear results directly documenting release of lysosomal Ca2+, or single bacterial tracking, or clear distinction between ASM dependent and independent processes dampen the enthusiasm.
I acknowledge the author's argument of different ASM inhibitors showing similar phenotypes across different assays as pointing to a role for ASM, but the lack of phenotype in ASM KO cells is concerning. The author's argument that altered lipid composition in ASM KO cells could be overcoming the ASM-mediated infection effects by other ASM-independent mechanisms is speculative, as they acknowledge, and moderates the importance of ASM-dependent pathway. The SM accumulation in ASM KO cells does not distinguish between localized alterations within the cells. If this pathway can be compensated, how central is it likely to be ?
The authors allude to lower phagosomal escape rate in ASM KO cells compared to inhibitor treatment, which appears to contradict the notion of uptake and intracellular trafficking phenotype being tightly linked. As they point out, these results might be hard to interpret. Could an inducible KD system recapitulate (some of) the phenotype of inhibitor treatment? If S. aureus does not escape phagosome in macrophages, could it provide a system to potentially decouple the uptake and intracellular trafficking effects by ASM (or its inhibitor treatment) ?
The role of ASM on cell surface remains unclear. The hypothesis proposed by the authors that the localized generation of Cer on the surface by released ASM leads to generation of Cer-enriched platforms could be plausible, but is not backed by data, technical challenges to visualize these platforms notwithstanding. These results do not rule out possible SM independent effects of ASM on the cell surface, if indeed the role of ASM is confirmed by controlled genetic depletion studies.
The reviewer acknowledges technical challenges in directly visualizing lysosomal Ca2+ using the methods outlined. Genetically encoded lysosomal Ca2+ sensor such as Gcamp3-ML1 might provide better ways to directly visualize this during inhibitor treatment, or S. aureus infection.
-
Author response:
The following is the authors’ response to the previous reviews
Public Reviews:
Reviewer #2 (Public review):
In the manuscript, Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry. Overall, this is manuscript argues for an important mechanism of a 'rapid' cellular entry pathway of S.aureus that is dependent on lysosomal exocytosis and acid sphingomyelinase and links the intracellular fate of bacterium including phagosomal dynamics, cytosolic replication and host cell death to different modes of uptake.
Key strength is the nature of the idea proposed, while continued reliance on inhibitor treatment combined with lack of phenotype for …
Author response:
The following is the authors’ response to the previous reviews
Public Reviews:
Reviewer #2 (Public review):
In the manuscript, Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry. Overall, this is manuscript argues for an important mechanism of a 'rapid' cellular entry pathway of S.aureus that is dependent on lysosomal exocytosis and acid sphingomyelinase and links the intracellular fate of bacterium including phagosomal dynamics, cytosolic replication and host cell death to different modes of uptake.
Key strength is the nature of the idea proposed, while continued reliance on inhibitor treatment combined with lack of phenotype for genetic knock out is a major weakness.
We agree with the reviewer that a S. aureus invasion phenotype in ASM K.O. cells would unequivocally demonstrate the importance of ASM for the process. In the revised manuscript, we report an invasion phenotype in ASM K.O. cells. The absence of an invasion phenotype in ASM K.O. cells in our original experiments was likely caused by SM accumulation in ASM-depleted cells originating from FBS (see Figure 2I, in the revised manuscript).
We thus cultured cells for up to three days in 2% FBS and then reduced the concentration to 1% FBS one day prior to experimentation. Under these conditions reduced S. aureus invasion in ASM K.O.s was observed when compared to wildtype cells.
This was not detected when we cultured the cells in medium containing the common concentration of 10% FBS. Our new data supports the results we acquired with three different ASM inhibitors.
The invasion defect in ASM K.O.s cultured in low FBS was more pronounced at 10 min p.i. when compared to the 30 minute time point (Figure 2K), further corroborating that the ASM-dependent invasion pathway is relevant early in infection. This is consistent with the invasion dynamics we observed upon interference with lysosomal Ca2+ signaling [TPC1 K.O. (Figure 1C), BAPTA-AM (Figure 3D)], lysosomal exocytosis [Syt7 K.O. (Figure 2F), Ionomycin (Figure 3D)] and ASM activity by inhibitor treatment (Figure 3D).
Originally, we had hypothesized that changes in the sphingolipidome induced by absence of ASM may have caused the lack of an S. aureus invasion phenotype. We thus compared the sphingolipidome of ASM K.O.s cultured in 1% and 10% FBS. Indeed, SM accumulation was less severe when we cultured the cells in 1% FBS (Figure 2M and Supp. Figure 3). Hence, we think that strong SM accumulations in ASM K.O. cells cultured in 10% FBS may facilitate ASM-independent invasion mechanisms and thus, the absence of ASM-dependent invasion could not be detected by analyzing the number of invaded bacteria. This is supported by experiments, where we treated ASM K.O.s with the ASM inhibitor ARC39, which only slightly affected S. aureus invasion, whereas we detected a strong reduction of internalized bacteria by ARC39 treatment of WT cells (Figure 2 J). We think that this experiment and the reduced invasion in ASM K.O.s rule out an ASM/SM-independent effect of the inhibitors.
- While the authors argue a role for undetectable nano-scale Cer platforms on the cell surface caused by ASM activity, results do not rule out a SM independent role in the cellular uptake phenotype of ASM inhibitors.
We agree with reviewer that we do not show formation of ceramide-enriched platforms, and we thus changed the manuscript accordingly (see below).
- The authors have attempted to address many of the points raised in the previous revision. While the new data presented provide partial evidence, the reliance on chemical inhibitors and lack of clear results directly documenting release of lysosomal Ca2+, or single bacterial tracking, or clear distinction between ASM dependent and independent processes dampen the enthusiasm.
We shared the reviewer’s desire to discriminate between ASM-dependent and ASM-independent processes, but we are limited by cell biology and the simultaneous occurrence of processes - here the uptake of bacteria by multiple pathways.
However, we were able to address ASM-dependency of our rapid uptake mechanism by observing a genetic phenotype in SMPD1 knockout-cells.
We here do not make any assumptions on the centrality of the pathway and its importance in vivo. As scientists we were interested in the fact that such an ASM dependent pathway existed. In different as of yet still unidentified cell lines such a pathway may pose the main entry point for bacteria. Or maybe it represent an ASM-dependent mode of receptor uptake which we have identified with the bacteria piggy-backing into the cells.
- I acknowledge the author's argument of different ASM inhibitors showing similar phenotypes across different assays as pointing to a role for ASM, but the lack of phenotype in ASM KO cells is concerning. The author's argument that altered lipid composition in ASM KO cells could be overcoming the ASM-mediated infection effects by other ASM-independent mechanisms is speculative, as they acknowledge, and moderates the importance of ASM-dependent pathway. The SM accumulation in ASM KO cells does not distinguish between localized alterations within the cells. If this pathway can be compensated, how central is it likely to be?
We are convinced that our new genetic evidence of an S. aureus invasion phenotype in ASM K.O.s will eliminate the reviewer’s concerns about the role of ASM during the bacterial invasion.
The new lipidomics data of ASM K.O.s cultured in 1% and 10% FBS (Figure 2, M, Supp. Figure 3) and inhibitor-treated WT cells (Figure 2L, Supp. Figure 3) show a correlation between SM accumulation and the invasion phenotype.
We agree with the reviewer, however, that the reason why changes in sphingolipidome increase ASM-independent S. aureus internalization by host cells remains elusive. One possible explanation is a dysfunction of the lipid raft-associated protein caveolin-1 upon strong SM accumulation, which was previously shown to appear in ASM-deficient cells (1, 2). A lack of caveolin-1 results in strongly increased host cell entry of S. aureus (3, 4). Characterization of the mechanism behind these observations requires further experimentation and is beyond the scope of the current manuscript.
Host cells possess mechanisms to prevent infections, while pathogens developed strategies to circumvent these defense processes. In the present scenario, a physiological membrane composition of the host cell represents such a pathogen defense mechanism (as shown e.g. for caveolin-1 that restricts invasion of S. aureus in healthy cells). If a defense mechanism is disabled (as we speculate it is the case upon strong SM accumulation in ASM K.O.s cultured in 10%FBS), infection is facilitated. In healthy WT cells, these mechanisms (e.g. caveolin-1) are functional and, hence, we would not expect a “compensation” of ASM-dependent invasion. We here analyze invasion events that cannot be prevented by host defense mechanisms as they occur in untreated WT cells and are absent upon interfering with the ASM-dependent invasion pathway (by inhibitors and genetic K.O.). Thus, we think the ASM-dependent pathway, which mediates 50-70% of bacteria internalized by healthy WT cells 10 min p.i., is central for the infection.
- The authors allude to lower phagosomal escape rate in ASM KO cells compared to inhibitor treatment, which appears to contradict the notion of uptake and intracellular trafficking phenotype being tightly linked. As they point out, these results might be hard to interpret.
We measured phagosomal escape of S. aureus JE2 in ASM K.O. cells cultured in 1% FBS. Again, we infected cells for 10 or 30 min and determined the escape rates 3h p.i. However, the results are similar to escape rates determined with 10% FBS (Author response image 1).
Escape rates of S. aureus were significantly decreased in absence of ASM regardless of the FBS concentration in the medium. We therefore think that prolonged absence of ASM has other side effects. For instance, certain endocytic pathways could be up- or down-regulated to adapt for the absence of ASM or could be affected by other changes in the lipidome (that can be minimized but not completely prevented by culturing cells in 1% FBS). This could, for instance, affect maturation of S. aureus-containing phagosomes and hence phagosomal escape.
Author response image 1.
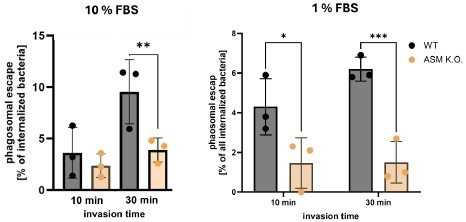
As it is unclear how prolonged absence of ASM can affect cellular processes, we think other experiments investigating the role of ASM-dependent invasion for phagosomal escape are more reliable. Most importantly, bacteria that enter host cell early during infection (and thus, predominantly via the “rapid” ASM-dependent pathway) possess lower phagosomal escape rates than bacteria that entered host cells later during infection (Figure 5, D and E). This is confirmed by higher escapes rates upon blocking ASM-dependent invasion with Vacuolin-1 (Figure 4E) and three different ASM inhibitors (Figure 4C and D). We further demonstrate that sphingomyelin on the plasma membrane during invasion influences phagosomal escape, while sphingomyelin levels in the phagosomal membrane did not change phagosomal escape (Figure5 a and b). This is summarized in Figure 5F.
- Could an inducible KD system recapitulate (some of) the phenotype of inhibitor treatment ? If S. aureus does not escape phagosome in macrophages, could it provide a system to potentially decouple the uptake and intracellular trafficking effects by ASM (or its inhibitor treatment)?
Inducible knock-downs in our laboratory are based on the vector pLVTHM in cells co-expressing the repressor TetR fused to a KRAB domain. It needs to be stated that for optimal knock-downs the induction has to be performed by doxycycline supplementation in the medium for 7 days thus leading to several days of growth of the cells, which will allow the cells to adapt their lipid metabolism thus reflecting a situation that we encounter for the K.O.s.
ASM-dependent uptake of S. aureus in macrophages has been demonstrated before (5). However, the course of infection in macrophages differs from non-professional phagocytes (6). E.g. in macrophages, S. aureus replicates within phagosomes, whereas in non-professional phagocytes replicates in the host cytosol. Absence of ASM therefore may influence the intracellular infection of macrophages with S. aureus in a distinct manner.
- The role of ASM on cell surface remains unclear. The hypothesis proposed by the authors that the localized generation of Cer on the surface by released ASM leads to generation of Cer-enriched platforms could be plausible, but is not backed by data, technical challenges to visualize these platforms notwithstanding. These results do not rule out possible SM independent effects of ASM on the cell surface, if indeed the role of ASM is confirmed by controlled genetic depletion studies.
We agree with the reviewer that we do not show generation of ceramide-enriched platforms. We thus changed Figure 6F in the revised manuscript to make clear that it remains elusive whether ceramide-enriched platforms are formed. We also added a sentence to the discussion (line 615) to emphasize that the existence of these microdomains is still debated in lipid research.
We think that the following observations support SM-dependent effects of ASM during S. aureus invasion:
(i) reduced invasion upon removing SM from the plasma membrane (Figure 2N, Supp. Figure 2M)
(ii) increased invasion in TPC1 and Syt7 K.O. (Figure 2, P) in presence of exogenously added SMase.
However, we agree with the reviewer that we do not directly demonstrate ASM-mediated SM cleavage during S. aureus invasion. Hence, we added a sentence to the discussion that mentions a possible SM-independent role of ASM for invasion (line 556) that reads:
“Since it remains elusive to which extent ASM processes SM on the plasma membrane during S. aureus invasion, one may speculate that ASM could also have functions other than SM metabolization during host cell entry of the pathogen. However, we did not detect a direct interaction between S. aureus and ASM in an S. aureus-host interactome screen (7).”
- The reviewer acknowledges technical challenges in directly visualizing lysosomal Ca2+ using the methods outlined. Genetically encoded lysosomal Ca2+ sensor such as Gcamp3-ML1 might provide better ways to directly visualize this during inhibitor treatment, or S. aureus infection.
We thank the reviewer for this suggestion. We included the following section in our discussion (line 593):
“Since fluorescent calcium reporters allow to monitor this process microscopically (8, 9) ,future experiments may visualize this process in more detail and contribute to our understanding of the underlying signaling. mechanisms.”
References
(1) J. Rappaport, C. Garnacho, S. Muro, Clathrin-mediated endocytosis is impaired in type A-B Niemann-Pick disease model cells and can be restored by ICAM-1-mediated enzyme replacement. Mol Pharm 11, 2887-2895 (2014).
(2) J. Rappaport, R. L. Manthe, C. Garnacho, S. Muro, Altered Clathrin-Independent Endocytosis in Type A Niemann-Pick Disease Cells and Rescue by ICAM-1-Targeted Enzyme Delivery. Mol Pharm 12, 1366-1376 (2015).
(3) C. Hoffmann et al., Caveolin limits membrane microdomain mobility and integrin-mediated uptake of fibronectin-binding pathogens. J Cell Sci 123, 4280-4291 (2010).
(4) L.-P. Tricou et al., Staphylococcus aureus can use an alternative pathway to be internalized by osteoblasts in absence of β1 integrins. Scientific Reports 14, 28643 (2024).
(5) C. Li et al., Regulation of Staphylococcus aureus Infection of Macrophages by CD44, Reactive Oxygen Species, and Acid Sphingomyelinase. Antioxid Redox Signal 28, 916-934 (2018).
(6) A. Moldovan, M. J. Fraunholz, In or out: Phagosomal escape of Staphylococcus aureus. Cell Microbiol 21, e12997 (2019).
(7) M. Rühling, F. Schmelz, A. Kempf, K. Paprotka, J. Fraunholz Martin, Identification of the Staphylococcus aureus endothelial cell surface interactome by proximity labeling. mBio 0, e03654-03624 (2025).
(8) D. Shen et al., Lipid storage disorders block lysosomal trafficking by inhibiting a TRP channel and lysosomal calcium release. Nat Commun 3, 731 (2012).
(9) L. C. Davis, A. J. Morgan, A. Galione, NAADP-regulated two-pore channels drive phagocytosis through endo-lysosomal Ca(2+) nanodomains, calcineurin and dynamin. EMBO J 39, e104058 (2020).
-
-
eLife Assessment
This valuable work proposes a novel, rapid S. aureus entry mechanism via Ca²⁺-dependent lysosomal exocytosis and acid sphingomyelinase release, which influences bacterial sub-cellular fate. However, reliance on chemical inhibitors and the absence of a knockout phenotype weakens the overall impact, making the study incomplete.
-
Reviewer #2 (Public review):
In the manuscript, Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry. Overall, this is manuscript argues for an important mechanism of a 'rapid' cellular entry pathway of S.aureus that is dependent on lysosomal exocytosis and acid sphingomyelinase and links the intracellular fate of bacterium including phagosomal dynamics, cytosolic replication and host cell death to different modes of uptake.
Key strength is the nature of the idea proposed, while continued reliance on inhibitor treatment combined with lack of phenotype for genetic knock out is a major weakness. While the authors argue a role for undetectable …
Reviewer #2 (Public review):
In the manuscript, Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry. Overall, this is manuscript argues for an important mechanism of a 'rapid' cellular entry pathway of S.aureus that is dependent on lysosomal exocytosis and acid sphingomyelinase and links the intracellular fate of bacterium including phagosomal dynamics, cytosolic replication and host cell death to different modes of uptake.
Key strength is the nature of the idea proposed, while continued reliance on inhibitor treatment combined with lack of phenotype for genetic knock out is a major weakness. While the authors argue a role for undetectable nano-scale Cer platforms on the cell surface caused by ASM activity, results do not rule out a SM independent role in the cellular uptake phenotype of ASM inhibitors.
The authors have attempted to address many of the points raised in the previous revision. While the new data presented provide partial evidence, the reliance on chemical inhibitors and lack of clear results directly documenting release of lysosomal Ca2+, or single bacterial tracking, or clear distinction between ASM dependent and independent processes dampen the enthusiasm.
I acknowledge the author's argument of different ASM inhibitors showing similar phenotypes across different assays as pointing to a role for ASM, but the lack of phenotype in ASM KO cells is concerning. The author's argument that altered lipid composition in ASM KO cells could be overcoming the ASM-mediated infection effects by other ASM-independent mechanisms is speculative, as they acknowledge, and moderates the importance of ASM-dependent pathway. The SM accumulation in ASM KO cells does not distinguish between localized alterations within the cells. If this pathway can be compensated, how central is it likely to be ?
The authors allude to lower phagosomal escape rate in ASM KO cells compared to inhibitor treatment, which appears to contradict the notion of uptake and intracellular trafficking phenotype being tightly linked. As they point out, these results might be hard to interpret. Could an inducible KD system recapitulate (some of) the phenotype of inhibitor treatment ? If S. aureus does not escape phagosome in macrophages, could it provide a system to potentially decouple the uptake and intracellular trafficking effects by ASM (or its inhibitor treatment) ?
The role of ASM on cell surface remains unclear. The hypothesis proposed by the authors that the localized generation of Cer on the surface by released ASM leads to generation of Cer-enriched platforms could be plausible, but is not backed by data, technical challenges to visualize these platforms notwithstanding. These results do not rule out possible SM independent effects of ASM on the cell surface, if indeed the role of ASM is confirmed by controlled genetic depletion studies.
The reviewer acknowledges technical challenges in directly visualizing lysosomal Ca2+ using the methods outlined. Genetically encoded lysosomal Ca2+ sensor such as Gcamp3-ML1 might provide better ways to directly visualize this during inhibitor treatment, or S. aureus infection.
-
Author response:
The following is the authors’ response to the original reviews
Public Reviews:
Reviewer #1 (Public review):
Summary:
The manuscript by Rühling et al analyzes the mode of entry of S. aureus into mammalian cells in culture. The authors propose a novel mechanism of rapid entry that involves the release of calcium from lysosomes via NAADP-stimulated activation of TPC1, which in turn causes lysosomal exocytosis; exocytic release of lysosomal acid sphingomyelinase (ASM) is then envisaged to convert exofacial sphingomyelin to ceramide. These events not only induce the rapid entry of the bacteria into the host cells but are also described to alter the fate of the intracellular S. aureus, facilitating escape from the endocytic vacuole to the cytosol.
Strengths:
The proposed mechanism is novel and could have important biological …
Author response:
The following is the authors’ response to the original reviews
Public Reviews:
Reviewer #1 (Public review):
Summary:
The manuscript by Rühling et al analyzes the mode of entry of S. aureus into mammalian cells in culture. The authors propose a novel mechanism of rapid entry that involves the release of calcium from lysosomes via NAADP-stimulated activation of TPC1, which in turn causes lysosomal exocytosis; exocytic release of lysosomal acid sphingomyelinase (ASM) is then envisaged to convert exofacial sphingomyelin to ceramide. These events not only induce the rapid entry of the bacteria into the host cells but are also described to alter the fate of the intracellular S. aureus, facilitating escape from the endocytic vacuole to the cytosol.
Strengths:
The proposed mechanism is novel and could have important biological consequences.
Weaknesses:
Unfortunately, the evidence provided is unconvincing and insufficient to document the multiple, complex steps suggested. In fact, there appear to be numerous internal inconsistencies that detract from the validity of the conclusions, which were reached mostly based on the use of pharmacological agents of imperfect specificity.
We thank the reviewer for the detailed evaluation of our manuscript. We will address the criticism below.
We agree with the reviewer that many of the experiments presented in our study rely on the usage of inhibitors. However, we want to emphasize that the main conclusion (invasion pathway affects the intracellular fate/phagosomal escape) was demonstrated without the use of inhibitors or genetic ablation in two key experiments (Figure5 D/E). These experiments were in line with the results we obtained with inhibitors (amitriptyline [Figure 4D], ARC39, PCK310, [Figure 4C] and Vacuolin-1 [Figure4E]). Importantly, the hypothesis was also supported by another key experiment, in which we showed the intracellular fate of bacteria is affected by removal of SM from the plasma membrane before invasion, but not by removal of SM from phagosomal membranes after bacteria internalization (Figure5A-C). Taken together, we thus believe that the main hypothesis is strongly supported by our data.
Moreover, we either used different inhibitors for the same molecule (ASM was inhibited by ARC39, amitriptyline and PCK310 with similar outcome) or supported our hypothesis with gene-ablated cell pools (TPC1, Syt7, SARM1), as we will point out in more detail below.
Firstly, the release of calcium from lysosomes is not demonstrated. Localized changes in the immediate vicinity of lysosomes need to be measured to ascertain that these organelles are the source of cytosolic calcium changes. In fact, 9-phenantrol, which the authors find to be the most potent inhibitor of invasion and hence of the putative calcium changes, is not a blocker of lysosomal calcium release but instead blocks plasmalemmal TRPM4 channels. On the other hand, invasion is seemingly independent of external calcium. These findings are inconsistent with each other and point to non-specific effects of 9-phenantrol. The fact that ionomycin decreases invasion efficiency is taken as additional evidence of the importance of lysosomal calcium release. It is not clear how these observations support involvement of lysosomal calcium release and exocytosis; in fact treatment with the ionophore should itself have induced lysosomal exocytosis and stimulated, rather than inhibited invasion. Yet, manipulations that increase and others that decrease cytosolic calcium both inhibited invasion.
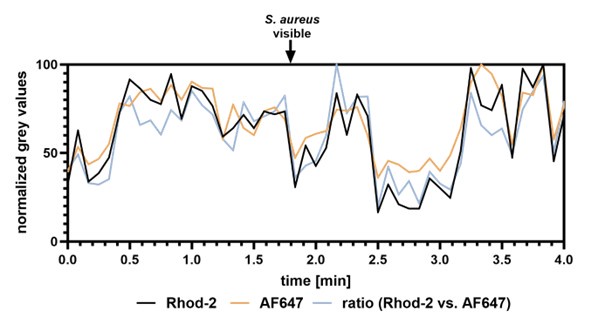
With respect to lysosomal Ca2+ release, we agree with the reviewer that direct visual demonstration of lysosomal Ca2+ release upon infection will improve the manuscript. We therefore performed live cell imaging to visualize lysosomal Ca2+ release by a previously published method.1 The approach is based on two dextran-coupled fluorophores that were incubated with host cells. The dyes are endocytosed and eventually stain the lysosomes. One of the dyes, Rhod-2, is Ca2+-sensitive and can be used to estimate the lysosomal Ca2+ content. The second dye, AF647, is Ca2+-insensitive and is used to visualize the lysosomes. If the ratio Rhod-2/AF647 within the lysosomes is decreasing, lysosomal Ca2+ release is indicated. We monitored lysosomal Ca2+ content during S. aureus infection with this method (Author response image 1 and Author response video 1). However, the lysosomes are very dynamic, and it is challenging to monitor the fluorescence intensities over time. Thus, quantitative measurements are not possible with our methodology, and we decided to not include these data in the main manuscript. However, one could speculate that lysosomal Ca2+ content in the selected ROI (Author response image 1 and Author response video 1) is decreased upon attachment of S. aureus to the host cells as indicated by a decrease in Rhod-2/AF647 ratio.
Author response image 1.
Lysosomal Ca2+ imaging during S. aureus infection. The lysosomes of HuLEC were stained with two dextran-coupled fluorescent dyes. A Ca2+-sensitive dye Rhod-2 as well as Ca2+insensitive AF647. Cells were infected with fluorescent S. aureus JE2 and monitored by live cell imaging (see Author response video 1). The intensity of Rhod-2/AF647 was measured close to a S. aureus-host contact site. Ratio of Rhod-2 vs. AF647 fluorescence intensity was calculated
As to the TRPM4 involvement in S. aureus host cell internalization, it has been reported that TRPM4 is activated by cytosolic Ca2+. However, the channel conducts monovalent cations such as K+ or Na+ but is impermeable for Ca2+ [2, 3]. The following of our observations are supporting this:
i) S. aureus invasion is dependent on intracellular Ca2+, but is independent from extracellular Ca2+ (Figure 1A).
ii) 9-phenantrol treatment reduces S. aureus internalization by host cells, illustrating the dependence of this process on TRPM4 (data removed from the manuscript) . We therefore hypothesize that TRPM4 is activated by Ca2+ released from lysosomes (see above).
TRPM4 is localized to focal adhesions and is connected to actin cytoskeleton[4, 5] – a requisite of host cell entry of S. aureus.[6, 7] This speaks for an important function of TRPM4 in uptake of S. aureus in general, but does not necessarily have to be involved exclusively in the rapid uptake pathway.
TRPM4 itself is not permeable for Ca2+ but is activated by the cation. Thus, it is unlikely to cause lysosomal exocytosis. The stronger bacterial uptake reduction by treatment with 9-phenantrol when compared to Ned19 thus may be caused by the involvement of TRPM4 in additional pathways of S. aureus host cell entry involving that association of TRPM4 with focal adhesions or as pointed out by the reviewer, unspecific side effects of 9-phenantrol that we currently cannot exclude. However, we think that experiments with 9-phenantrol distract from the main story (lysosomal Ca2+ and exocytosis) and might be confusing for the reader. We thus removed all data and discussion concerning 9phenantrol in the revised manuscript.
Regarding the reduced S. aureus invasion after ionomycin treatment, we agree with the reviewer that ionomycin is known to lead to lysosomal exocytosis as was previously shown by others8 as well as our laboratory[9}.
We hypothesized that pretreatment with ionomycin would trigger lysosomal exocytosis and thus would reduce the pool of lysosomes that can undergo exocytosis before host cells are contacted by S. aureus. As a result, we should observe a marked reduction of S. aureus internalization in such “lysosome-depleted cells”, if the lysosomal exocytosis is coupled to bacterial uptake. Our observation of reduced bacterial internalization after ionomycin treatment supports this hypothesis.
However, ionomycin treatment and S. aureus infection of host cells are distinct processes.
While ionomycin results in strong global and non-directional lysosomal exocytosis of all “releasable” lysosomes (~5-10 % of all lysosomes according to previous observations)8, we hypothesize that lysosomal exocytosis upon contact with S. aureus only involves a small proportion of lysosomes at host-bacteria contact sites. This is supported by experiments that demonstrate that ~30% of the lysosomes that are released by ionomycin treatment are exocytosed during S. aureus infection (see below and Figure 2, A-C). We added this new data as well as an according section to the discussion (line 563 ff). Moreover, we moved the data obtained with ionomycin to Figure 2E and described our idea behind this experiment more precisely (line 166 ff).
The proposed role of NAADP is based on the effects of "knocking out" TPC1 and on the pharmacological effects of Ned-19. It is noteworthy that TPC2, rather than TPC1, is generally believed to be the primary TPC isoform of lysosomes. Moreover, the gene ablation accomplished in the TPC1 "knockouts" is only partial and rather unsatisfactory. Definitive conclusions about the role of TPC1 can only be reached with proper, full knockouts. Even the pharmacological approach is unconvincing because the high doses of Ned-19 used should have blocked both TPC isoforms and presumably precluded invasion. Instead, invasion is reduced by only ≈50%. A much greater inhibition was reported using 9-phenantrol, the blocker of plasmalemmal calcium channels. How is the selective involvement of lysosomal TPC1 channels justified?
As to partial gene ablation of TPC1: To avoid clonal variances, we usually perform pool sorting to obtain a cell population that predominantly contains cells -here- deficient in TPC1, but also a small proportion of wildtype cells as seen by the residual TPC1 protein on the Western blot. We observe a significant reduction in bacterial uptake in this cell pool suggesting that the uptake reduction in a pure K.O. population may be even more pronounced.
As to the inhibition by Ned19:
The scale of invasion reduction upon Ned19 treatment (50%, Figure 1B) is comparable with the reduction caused by other compounds that influence the ASM-dependent pathway (such as amitriptyline, ARC39 [Figure 2G], BAPTA-AM [Figure 1A], Vacuolin-1 [Figure 2D], β-toxin [Figure 2L] and ionomycin [Figure 2E]). Further, the partial reduction of invasion is most likely due to the concurrent activity of multiple internalization pathways which are not all targeted by the used compounds and which we briefly discuss in the manuscript.
We agree with the reviewer that Ned19 inhibits TPC1 and TPC2. Since ablation of TPC1 reduced invasion of S. aureus, we concluded that TPC1 is important for S. aureus host cell invasion. We thus agree with the reviewer that a role for TPC2 cannot be excluded. We clarified this in the revised manuscript (Lines 552). It needs to be noted, however, that deficiency in either TPC1 or TPC2 alone was sufficient to prevent Ebola virus infection10, which is in line with our observations.
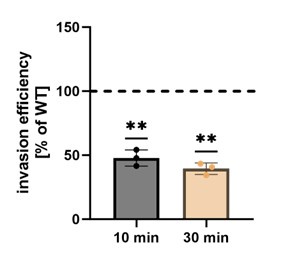
In order to address the role of TPC2 for this review process, we kindly were gifted TPCN1/TPCN2 double knock-out HeLa cells by Norbert Klugbauer (Freiburg, Germany), which we tested for S. aureus internalization. We found that invasion was reduced in these cell lines supporting a role of lysosomal Ca2+ release in S. aureus host cell entry and a role for both TPC channels (Author response image 2, see end of the document). Since we did not have a single TPCN2 knock-out available we decided to exclude these data from the main manuscript.
Author response image 2.
Invasion efficiency is reduced in TPC1/TPC2 double K.O. HeLa cells. Invasion efficiency of S. aureus JE2 was determined in TPC1/TPC2 double K.O. cells after 10 and 30 min. Results were normalized to the parental HeLa WT cell line (set to 100 %).
Invoking an elevation of NAADP as the mediator of calcium release requires measurements of the changes in NAADP concentration in response to the bacteria. This was not performed. Instead, the authors analyzed the possible contribution of putative NAADP-generating systems and reported that the most active of these, CD38, was without effect, while the elimination of SARM1, another potential source of NAADP, had a very modest (≈20%) inhibitory effect that may have been due to clonal variation, which was not ruled out. In view of these data, the conclusion that NAADP is involved in the invasion process seems unwarranted.
Our results from two independent experimental set-ups (Ned19 [Figure 1B] and TPC1 K.O. [Figure 1C & Figure 2N]) indicate the involvement of NAADP in the process. Together with the metabolomics unit at the Biocenter Würzburg, we attempted to measure cellular NAADP levels, however, this proved to be non-trivial and requires further optimization. However, we can rule out clonal variation in the SARM1 mutant since experiments were conducted with a cell pool as described above in order to avoid clonal variation of single clones.
The mechanism behind biosynthesis of NAADP is still debated. CD38 was the first enzyme discovered to possess the ability of producing NAADP. However, it requires acidic pH to produce NAADP[11] -which does not match the characteristics of a cytosolic NAADP producer. HeLa cells do not express CD38 and hence, it is not surprising that inhibition of CD38 had no effect on S. aureus invasion in HeLa cells. However, NAADP production by HeLa cells was observed in absence of CD38[12]. Thus CD38independent NAADP generation is likely. SARM1 can produce NAADP at neutral pH[13] and is expressed in HeLa, thus providing a more promising candidate.
We agree with the reviewer that the reduction of S. aureus internalization after ablation of SARM1 is less pronounced than in other experiments of ours. This may be explained by NAADP originating from other enzymes, such as the recently discovered DUOX1, DUOX2, NOX1 and NOX2[14], which – with exception of DUOX2- possess a low expression even in HeLa cells. We add this to the discussion in the revised manuscript (line 579).
We can, however, rule out clonal variation for the inhibitory effect. As stated above we generated K.O. cell pools specifically to avoid inherent problems of clonality. Thus, we also detect some residual wildtype cells within our cell pools.
The involvement of lysosomal secretion is, again, predicated largely on the basis of pharmacological evidence. No direct evidence is provided for the insertion of lysosomal components into the plasma membrane, or for the release of lysosomal contents to the medium. Instead, inhibition of lysosomal exocytosis by vacuolin-1 is the sole source of evidence. However, vacuolin-1 is by no means a specific inhibitor of lysosomal secretion: it is now known to act primarily as a PIKfyve inhibitor and to cause massive distortion of the endocytic compartment, including gross swelling of endolysosomes. The modest (20-25%) inhibition observed when using synaptotagmin 7 knockout cells is similarly not convincing proof of the requirement for lysosomal secretion.
We agree with the reviewer that the manuscript will benefit from a functional analysis of lysosomal exocytosis and therefore conducted assays to investigate exocytosis in the revised manuscript. We previously showed i) by addition of specific antisera that LAMP1 transiently is exposed on the plasma membrane during ionomycin and pore-forming toxin challenge and ii) demonstrated the release of ASM activity into the culture medium under these conditions.[9] However, both measurements are not compatible with S. aureus infection, since LAMP1 antibodies also are non-specifically bound by protein A and another IgG-binding proteins on the S. aureus surface, which would bias the results. Since protein A also may serve as an adhesin in the investigated pathway, we cannot simply delete the ORF without changing other aspects of staphylococcal virulence. Further, FBS contains a ASM background activity that impedes activity measurements of cell culture medium. We previously removed this background activity by a specific heat-inactivation protocol.[9] However, S. aureus invasion is strongly reduced in culture medium containing this heat-inactivated FBS.
We therefore developed a luminescence assay based on split NanoLuc luciferase that enables detection of LAMP1 exposed on the plasma membrane without usage of antibodies (Figure 2, A-C). We added a section on the assay in the revised manuscript. Briefly, we generated reporter cells by fusing a short peptide fragment of NanoLuc called HiBiT between the signal peptide and the mature luminal domain of LAMP1 and stably expressed the resulting protein in HeLa cells by lentiviral transduction. The LgBiT protein domain of NanoLuc luciferase (Promega) as well as the substrate Furimazine are added to the culture medium. HiBiT can reconstitute a functional NanoLuc with LgBiT and process Furimazine when lysosomes are exocytosed thereby generating luminescence measurable in a suitable plate reader.
With this assay we detected that about 30% of lysosomes that were “releasable” by treatment with ionomycin are exocytosed during S. aureus infection. Lysosomal exocytosis was strongly reduced (even below the levels of untreated controls), if we treated cells with Vacuolin-1 or Ned19.
We agree with the reviewer that Vacuolin-1 to some extent has unspecific side effects as has been shown by others and which we addressed in the revised version of the manuscript (line 541 ff). However, our new results with the HiBiT reporter cell line clearly demonstrate a reduction of lysosomal exocytosis after Vacuolin-1 treatment. Supported by this and our other results we hypothesize that Vacuolin-1 decreases S. aureus internalization due to the inhibition of lysosomal exocytosis.
As to the involvement of synaptotagmin 7: The effect of Syt7 K.O. on invasion was moderate in initial experiments, likely due to a high culture passage and presumably overgrowth of WT cells. However, reduction of invasion in Syt7 K.O.s was more pronounced in experiments with β-toxin complementation (Figure 2, N) and hence, we combined the two data sets (Figure 2, F). This demonstrates the reduction of bacterial invasion by ~40% in Syt7 K.O. cell pools. Moreover, Syt7 is not the only protein possibly involved in Ca2+-dependent exocytosis. For instance, Syt1 has been shown to possess an overlapping function.[15] This may explain the differences between our Vacuolin-1 and Syt7 ablation experiments. We added this information to the discussion.
ASM is proposed to play a central role in the rapid invasion process. As above, most of the evidence offered in this regard is pharmacological and often inconsistent between inhibitors or among cell types. Some drugs affect some of the cells, but not others. It is difficult to reach general conclusions regarding the role of ASM. The argument is made even more complex by the authors' use of exogenous sphingomyelinase (beta-toxin). Pretreatment with the toxin decreased invasion efficiency, a seemingly paradoxical result. Incidentally, the effectiveness of the added toxin is never quantified/validated by directly measuring the generation of ceramide or the disappearance of SM.
Although pharmacological inhibitors can have unspecific side effects, we want to emphasize that the inhibitors used in our study act on the enzyme ASM by completely different mechanisms. Amitriptyline is a so called functional inhibitor of ASM (FIASMA) which induces the detachment of ASM from lysosomal membranes resulting in degradation of the enzyme.[16] By contrast, ARC39 is a competitive inhibitor.[17, 18]
There are no inconsistencies in our data obtained with ASM inhibitors. Amitriptyline and ARC39 both reduce the invasion of S. aureus in HuLEC, HuVEC and HeLa cells (Figure 2G). ARC39 needs a longer pre-incubation, since its uptake by host cells is slower (to be published elsewhere). We observe a different outcome in 16HBE14o- and Ea.Hy 926 cells, with 16HBE14o- even demonstrating a slightly increased invasion of S. aureus upon ARC39 treatment. Amitriptyline had no effect (Figure 2G).
Thus, the ASM-dependent S. aureus internalization is cell type/line specific, which we state in the manuscript. The molecular origin of these differences is unclear and will require further investigation, e.g. in testing cell lines for potential differences in surface receptors. In a separate study we have already developed a biotinylation-based approach to identify potential novel host cell surface interaction partners during S. aureus infection.[19]
Moreover, both inhibitors affected the invasion dynamics (Figure 3D), phagosomal escape (Figure 4C and Figure 4D) and Rab7 recruitment (Figure 4A and Supp. Figure 4A-C) in a similar fashion. Proper inhibition of ASM by both compounds in all cell lines used was validated by enzyme assays (Supp. Figure 2H), which again suggests that the ASM-dependent pathway does only exist in specific cell lines and also supports that we do not observe unspecific side effects of the compounds. We clarified this in the revised manuscript.
ASM is a key player for SM degradation and recycling. In clinical context, deficiency in ASM results in the so-called Niemann Pick disease type A/B. The lipid profile of ASM-deficient cells is massively altered[20], which will result in severe side effects. Short-term inhibition by small molecules therefore poses a clear benefit when compared to the usage of ASM K.O. cells. In order to satisfy the query of the reviewer, we generated two ASM K.O. cell pools (generated with two different sgRNAs) and tested these for S. aureus invasion efficiency (Figure 2, I). We did not observe bacterial invasion differences between WT and K.O. cells. However, when we treated the cells additionally with ASM inhibitor, we observed a strongly reduced invasion in WT cells, while invasion efficiency in ASM K.O. was only slightly affected (Figure 2, J). We concluded that the reduced invasion observed in inhibitor-treated WT cells predominantly is due to absence of ASM, while the small reduction observed in ARC39treated ASM K.O.s is likely due to unspecific side effects.
We performed lipidomics on these cells and demonstrated a strongly altered sphingolipid profile in ASM K.O. cells compared to untreated and inhibitor-treated WT cells (Figure 2, K). We speculate that other ASM-independent bacterial invasion pathways are upregulated in ASM K.O.s., thereby obscuring the effect contributed by absence of ASM. We discussed this in the revised manuscript (line 518 ff).
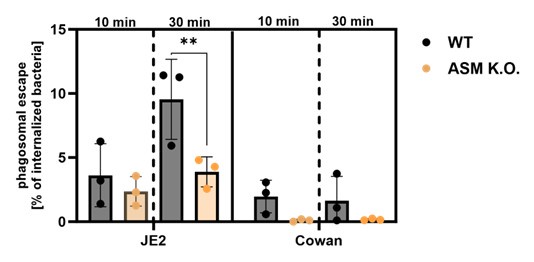
Moreover, we introduced the RFP-CWT escape marker into the ASM K.O. cells and measured phagosomal escape of S. aureus JE2 and Cowan I. The latter strain is non-cytotoxic and serves as negative control, since it is known to possess a very low escape rate, due to its inability to produce toxin. Again, we compared early invaders (infection for 10 min) with early+late invaders (infection for 30 min). As observed for JE2, “early invaders” possess lower escape rates than “early+late invaders”.
We did not observe differences between WT and ASM K.O. cells, if we infected for only 10 min. By contrast, we observed a lower escape rate in ASM K.O (Author response image 3, see end of the document). compared to WT cells, when we infected for 30 min.
However, we usually observe an increased phagosomal escape, when we treated host cells with ASM inhibitors (Figure 4C and D). Reduced phagosomal escape of intracellular S. aureus in ASM K.O. cells may be caused by the altered sphingolipid profile(e.g., by interference with binding of bacterial toxins to phagosomal membranes or altered vesicular acidification). We hence think that these data are difficult to interpret, and clarification would require intense additional experimentation. Thus, we did not include this data in the manuscript.
Author response image 3.
Phagosomal escape rates were established in either HeLa wild-type or ASM K.O. cells expressing the phagosomal escape reporter RFP-CWT. Host cells that were infected with the cytotoxic S. aureus strain JE2 or the non-cytotoxic strain Cowan I for 10 or 30 minutes and escape rates were determined by microscopy 3h p.i.
As to the treatment with a bacterial sphingomyelinase:
Treatment with the bacterial SMase (bSMase, here: β-toxin) was performed in two different ways:
i) Pretreatment of host cells with β-toxin to remove SM from the host cell surface before infection. This removes the substrate of ASM from the cell surface prior to addition of the bacteria (Figure 2L, Figure 4A-C). Since SM is not present on the extracellular plasma membrane leaflet after treatment, a release of ASM cannot cause localized ceramide formation at the sites of lysosomal exocytosis. Similar observations were made by others.[21]
ii) Addition of bSMase to host cells together with the bacteria to complement for the absence of ASM (Figure 2N).
Removal of the ASM substrate before infection (i) prevents localized ASM-mediated conversion of SM to Cer during infection and resulted in a decreased invasion, while addition of the SMase during infection resulted in an increased invasion in TPC1 and Syt7 ablated cells. Thus, both experiments are consistent with each other and in line with our other observations.
Removal of SM from the plasma membrane by β-toxin was indirectly demonstrated by the absence of Lysenin recruitment to phagosomes/escaped bacteria when host cells were pretreatment with the toxin before infection (Figure5C). We also added another data set that demonstrates degradation of a fluorescence SM derivative upon β-toxin treatment of host cells (Supp Figure 2, M). In another publication, we recently quantified the effectiveness of β-toxin treatment, even though with slightly longer treatment times (75 min vs. 3h).[22]
To clarify our experimental approaches to the readership we added an explanatory section to the revised manuscript (line 287 ff) and we also added a scheme to in Figure 2M describing the experimental settings.
As to the general conclusions regarding the role of ASM: ASM and lysosomal exocytosis has been shown to be involved in uptake of a variety of pathogens[21, 23-27] supporting its role in the process.
The use of fluorescent analogs of sphingomyelin and ceramide is not well justified and it is unclear what conclusions can be derived from these observations. Despite the low resolution of the images provided, it appears as if the labeled lipids are largely in endomembrane compartments, where they would presumably be inaccessible to the secreted ASM. Moreover, considering the location of the BODIPY probe, the authors would be unable to distinguish intact sphingomyelin from its breakdown product, ceramide. What can be concluded from these experiments? Incidentally, the authors report only 10% of BODIPY-positive events after 10 min. What are the implications of this finding? That 90% of the invasion events are unrelated to sphingomyelin, ASM, and ceramide?
During the experiments with fluorescent SM analogues (Figure 3a,b), S. aureus was added to the samples immediately before the start of video recording. Hence, bacteria are slowly trickling onto the host cells, and we thus can image the initial contact between them and the bacteria, for instance, the bacteria depicted in Figure 3A contact the host cell about 9 min before becoming BODIPY-FL-positive (see Supp. Video 1, 55 min). Hence, in these cases we see the formation of phagosomes around bacteria rather than bacteria in endomembrane compartments. Since generation of phagosomes happens at the plasma membrane, SM is accessible to secreted ASM.
The “trickling” approach for infection is an experimental difference to our invasion measurements, in which we synchronized the infection by centrifugation. This ensures that all bacteria have contact to host cells and are not just floating in the culture medium. However, live cell imaging of initial bacterialhost contact and synchronization of infection is hard to combine technically.
In our invasion measurements -with synchronization-, we typically see internalization of ~20% of all added bacteria after 30 min. Hence, most bacteria that are visible in our videos likely are still extracellular and only a small proportion was internalized. This explains why only 10% of total bacteria are positive for BODIPY-FL-SM after 10 min. The proportion of internalized bacteria that are positive for BODIPY-FL-SM should be way higher but cannot be determined with this method.
We agree with the reviewer that we cannot observe conversion of BODIPY-FL-SM by ASM. In order to do that, we attempted to visualize the conversion of a visible-range SM FRET probe (Supp. Figure 3), but the structure of the probe is not compatible with measurement of conversion on the plasma membrane, since the FITC fluorophore released into the culture medium by the ASM activity thereby gets lost for imaging. In general, the visualization of SM conversion with subcellular resolution is challenging and even with novel tools developed in our lab[28] visualization of SM on the plasma membrane is difficult.
The conclusions we draw from these experiments are that i.) S. aureus invasion is associated with SM and ii.) SM-associated invasion can be very fast, since bacteria are rapidly engulfed by BODIPY-FL-SM containing membranes.
It is also unclear how the authors can distinguish lysenin entry into ruptured vacuoles from the entry of RFP-CWT, used as a criterion of bacterial escape. Surely the molecular weights of the probes are not sufficiently different to prevent the latter one from traversing the permeabilized membrane until such time that the bacteria escape from the vacuole.
We here want to clarify that both Lysenin as well as the CWT reporter have access to ruptured vacuoles (Figure 4B). We used the Lysenin reporter in these experiments for estimation of SM content of phagosomal membranes. If a vacuole is ruptured, both the bacteria and the luminal leaflet of the phagosomal membrane remnants get in contact with the cytosol and hence with the cytosolically expressed reporters YFP-Lysenin as well as RFP-CWT resulting in “Lysenin-positive escape” when phagosomes contained SM (see Figure 5C). By contrast, either β-toxin expression by S. aureus or pretreatment with the bSMase resulted in absence of Lysenin recruitment suggesting that the phagosomal SM levels were decreased/undetectable (Figure 5C, Supp Figure 6F, G, I, J).
Although this approach does not enable a quantitative measurement of phagosomal SM, this method is sufficient to show that β-toxin expression and pretreatment result in markedly decreased phagosomal SM levels in the host cells.
The approach we used here to analyze “Lysenin-positive escape” can clearly be distinguished from Lysenin-based methods that were used by others.29 There Lysenin was used to show trans-bilayer movement of SM before rupture of bacteria-containing phagosomes.
To clarify the function of Lysenin in our approach we added additional figures (Figure 4F, Supp. Figure 5) and a movie (Supp. Video 4) to the revised manuscript.
Both SMase inhibitors (Figure 4C) and SMase pretreatment increased bacterial escape from the vacuole. The former should prevent SM hydrolysis and formation of ceramide, while the latter treatment should have the exact opposite effects, yet the end result is the same. What can one conclude regarding the need and role of the SMase products in the escape process?
As pointed out above, pretreatment of host cells with SMase removes SM from the plasma membrane and hence, ASM does not have access to its substrate. Hence, both treatment with either ASM inhibitors or pretreatment with bacterial SMase prevent ASM from being active on the plasma membrane and hence block the ASM-dependent uptake (Figure 2 G, L). Although overall less bacteria were internalized by host cells under these conditions, the bacteria that invaded host cells did so in an ASM-independent manner.
Since blockage of the ASM-dependent internalization pathway (with ASM inhibitor [Figure 4C, D], SMase pretreatment [Figure 5B] and Vacuolin-1[Figure.4E]) always resulted in enhanced phagosomal escape, we conclude that bacteria that were internalized in an ASM-independent fashion cause enhanced escape. Vice versa, bacteria that enter host cells in an ASM-dependent manner demonstrate lower escape rates.
This is supported by comparing the escape rates of “early” and “late” invaders [Figure 5D, E], which in our opinion is a key experiment that supports this hypothesis. The “early” invaders are predominantly ASM-dependent (see e.g. Figure 3E) and thus, bacteria that entered host cell in the first 10 min of infection should have been internalized predominantly in an ASM-dependent fashion, while slower entry pathways are active later during infection. The early ASM dependent invaders possessed lower escape rates, which is in line with the data obtained with inhibitors (e.g. Figure 4C, D).
We hypothesize that the activity of ASM on the plasma membrane during invasion mediates the recruitment of a specific subset of receptors, which then influences downstream phagosomal maturation and escape. This hypothesis is supported by the fact that the subset of receptors interacting with S. aureus is altered upon inhibition of the ASM-dependent uptake pathway. We describe this in another study that is currently under evaluation elsewhere.
Reviewer #2 (Public review):
Summary:
In this manuscript, Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry.
The evidence provided is solid, methods used are appropriate and results largely support their conclusions, but can be substantiated further as detailed below. The weakness is a reliance on chemical inhibitors that can be non-specific to delineate critical steps.
Specific comments:
A large number of experiments rely on treatment with chemical inhibitors. While this approach is reasonable, many of the inhibitors employed such as amitriptyline and vacuolin1 have other or nondefined cellular targets and pleiotropic effects cannot be ruled out. Given the centrality of ASM for the manuscript, it will be important to replicate some key results with ASM KO cells.
We thank the reviewer for the critical evaluation of our manuscript and plenty of constructive comments.
We agree with the reviewer, that ASM inhibitors such as functional inhibitors of ASM (FIASMA) like amitriptyline used in our study have unspecific side effects given their mode-of-action. FIASMAs induce the detachment of ASM from lysosomal membranes resulting in degradation of the enzyme.[16] However, we want to emphasize that we also used the competitive inhibitor ARC39 in our study[17, 18] which acts on the enzyme by a completely different mechanism. All phenotypes (reduced invasion [Figure 2G], effect on invasion dynamics [Figure 3D], enhanced escape [Figure 4C, D] and differential recruitment of Rab7 [Supp. Figure 4A-C]) were observed with both inhibitors thereby supporting the role of ASM in the process.
We further agree that experiments with genetic evidence usually support and improve scientific findings. However, ASM is a cellular key player for SM degradation and recycling. In a clinical context, deficiency in ASM results in a so-called Niemann Pick disease type A/B. The lipid profile of ASMdeficient cells is massively altered[20], which in itself will result in severe side effects. Thus, the usage of inhibitors provides a clear benefit when compared to ASM K.O. cells, since ASM activity can be targeted in a short-term fashion thereby preventing larger alterations in cellular lipid composition.
We nevertheless generated two ASM K.O. cell pools (generated with two different sgRNAs) and tested for invasion efficiency (Figure 2, I). Here, we did not observe differences between WT and mutants. However, if we treated the cells additionally with ASM inhibitor, we observed a strongly reduced invasion in WT cells, while invasion efficiency in ASM K.O. was only slightly affected (Figure 2, J). We concluded that the reduced invasion observed in WT cells upon inhibitor treatment predominantly is due to inhibition of ASM, whereas the small reduction observed in ARC39-treated ASM K.O.s is likely due to unspecific side effects. We also demonstrated a strongly altered sphingolipid profile in ASM K.O. cells when compared to untreated and inhibitor-treated WT cells (new Figure 2, K). We speculate that other ASM-independent invasion pathways are upregulated in ASM K.O.s., thereby making up for the absence of ASM. We discuss this in the revised manuscript (line 518 ff).
We introduced the RFP-CWT escape marker into the ASM K.O. cells and measured phagosomal escape of S. aureus JE2 and Cowan I (Author response image 3). The latter serves as negative control, since it is known to possess a very low escape rate, due to its inability of toxin production. Again, we compared early invaders (infection for 10 min) with early+late invaders (infection for 30 min). As seen before for JE2, early invaders possess lower escape rates than early+late invaders. We did not observe differences between WT and K.O. cells, if we infected for 10 min. By contrast, we observed a lower escape rate in ASM K.O. compared to WT cells, when we infected for 30 min. However, we usually observe an increased phagosomal escape, when we treated host cells with ASM inhibitors (Figure 4C and D). We think that the reduced phagosomal escape in ASM K.O. is caused by the altered sphingolipid profile, which could have versatile effects (e.g., inference with binding of bacterial toxins to phagosomal membranes or changes in acidification). We hence think that these data are difficult to interpret, and clarification would require intense additional experimentation. Thus, we did not include this data in the manuscript.
Most experiments are done in HeLa cells. Given the pathway is projected as generic, it will be important to further characterize cell type specificity for the process. Some evidence for a similar mechanism in other cell types S. aureus infects, perhaps phagocytic cell type, might be good.
Whenever possible we performed the experiments not only in HeLa but also in HuLECs. For example, we refer to experiments concerning the role of Ca2+ (Figure 1A/Supp.Figure1A), lysosomal Ca2+/Ned19 (Figure1B/Supp Figure 1C), lysosomal exocytosis/Vacuolin-1 (Figure 2D/Supp. Figure2D), ASM/ARC39 and amitriptyline (Figure 2G), surface SM/β-toxin (Figure 2L/Supp. Figure 2L), analysis of invasion dynamics (complete Figure 3) and measurement of cell death during infection (Figure 6C+E, Supp. Figure 8A+B).
HuLECs, however, are not really genetically amenable and hence we were not able to generate gene deletions in these cells and upon introduction of the fluorescence escape reporter the cells are not readily growing.
As to ASM involvement in phagocytic cells: a role for ASM during the uptake of S. aureus by macrophages was previously reported by others.[25] However, in professional phagocytes S. aureus does not escape from the phagosome and replicates within the phagosome.[30]
I'm a little confused about the role of ASM on the surface. Presumably, it converts SM to ceramide, as the final model suggests. Overexpression of b-toxin results in the near complete absence of SM on phagosomes (having representative images will help appreciate this), but why is phagosomal SM detected at high levels in untreated conditions? If bacteria are engulfed by SM-containing membrane compartments, what role does ASM play on the surface? If surface SM is necessary for phagosomal escape within the cell, do the authors imply that ASM is tuning the surface SM levels to a certain optimal range? Alternatively, can there be additional roles for ASM on the cell surface? Can surface SM levels be visualized (for example, in Figure 4 E, F)?
We initially hypothesized that we would detect higher phagosomal SM levels upon inhibition of ASM, since our model suggests SM cleavage by ASM on the host cell surface during bacterial cell entry. However, we did not detect any changes in our experiments (Supp. Figure 4F). We currently favor the following explanation: SM is the most abundant sphingolipid in human cells.[31] If peripheral lysosomes are exocytosed and thereby release ASM, only a localized and relative small proportion of SM may get converted to Cer, which most likely is below our detection limit. In addition, the detection of cytosolically exposed phagosomal SM by YFP-Lysenin is not quantitative and provides a “Yes or No” measurement. Hence, we think that the rather limited SM to Cer conversion in combination with the high abundance of SM in cellular membranes does not visibly affect the recruitment of the Lysenin reporter.
In our experiments that employ BODIPY-FL-SM (Figure 3a+b), we cannot distinguish between native SM and downstream metabolites such as Cer. Hence, again we cannot make any assumptions on the extent to which SM is converted on the surface during bacterial internalization. Although our laboratory recently used trifunctional sphingolipid analogs to analyze the SM to Cer conversion[22], the visualization of this process on the plasma membrane is currently still challenging.
Overall, we hypothesize that the localized generation of Cer on the surface by released ASM leads to generation of Cer-enriched platforms. Subsequently, a certain subset of receptors may be recruited to these platforms and influence the uptake process. These platforms are supposed to be very small, which also would explain that we did not detect changes in Lysenin recruitment.
Related to that, why is ASM activity on the cell surface important? Its role in non-infectious or other contexts can be discussed.
ASM release by lysosomal exocytosis is implied in plasma membrane repair upon injury. We added a short description of the role of extracellular ASM in the introduction (line 35).
If SM removal is so crucial for uptake, can exocytosis of lysosomes alone provide sufficient ASM for SM removal? How much or to what extent is lysosomal exocytosis enhanced by initial signaling events? Do the authors envisage the early events in their model happening in localized confines of the PM, this can be discussed.
Ionomycin treatment led to a release of ~10 % of all lysosomes and also increased extracellular ASM activity.[8, 9] In the revised manuscript, we developed an assay to determine lysosomal exocytosis during S. aureus infection (Figure 2, A-C). We detected lysosomal exocytosis of ~30% when compared to ionomycin treatment during infection. Since this is only a fraction of the “releasable lysosomes”, we assume that the effects (lysosomal Ca2+ liberation, lysosomal exocytosis and ASM activity) are very localized and take place only at host-pathogen contact sites (see also above). We discuss this in the revised manuscript (line 563 ff). To our knowledge it is currently unclear to which extent the released ASM affects surface SM levels. We attempted to visualize the local ASM activity on the cell surface by using a visible range FRET probe (Supp. Fig. 3). Cleavage of the probe by ASM on the surface leads to release of FITC into the cell culture medium, which does not contribute a measurable signal at the surface.
How are inhibitor doses determined? How efficient is the removal of extracellular bacteria at 10 min? It will be good to substantiate the cfu experiments for infectivity with imaging-based methods. Are the roles of TPC1 and TPC2 redundant? If so, why does silencing TPC1 alone result in a decrease in infectivity? For these and other assays, it would be better to show raw values for infectivity. Please show alterations in lysosomal Ca2+ at the doses of inhibitors indicated. Is lysosomal Ca2+ released upon S. aureus binding to the cell surface? Will be good to directly visualize this.
Concerning the inhibitor concentrations, we either used values established in published studies or recommendations of the suppliers (e.g. 2-APB, Ned19, Vacuolin-1). For ASM inhibitors, we determined proper inhibition of ASM by activity assays. Concentrations of ionomycin resulting in Ca2+ influx and lysosomal exocytosis was determined in earlier studies of our lab.[9, 32]
As to the removal of bacteria at 10 min p.i.: Lysostaphin is very efficient for removal of extracellular S. aureus and sterilizes the tissue culture supernatant. It significantly lyses bacteria within a few minutes, as determined by turbidity assays.[33]
As to imaging-based infectivity assays: We performed imaging-based invasion assays to show reduced invasion efficiency with two ASM inhibitors in the revised manuscript with similar results as obtained by CFU counts (Supp. Figure 2, J).
Regarding the roles of TPC1 and TPC2: from our data we cannot conclude whether the roles of TPC1 and TPC2 are redundant. One could speculate that since blockage of TPC1 alone is sufficient to reduce internalization of bacteria, that both channels may have distinct roles. On the other hand, there might be a Ca2+ threshold in order to initiate lysosomal exocytosis that can only be attained if TPC1 and TPC2 are activated in parallel. Thus, our observations are in line with another study that shows reduced Ebola virus infection in absence of either TPC1 or TPC2.[34] In order to address the role of TPC2 for this review process, we kindly were gifted TPCN1/TPCN2 double knock-out HeLa cells by Norbert Klugbauer (Freiburg, Germany), which we tested for S. aureus internalization. We found that invasion was reduced in these double KO cell lines even further supporting a role of lysosomal Ca2+ release in S. aureus host cell entry (Author response image 2, see end of the document). Since we did not have a single TPCN2 knockout available, we decided to exclude these data from the main manuscript.
As to raw CFU counts: whereas the observed effects upon blocking the invasion of S. aureus are stable, the number of internalized bacteria varies between individual biological replicates, for instance, by differences in host cell fitness or growth differences in bacterial cultures, which are prepared freshly for each experiment.
With respect to visualization of lysosomal Ca2+ release: we agree with the reviewer that direct visual demonstration of lysosomal Ca2+ release upon infection would improve the manuscript. We therefore performed live cell imaging to visualize lysosomal Ca2+ release by a previously published method.[1] The approach is based on two dextran-coupled fluorophores that were incubated with host cells. The dyes are endocytosed and eventually stain the lysosomes. One of the dyes, Rhod-2, is Ca2+-sensitive and can be used to estimate the lysosomal Ca2+ content. The second dye, AF647, is Ca2+-insensitive and is used to visualize the lysosomes. If the ratio Rhod-2/AF647 within the lysosomes is decreasing, lysosomal Ca2+ release is indicated. We monitored lysosomal Ca2+ content during S. aureus infection with this method (Author response image 1 and Author response video 1). However, the lysosomes are very dynamic, and it is challenging to monitor the fluorescence intensities over time. Thus, quantitative measurements are not possible with our methodology, and we decided to not include these data in the final manuscript. However, one could speculate that lysosomal Ca2+ content in the selected ROI (Author response image 1 and Author response video 1) is decreased upon attachment of S. aureus to the host cells as indicated by a decrease in Rhod-2/AF647 ratio.
The precise identification of cytosolic vs phagosomal bacteria is not very easy to appreciate. The methods section indicates how this distinction is made, but how do the authors deal with partial overlaps and ambiguities generally associated with such analyses? Please show respective images.
The number of events (individual bacteria) for the live cell imaging data should be clearly mentioned.
We apologize for not having sufficiently explained the technology to detect escaped S. aureus. The cytosolic location of S. aureus is indicated by recruitment of RFP-CWT.[35] CWT is the cell wall targeting domain of lysostaphin, which efficiently binds to the pentaglycine cross bridge in the peptidoglycan of S. aureus. This reporter is exclusively and homogenously expressed in the host cytosol. Only upon rupture of phagoendosomal membranes, the reporter can be recruited to the cell wall of now cytosolically located bacteria. S. aureus mutants, for instance in the agr quorum sensing system, cannot break down the phagosomal membrane in non-professional phagocytes and thus stay unlabeled by the CWT-reporter.[35] We include several images (Figure 4, F, Supp. Figure 5) /movies (Supp. Video 4) of escape events in the revised manuscript. The bacteria numbers for live cell experiments are now shown in Supp. Figure 7.
In the phagosome maturation experiments, what is the proportion of bacteria in Rab5 or Rab7 compartments at each time point? Will the decreased Rab7 association be accompanied by increased Rab5? Showing raw values and images will help appreciate such differences. Given the expertise and tools available in live cell imaging, can the authors trace Rab5 and Rab7 positive compartment times for the same bacteria?
We included the proportion of Rab7-associated bacteria in the revised manuscript (Supp. Figure 4A and C) and also shortly mention these proportions in the text (line 353). Usually, we observe that Rab5 is only transiently (for a few minutes) present on phagosomes and only afterwards the phagosomes become positive for Rab7. We do not think that a decrease in Rab7-positive phagosomes would increase the proportion of Rab5-positive phagosomes. However, we cannot exclude this hypothesis with our data.
We can achieve tracing of individual bacteria for recruitment of Rab5/Rab7 only manually, which impedes a quantitative evaluation. However, we included a Video (Supp. Video 3) that illustrates the consecutive recruitment of the GTPases.
The results with longer-term infection are interesting. Live cell imaging suggests that ASM-inhibited cells show accelerated phagosomal escape that reduces by 6 hpi. Where are the bacteria at this time point ? Presumably, they should have reached lysosomes. The relationship between cytosolic escape, replication, and host cell death is interesting, but the evidence, as presented is correlative for the populations. Given the use of live cell imaging, can the authors show these events in the same cell?
We think that most bacteria-containing phagoendosomes should have fused with lysosomes 6 h p.i. as we have previously shown by acidification to pH of 5 and LAMP1 decoration.[36]
The correlation between phagosomal escape and replication in the cytosol of non-professional phagocytes has been observed by us and others. In the revised manuscript we also provide images (Supp. Figure 5)/videos (Supp. Video 4) to show this correlation in our experiments.
Given the inherent heterogeneity in uptake processes and the use of inhibitors in most experiments, the distinction between ASM-dependent and independent pathways might not be as clear-cut as the authors suggest. Some caution here will be good. Can the authors estimate what fraction of intracellular bacteria are taken up ASM-dependent?
We agree with the reviewer that an overlap between internalization pathways is likely. A clear distinction is therefore certainly non-trivial. Alternative to ASM-dependent and ASM-independent pathways, the ASM activity may also accelerate one or several internalization pathways. We address this limitation in the discussion of the revised manuscript (line 596 ff).
Early in infection (~10 min after contact with the cells), the proportion of bacteria that enter host cells ASM-dependently is relatively high amounting to roughly 75-80% in HuLEC. After 30 min, this proportion is decreasing to about 50%. We included a paragraph in the discussion of the revised manuscript (line 593 ff).
Reviewer #2 (Recommendations for the authors):
(1) The experiment in Figure 4H is interesting. Details on what proportion of the cell is double positive, and if only this fraction was used for analysis will be good.
We did use all bacteria found in the images independently from whether host cells were infected with only one or both strains. We unfortunately cannot properly determine the proportion of cells that are double infected, since i) we record the samples with CLSM and hence, cannot exclude that there are intracellular bacteria found in higher or lower optical sections. ii) we visualized cells by staining Nuclei and did not stain the cell borders, thus we cannot precisely tell to which host cell the bacteria localize.
(2) Data is sparse for steps 5 and 6 of the model (line 330).
We apologize for the inconvenience. There is a related study published elsewhere[19], in which we identified NRCAM and PTK7 as putative receptors involved in this invasion pathway. We included a section in the discussion with the corresponding citation (line 569).
(3) Data for the reduced number of intracellular bacteria upon blocking ASM-dependent uptake (line 235) is not clear. Do they mean decreased invasion efficiency? These two need not be the same.
We changed “reduced number of intracellular bacteria” to “invasion efficiency”.
(4) b-toxin added to the surface can get endocytosed. Can its surface effect be delineated from endo/phagosomal effect?
We attempted to delineate effects contributed by the toxin activity on the surface vs. within phagosomes (Figure 5 A-C). We see an increased phagosomal escape, when we pretreated host cells with β-toxin (removal of SM form the surface) and infected either in presence (toxin will be taken up together with the bacteria into the phagosome) or in absence (toxin was washed away shortly before infection) of β-toxin. By contrast, overexpression of β-toxin by S. aureus did not affect phagosomal escape rates. The proper activity of β-toxin was confirmed by absence of Lysenin recruitment during phagosomal escape in all three conditions. We concluded that the activity on the surface and not the activity in the phagosome is important.
(5) The potential role(s) of bacterial factors in the uptake and subsequent intracellular stages can be discussed.
There are multiple bacterial adhesins known in S. aureus. These usually are either covalently attached to the bacterial cell wall such as the sortase-dependently anchored Fibronectin-binding Proteins A and B but also secreted and “cell wall binding” proteins as well at non proteinaceous factor such as wall-teichoic acids. A discussion of these factors would thus be out of the scope of this manuscript, and we here suggest reverting to specialized reviews on that topic.
(6) The manuscript is not very easy to read. The abstract could be rephrased for better clarity and succinctness, with a clearly stated problem statement. The introduction is somewhat haphazard, I feel it can be better structured.
We apologize for the inconvenience. We stated the problem/research question in the abstract and tried to improve the introduction without adding too much unnecessary detail. In general, we tried to improve the readability of the manuscript and hope that our results and conclusions can be easier understood by the reader in the revised version.
(7) Typo in Figure 5F. Step 6 should read "accessory receptors"
The typo was corrected.
References
(1) Lloyd-Evans, E. et al. Niemann-Pick disease type C1 is a sphingosine storage disease that causes deregulation of lysosomal calcium. Nature Medicine 14, 1247-1255 (2008).
(2) Launay, P. et al. TRPM4 Is a Ca2+-Activated Nonselective Cation Channel Mediating Cell Membrane Depolarization. Cell 109, 397-407 (2002).
(3) Nilius, B. et al. The Ca2+‐activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5‐biphosphate. The EMBO Journal 25, 467-478-478 (2006).
(4) Cáceres, M. et al. TRPM4 Is a Novel Component of the Adhesome Required for Focal Adhesion Disassembly, Migration and Contractility. PLoS One 10, e0130540 (2015).
(5) Silva, I., Brunett, M., Cáceres, M. & Cerda, O. TRPM4 modulates focal adhesion-associated calcium signals and dynamics. Biophysical Journal 123, 390a (2024).
(6) Schlesier, T., Siegmund, A., Rescher, U. & Heilmann, C. Characterization of the Atl-mediated staphylococcal internalization mechanism. International Journal of Medical Microbiology 310, 151463 (2020).
(7) Jevon, M. et al. Mechanisms of Internalization ofStaphylococcus aureus by Cultured Human Osteoblasts. Infection and Immunity 67, 2677-2681 (1999).
(8) Rodriguez, A., Webster, P., Ortego, J. & Andrews, N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol 137, 93-104 (1997).
(9) Krones & Rühling et al. Staphylococcus aureus alpha-Toxin Induces Acid Sphingomyelinase Release From a Human Endothelial Cell Line. Front Microbiol 12, 694489 (2021).
(10) Sakurai, Y. et al. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347, 995-998 (2015).
(11) Aarhus, R., Graeff, R.M., Dickey, D.M., Walseth, T.F. & Lee, H.C. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J Biol Chem 270, 3032730333 (1995).
(12) Schmid, F., Fliegert, R., Westphal, T., Bauche, A. & Guse, A.H. Nicotinic acid adenine dinucleotide phosphate (NAADP) degradation by alkaline phosphatase. J Biol Chem 287, 32525-32534 (2012).
(13) Angeletti, C. et al. SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated bymultiple physiologically relevant NAD metabolites. iScience 25, 103812 (2022).
(14) Gu, F. et al. Dual NADPH oxidases DUOX1 and DUOX2 synthesize NAADP and are necessary for Ca(2+) signaling during T cell activation. Sci Signal 14, eabe3800 (2021).
(15) Schonn, J.-S., Maximov, A., Lao, Y., Südhof, T.C. & Sørensen, J.B. Synaptotagmin-1 and -7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proceedings of the National Academy of Sciences 105, 3998-4003 (2008).
(16) Kornhuber, J. et al. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cell Physiol Biochem 26, 9-20 (2010).
(17) Naser, E. et al. Characterization of the small molecule ARC39, a direct and specific inhibitor of acid sphingomyelinase in vitro. J Lipid Res 61, 896-910 (2020).
(18) Roth, A.G. et al. Potent and selective inhibition of acid sphingomyelinase by bisphosphonates. Angew Chem Int Ed Engl 48, 7560-7563 (2009).
(19) Rühling, M., Schmelz, F., Kempf, A., Paprotka, K. & Fraunholz Martin, J. Identification of the Staphylococcus aureus endothelial cell surface interactome by proximity labeling. mBio 0, e03654-03624 (2025).
(20) Schuchman, E.H. & Desnick, R.J. Types A and B Niemann-Pick disease. Mol Genet Metab 120, 27-33 (2017).
(21) Miller, M.E., Adhikary, S., Kolokoltsov, A.A. & Davey, R.A. Ebolavirus Requires Acid Sphingomyelinase Activity and Plasma Membrane Sphingomyelin for Infection. Journal of Virology 86, 7473-7483 (2012).
(22) M. Rühling, L.K., F. Wagner, F. Schumacher, D. Wigger, D. A. Helmerich, T. Pfeuffer, R. Elflein, C. Kappe, M. Sauer, C. Arenz, B. Kleuser, T. Rudel, M. Fraunholz, J. Seibel Trifunctional sphingomyelin derivatives enable nanoscale resolution of sphingomyelin turnover in physiological and infection processes via expansion microscopy. Nat Commun accepted in principle (2024).
(23) Peters, S. et al. Neisseria meningitidis Type IV Pili Trigger Ca(2+)-Dependent Lysosomal Trafficking of the Acid Sphingomyelinase To Enhance Surface Ceramide Levels. Infect Immun 87 (2019).
(24) Grassmé, H. et al. Acidic sphingomyelinase mediates entry of N. gonorrhoeae into nonphagocytic cells. Cell 91, 605-615 (1997).
(25) Li, C. et al. Regulation of Staphylococcus aureus Infection of Macrophages by CD44, Reactive Oxygen Species, and Acid Sphingomyelinase. Antioxid Redox Signal 28, 916-934 (2018).
(26) Fernandes, M.C. et al. Trypanosoma cruzi subverts the sphingomyelinase-mediated plasma membrane repair pathway for cell invasion. J Exp Med 208, 909-921 (2011).
(27) Luisoni, S. et al. Co-option of Membrane Wounding Enables Virus Penetration into Cells. Cell Host & Microbe 18, 75-85 (2015).
(28) Rühling, M. et al. Trifunctional sphingomyelin derivatives enable nanoscale resolution of sphingomyelin turnover in physiological and infection processes via expansion microscopy. Nature Communications 15, 7456 (2024).
(29) Ellison, C.J., Kukulski, W., Boyle, K.B., Munro, S. & Randow, F. Transbilayer Movement of Sphingomyelin Precedes Catastrophic Breakage of Enterobacteria-Containing Vacuoles. Curr Biol 30, 2974-2983 e2976 (2020).
(30) Moldovan, A. & Fraunholz, M.J. In or out: Phagosomal escape of Staphylococcus aureus. Cell Microbiol 21, e12997 (2019).
(31) Slotte, J.P. Biological functions of sphingomyelins. Progress in Lipid Research 52, 424-437 (2013).
(32) Stelzner, K. et al. Intracellular Staphylococcus aureus Perturbs the Host Cell Ca(2+) Homeostasis To Promote Cell Death. mBio 11 (2020).
(33) Kunz, T.C. et al. The Expandables: Cracking the Staphylococcal Cell Wall for Expansion Microscopy. Front Cell Infect Microbiol 11, 644750 (2021).
(34) Sakurai, Y. et al. Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347, 995-998 (2015).
(35) Grosz, M. et al. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin alpha. Cell Microbiol 16, 451-465 (2014).
(36) Giese, B. et al. Staphylococcal alpha-toxin is not sufficient to mediate escape from phagolysosomes in upper-airway epithelial cells. Infect Immun 77, 3611-3625 (2009).
-
-
eLife Assessment
This valuable study proposes a novel rapid-entry mechanism of S. aureus that involves the rapid release of calcium from lysosomes. The strength of the paper lies in a very interesting hypothesis; what diminishes enthusiasm is the lack of appropriate methodology, thus making the study incomplete. The methods used are deficient: they are largely reliant on the use of chemical inhibitors and do not adequately support the conclusions.
-
Reviewer #1 (Public review):
Summary:
The manuscript by Rühling et al analyzes the mode of entry of S. aureus into mammalian cells in culture. The authors propose a novel mechanism of rapid entry that involves the release of calcium from lysosomes via NAADP-stimulated activation of TPC1, which in turn causes lysosomal exocytosis; exocytic release of lysosomal acid sphingomyelinase (ASM) is then envisaged to convert exofacial sphingomyelin to ceramide. These events not only induce the rapid entry of the bacteria into the host cells but are also described to alter the fate of the intracellular S. aureus, facilitating escape from the endocytic vacuole to the cytosol.
Strengths:
The proposed mechanism is novel and could have important biological consequences.
Weaknesses:
Unfortunately, the evidence provided is unconvincing and insufficient …
Reviewer #1 (Public review):
Summary:
The manuscript by Rühling et al analyzes the mode of entry of S. aureus into mammalian cells in culture. The authors propose a novel mechanism of rapid entry that involves the release of calcium from lysosomes via NAADP-stimulated activation of TPC1, which in turn causes lysosomal exocytosis; exocytic release of lysosomal acid sphingomyelinase (ASM) is then envisaged to convert exofacial sphingomyelin to ceramide. These events not only induce the rapid entry of the bacteria into the host cells but are also described to alter the fate of the intracellular S. aureus, facilitating escape from the endocytic vacuole to the cytosol.
Strengths:
The proposed mechanism is novel and could have important biological consequences.
Weaknesses:
Unfortunately, the evidence provided is unconvincing and insufficient to document the multiple, complex steps suggested. In fact, there appear to be numerous internal inconsistencies that detract from the validity of the conclusions, which were reached mostly based on the use of pharmacological agents of imperfect specificity.
Firstly, the release of calcium from lysosomes is not demonstrated. Localized changes in the immediate vicinity of lysosomes need to be measured to ascertain that these organelles are the source of cytosolic calcium changes. In fact, 9-phenantrol, which the authors find to be the most potent inhibitor of invasion and hence of the putative calcium changes, is not a blocker of lysosomal calcium release but instead blocks plasmalemmal TRPM4 channels. On the other hand, invasion is seemingly independent of external calcium. These findings are inconsistent with each other and point to non-specific effects of 9-phenantrol. The fact that ionomycin decreases invasion efficiency is taken as additional evidence of the importance of lysosomal calcium release. It is not clear how these observations support involvement of lysosomal calcium release and exocytosis; in fact treatment with the ionophore should itself have induced lysosomal exocytosis and stimulated, rather than inhibited invasion. Yet, manipulations that increase and others that decrease cytosolic calcium both inhibited invasion.
The proposed role of NAADP is based on the effects of "knocking out" TPC1 and on the pharmacological effects of Ned-19. It is noteworthy that TPC2, rather than TPC1, is generally believed to be the primary TPC isoform of lysosomes. Moreover, the gene ablation accomplished in the TPC1 "knockouts" is only partial and rather unsatisfactory. Definitive conclusions about the role of TPC1 can only be reached with proper, full knockouts. Even the pharmacological approach is unconvincing because the high doses of Ned-19 used should have blocked both TPC isoforms and presumably precluded invasion. Instead, invasion is reduced by only ≈50%. A much greater inhibition was reported using 9-phenantrol, the blocker of plasmalemmal calcium channels. How is the selective involvement of lysosomal TPC1 channels justified?
Invoking an elevation of NAADP as the mediator of calcium release requires measurements of the changes in NAADP concentration in response to the bacteria. This was not performed. Instead, the authors analyzed the possible contribution of putative NAADP-generating systems and reported that the most active of these, CD38, was without effect, while the elimination of SARM1, another potential source of NAADP, had a very modest (≈20%) inhibitory effect that may have been due to clonal variation, which was not ruled out. In view of these data, the conclusion that NAADP is involved in the invasion process seems unwarranted.
The involvement of lysosomal secretion is, again, predicated largely on the basis of pharmacological evidence. No direct evidence is provided for the insertion of lysosomal components into the plasma membrane, or for the release of lysosomal contents to the medium. Instead, inhibition of lysosomal exocytosis by vacuolin-1 is the sole source of evidence. However, vacuolin-1 is by no means a specific inhibitor of lysosomal secretion: it is now known to act primarily as a PIKfyve inhibitor and to cause massive distortion of the endocytic compartment, including gross swelling of endolysosomes. The modest (20-25%) inhibition observed when using synaptotagmin 7 knockout cells is similarly not convincing proof of the requirement for lysosomal secretion.
ASM is proposed to play a central role in the rapid invasion process. As above, most of the evidence offered in this regard is pharmacological and often inconsistent between inhibitors or among cell types. Some drugs affect some of the cells, but not others. It is difficult to reach general conclusions regarding the role of ASM. The argument is made even more complex by the authors' use of exogenous sphingomyelinase (beta-toxin). Pretreatment with the toxin decreased invasion efficiency, a seemingly paradoxical result. Incidentally, the effectiveness of the added toxin is never quantified/validated by directly measuring the generation of ceramide or the disappearance of SM.
The use of fluorescent analogs of sphingomyelin and ceramide is not well justified and it is unclear what conclusions can be derived from these observations. Despite the low resolution of the images provided, it appears as if the labeled lipids are largely in endomembrane compartments, where they would presumably be inaccessible to the secreted ASM. Moreover, considering the location of the BODIPY probe, the authors would be unable to distinguish intact sphingomyelin from its breakdown product, ceramide. What can be concluded from these experiments? Incidentally, the authors report only 10% of BODIPY-positive events after 10 min. What are the implications of this finding? That 90% of the invasion events are unrelated to sphingomyelin, ASM, and ceramide?
It is also unclear how the authors can distinguish lysenin entry into ruptured vacuoles from the entry of RFP-CWT, used as a criterion of bacterial escape. Surely the molecular weights of the probes are not sufficiently different to prevent the latter one from traversing the permeabilized membrane until such time that the bacteria escape from the vacuole.
Both SMase inhibitors (Figure 4C) and SMase pretreatment increased bacterial escape from the vacuole. The former should prevent SM hydrolysis and formation of ceramide, while the latter treatment should have the exact opposite effects, yet the end result is the same. What can one conclude regarding the need and role of the SMase products in the escape process?
-
Reviewer #2 (Public review):
Summary:
In this manuscript, Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry.
The evidence provided is solid, methods used are appropriate and results largely support their conclusions, but can be substantiated further as detailed below. The weakness is a reliance on chemical inhibitors that can be non-specific to delineate critical steps.
Specific comments:
A large number of experiments rely on treatment with chemical inhibitors. While this approach is reasonable, many of the inhibitors employed such as amitriptyline and vacuolin1 have other or non-defined cellular targets and pleiotropic effects cannot be ruled …
Reviewer #2 (Public review):
Summary:
In this manuscript, Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry.
The evidence provided is solid, methods used are appropriate and results largely support their conclusions, but can be substantiated further as detailed below. The weakness is a reliance on chemical inhibitors that can be non-specific to delineate critical steps.
Specific comments:
A large number of experiments rely on treatment with chemical inhibitors. While this approach is reasonable, many of the inhibitors employed such as amitriptyline and vacuolin1 have other or non-defined cellular targets and pleiotropic effects cannot be ruled out. Given the centrality of ASM for the manuscript, it will be important to replicate some key results with ASM KO cells.
Most experiments are done in HeLa cells. Given the pathway is projected as generic, it will be important to further characterize cell type specificity for the process. Some evidence for a similar mechanism in other cell types S. aureus infects, perhaps phagocytic cell type, might be good.
I'm a little confused about the role of ASM on the surface. Presumably, it converts SM to ceramide, as the final model suggests. Overexpression of b-toxin results in the near complete absence of SM on phagosomes (having representative images will help appreciate this), but why is phagosomal SM detected at high levels in untreated conditions? If bacteria are engulfed by SM-containing membrane compartments, what role does ASM play on the surface? If surface SM is necessary for phagosomal escape within the cell, do the authors imply that ASM is tuning the surface SM levels to a certain optimal range? Alternatively, can there be additional roles for ASM on the cell surface? Can surface SM levels be visualized (for example, in Figure 4 E, F)?
Related to that, why is ASM activity on the cell surface important? Its role in non-infectious or other contexts can be discussed.
If SM removal is so crucial for uptake, can exocytosis of lysosomes alone provide sufficient ASM for SM removal? How much or to what extent is lysosomal exocytosis enhanced by initial signaling events? Do the authors envisage the early events in their model happening in localized confines of the PM, this can be discussed.
How are inhibitor doses determined? How efficient is the removal of extracellular bacteria at 10 min? It will be good to substantiate the cfu experiments for infectivity with imaging-based methods. Are the roles of TPC1 and TPC2 redundant? If so, why does silencing TPC1 alone result in a decrease in infectivity? For these and other assays, it would be better to show raw values for infectivity. Please show alterations in lysosomal Ca2+ at the doses of inhibitors indicated. Is lysosomal Ca2+ released upon S. aureus binding to the cell surface? Will be good to directly visualize this.
The precise identification of cytosolic vs phagosomal bacteria is not very easy to appreciate. The methods section indicates how this distinction is made, but how do the authors deal with partial overlaps and ambiguities generally associated with such analyses? Please show respective images. The number of events (individual bacteria) for the live cell imaging data should be clearly mentioned.
In the phagosome maturation experiments, what is the proportion of bacteria in Rab5 or Rab7 compartments at each time point? Will the decreased Rab7 association be accompanied by increased Rab5? Showing raw values and images will help appreciate such differences. Given the expertise and tools available in live cell imaging, can the authors trace Rab5 and Rab7 positive compartment times for the same bacteria?
The results with longer-term infection are interesting. Live cell imaging suggests that ASM-inhibited cells show accelerated phagosomal escape that reduces by 6 hpi. Where are the bacteria at this time point ? Presumably, they should have reached lysosomes. The relationship between cytosolic escape, replication, and host cell death is interesting, but the evidence, as presented is correlative for the populations. Given the use of live cell imaging, can the authors show these events in the same cell?
Given the inherent heterogeneity in uptake processes and the use of inhibitors in most experiments, the distinction between ASM-dependent and independent pathways might not be as clear-cut as the authors suggest. Some caution here will be good. Can the authors estimate what fraction of intracellular bacteria are taken up ASM-dependent?
-
Author response:
Reviewer #1 (Public review):
Summary:
The manuscript by Rühling et al analyzes the mode of entry of S. aureus into mammalian cells in culture. The authors propose a novel mechanism of rapid entry that involves the release of calcium from lysosomes via NAADP-stimulated activation of TPC1, which in turn causes lysosomal exocytosis; exocytic release of lysosomal acid sphingomyelinase (ASM) is then envisaged to convert exofacial sphingomyelin to ceramide. These events not only induce the rapid entry of the bacteria into the host cells but are also described to alter the fate of the intracellular S. aureus, facilitating escape from the endocytic vacuole to the cytosol.
Strengths:
The proposed mechanism is novel and could have important biological consequences.
Weaknesses:
Unfortunately, the evidence provided is unconvincing …
Author response:
Reviewer #1 (Public review):
Summary:
The manuscript by Rühling et al analyzes the mode of entry of S. aureus into mammalian cells in culture. The authors propose a novel mechanism of rapid entry that involves the release of calcium from lysosomes via NAADP-stimulated activation of TPC1, which in turn causes lysosomal exocytosis; exocytic release of lysosomal acid sphingomyelinase (ASM) is then envisaged to convert exofacial sphingomyelin to ceramide. These events not only induce the rapid entry of the bacteria into the host cells but are also described to alter the fate of the intracellular S. aureus, facilitating escape from the endocytic vacuole to the cytosol.
Strengths:
The proposed mechanism is novel and could have important biological consequences.
Weaknesses:
Unfortunately, the evidence provided is unconvincing and insufficient to document the multiple, complex steps suggested. In fact, there appear to be numerous internal inconsistencies that detract from the validity of the conclusions, which were reached mostly based on the use of pharmacological agents of imperfect specificity.
We thank the reviewer for the detailed evaluation of our manuscript. We will address the criticism below.
We agree with the reviewer that many of the experiments presented in our study rely on the usage of inhibitors. However, we want to emphasize that the main conclusion (invasion pathway affects the intracellular fate/phagosomal escape) was demonstrated without the use of inhibitors or genetic ablation in two key experiments (Figure4 G/H). These experiments were in line with the results we obtained with inhibitors (amitriptyline [Supp. Figure 4E], ARC39, PCK310, [Figure 4c] and Vacuolin-1 [Supp. Figure4f]). Importantly, the hypothesis was also supported by another key experiment, in which we showed the intracellular fate of bacteria is affected by removal of SM from the plasma membrane before invasion, but not by removal of SM from phagosomal membranes after bacteria internalization (Figure4d-f). Taken together, we thus believe that the main hypothesis is strongly supported by our data.
Moreover, we either used different inhibitors for the same molecule (ASM was inhibited by ARC39, amitriptyline and PCK310 with similar outcome) or supported our hypothesis with gene-ablated cell pools (TPC1, Syt7, SARM1), as we will point out in more detail below.
Firstly, the release of calcium from lysosomes is not demonstrated. Localized changes in the immediate vicinity of lysosomes need to be measured to ascertain that these organelles are the source of cytosolic calcium changes. In fact, 9-phenantrol, which the authors find to be the most potent inhibitor of invasion and hence of the putative calcium changes, is not a blocker of lysosomal calcium release but instead blocks plasmalemmal TRPM4 channels. On the other hand, invasion is seemingly independent of external calcium. These findings are inconsistent with each other and point to non-specific effects of 9-phenantrol. The fact that ionomycin decreases invasion efficiency is taken as additional evidence of the importance of lysosomal calcium release. It is not clear how these observations support involvement of lysosomal calcium release and exocytosis; in fact treatment with the ionophore should itself have induced lysosomal exocytosis and stimulated, rather than inhibited invasion. Yet, manipulations that increase and others that decrease cytosolic calcium both inhibited invasion.
With respect to lysosomal Ca2+ release, we agree with the reviewer that direct visual demonstration of lysosomal Ca2+ release upon infection will improve the manuscript. We therefore will perform additional experimentation to show alterations of Ca2+ at the lysosomes during infection.
As to the TRPM4 involvement in S. aureus host cell internalization, it has been reported that TRPM4 is activated by cytosolic Ca2+. However, the channel conducts monovalent cations such as K+ or Na+ but is impermeable for Ca2+ 1, 2. The following of our observations are supporting this:
i) S. aureus invasion is dependent on intracellular Ca2+, but is independent from extracellular Ca2+ (Figure 1c).
ii) 9-phenantrol treatment reduces S. aureus internalization by host cells, illustrating the dependence of this process on TRPM4 (Figure 1b). We therefore hypothesize that TRPM4 is activated by Ca2+ released from lysosomes (see above).
TRPM4 is localized to focal adhesions and is connected to actin cytoskeleton3, 4 – a requisite of host cell entry of S. aureus.5, 6 This speaks for an important function of TRPM4 in uptake of S. aureus in general, but does not necessarily have to be involved exclusively in the rapid uptake pathway.
TRPM4 itself is not permeable for Ca2+ but is activated by the cation. Thus, it is unlikely to cause lysosomal exocytosis. The stronger bacterial uptake reduction by treatment with 9-phenantrol when compared to Ned19 thus may be caused by the involvement of TRPM4 in additional pathways of S. aureus host cell entry involving that association of TRPM4 with focal adhesions or, as pointed out by the reviewer, unspecific side effects of 9-phenantrol that we currently cannot exclude. We will include this information in the revised manuscript.
Regarding the reduced S. aureus invasion after ionomycin treatment, we agree with the reviewer that ionomycin is known to lead to lysosomal exocytosis as was previously shown by others7 as well as our laboratory8.
We hypothesized that pretreatment with ionomycin would trigger lysosomal exocytosis and thus would reduce the pool of lysosomes that can undergo exocytosis before host cells are contacted by S. aureus. As a result, we should observe a marked reduction of S. aureus internalization in such “lysosome-depleted cells”, if the lysosomal exocytosis is coupled to bacterial uptake. Our observation of reduced bacterial internalization after ionomycin treatment supports this hypothesis.
However, ionomycin treatment and S. aureus infection of host cells are distinct processes.
While ionomycin results in strong global and non-directional lysosomal exocytosis of all “releasable” lysosomes (~5-10 % of all lysosomes according to previous observations)7, we hypothesize that lysosomal exocytosis upon contact with S. aureus only involves a very small proportion of lysosomes at host-bacteria contact sites.
Since ionomycin disturbs the overall cellular Ca2+ homeostasis, we agree with the reviewer that this does not directly show lysosomal Ca2+ liberation. We will discuss this in more detail in the revised manuscript.
The proposed role of NAADP is based on the effects of "knocking out" TPC1 and on the pharmacological effects of Ned-19. It is noteworthy that TPC2, rather than TPC1, is generally believed to be the primary TPC isoform of lysosomes. Moreover, the gene ablation accomplished in the TPC1 "knockouts" is only partial and rather unsatisfactory. Definitive conclusions about the role of TPC1 can only be reached with proper, full knockouts. Even the pharmacological approach is unconvincing because the high doses of Ned-19 used should have blocked both TPC isoforms and presumably precluded invasion. Instead, invasion is reduced by only ≈50%. A much greater inhibition was reported using 9-phenantrol, the blocker of plasmalemmal calcium channels. How is the selective involvement of lysosomal TPC1 channels justified?
As to partial gene ablation of TPC1: To avoid clonal variances, we usually perform pool sorting to obtain a cell population that predominantly contains cells -here- deficient in TPC1, but also a small proportion of wildtype cells as seen by the residual TPC1 protein on the Western blot. We observe a significant reduction of bacterial uptake in this cell pool suggesting that the uptake reduction in a pure K.O. population may be even larger.
As to the inhibition by Ned19: We agree with the reviewer that Ned19 inhibits TPC1 and TPC2. Since ablation of TPC1 reduced invasion of S. aureus, we concluded that TPC1 is important for S. aureus host cell invasion. We thus agree with the reviewer that a role for TPC2 cannot be excluded. We will clarify this in the reviewed manuscript. It needs to be noted, however, that deficiency in either TPC1 or TPC2 alone was sufficient to prevent Ebola virus infection9, which is in line with our observations.
The 50% reduction of invasion upon Ned19 treatment (Figure 1d) is comparable with the reduction caused by other compounds that influence the ASM-dependent pathway (such as amitriptyline, ARC39 [Figure 2c], BAPTA-AM [Figure 1c], Vacuolin-1 [Figure 2a], β-toxin [Figure 2e] and ionomycin [Figure 1a]). Further, the partial reduction of invasion is most likely due to the concurrent activity of multiple internalization pathways which are not all targeted by the used compounds.
Invoking an elevation of NAADP as the mediator of calcium release requires measurements of the changes in NAADP concentration in response to the bacteria. This was not performed. Instead, the authors analyzed the possible contribution of putative NAADP-generating systems and reported that the most active of these, CD38, was without effect, while the elimination of SARM1, another potential source of NAADP, had a very modest (≈20%) inhibitory effect that may have been due to clonal variation, which was not ruled out. In view of these data, the conclusion that NAADP is involved in the invasion process seems unwarranted.
Our results from two independent experimental set-ups (Ned19 [Figure 1d] and TPC1 K.O. [Figure 1e & Figure 2f]) indicate the involvement of NAADP in the process. However, the measurement of NAADP concentration is non-trivial. However, we can rule out clonal variation in the SARM1 mutant since experiments were conducted with a cell pool as described above in order to avoid clonal variation of single clones.
The mechanism behind biosynthesis of NAADP is still debated. CD38 was the first enzyme discovered to possess the ability of producing NAADP. However, it requires acidic pH to produce NAADP10 -which does not match the characteristics of a cytosolic NAADP producer. HeLa cells do not express CD38 and hence, it is not surprising that inhibition of CD38 had no effect on S. aureus invasion in HeLa cells. However, NAADP production by HeLa cells was observed in absence of CD3811. Thus CD38-independent NAADP generation is likely. SARM1 can produce NAADP at neutral pH12 and is expressed in HeLa, thus providing a more promising candidate.
We agree with the reviewer that the reduction of S. aureus internalization after ablation of SARM1 is less pronounced than in other experiments of ours. This may be explained by NAADP originating from other enzymes, such as the recently discovered DUOX1, DUOX2, NOX1 and NOX213, which – with exception of DUOX2- possess a low expression even in HeLa cells. We will discuss this in the revised manuscript.
The involvement of lysosomal secretion is, again, predicated largely on the basis of pharmacological evidence. No direct evidence is provided for the insertion of lysosomal components into the plasma membrane, or for the release of lysosomal contents to the medium. Instead, inhibition of lysosomal exocytosis by vacuolin-1 is the sole source of evidence. However, vacuolin-1 is by no means a specific inhibitor of lysosomal secretion: it is now known to act primarily as a PIKfyve inhibitor and to cause massive distortion of the endocytic compartment, including gross swelling of endolysosomes. The modest (20-25%) inhibition observed when using synaptotagmin 7 knockout cells is similarly not convincing proof of the requirement for lysosomal secretion.
We agree that the manuscript will strongly benefit from a functional analysis of lysosomal exocytosis. We therefore will conduct assays to investigate exocytosis in the revision. However, we previously showed i) by addition of specific antisera that LAMP1 transiently is exposed on the plasma membrane during ionomycin and pore-forming toxin challenge and ii) demonstrated the release of ASM activity into the culture medium under these conditions.8 Both measurements are not compatible with S. aureus infection, since LAMP1 antibodies also are non-specifically bound by protein A and another IgG-binding protein on the S. aureus surface, which would bias the results. Since protein A also serves as an adhesin, we cannot simply delete the ORF without changing other aspects of staphylococcal virulence. Further, FBS contains a ASM background activity that impedes activity measurements of cell culture medium. We previously removed this background activity by a specific heat-inactivation protocol.8 However, S. aureus invasion is strongly reduced in culture medium containing this heat-inactivated FBS.
We agree with the reviewer that Vacuolin-1 has unspecific side effects. We will address this in the revised version of the manuscript.
As to the involvement of synaptotagmin 7:
Synaptotagmin 7 is not the only protein possibly involved in Ca-dependent exocytosis. For instance, SYT1 has been shown to possess an overlapping function.14 This may explain the discrepancy between our vacuolin-1 and SYT7 ablation experiments. We will add an according section to the discussion.
ASM is proposed to play a central role in the rapid invasion process. As above, most of the evidence offered in this regard is pharmacological and often inconsistent between inhibitors or among cell types. Some drugs affect some of the cells, but not others. It is difficult to reach general conclusions regarding the role of ASM. The argument is made even more complex by the authors' use of exogenous sphingomyelinase (beta-toxin). Pretreatment with the toxin decreased invasion efficiency, a seemingly paradoxical result. Incidentally, the effectiveness of the added toxin is never quantified/validated by directly measuring the generation of ceramide or the disappearance of SM.
Although pharmacological inhibitors can have unspecific side effects, we want to emphasize that the inhibitors used in our study act on the enzyme ASM by completely different mechanisms. Amitriptyline is a so called functional inhibitor of ASM (FIASMA) which induces the detachment of ASM from lysosomal membranes resulting in degradation of the enzyme.15 By contrast, ARC39 is a competitive inhibitor.16, 17
We do not see inconsistencies in our data obtained with ASM inhibitors. Amitriptyline and ARC39 both reduce the invasion of S. aureus in HuLEC, HuVEC and HeLa cells (Figure 2c). ARC39 needs a longer pre-incubation, since its uptake by host cells is slower (data not shown). We observe a different outcome in 16HBE14o- and Ea.Hy 926 cells, with 16HBE14o- even demonstrating a slightly increased invasion of S. aureus upon ARC39 treatment. Amitriptyline had no effect (Figure 2c). Moreover, both inhibitors affected the invasion dynamics (Figure 3d), phagosomal escape (Figure 4c and Supp. Figure 4e) and Rab7 recruitment (Figure 4a and Supp. Figure 4b) in a similar fashion. Proper inhibition of ASM by both compounds in all cell lines used was validated by enzyme assays (Supp. Figure 2e), which suggests that the ASM-dependent pathway does only exist in specific cell lines. This also may serve as an argument that we here do not observe unspecific side effects of the compounds. We will clarify this in the revised manuscript.
ASM is a key player for SM degradation and recycling. In clinical context, deficiency in ASM results in the so-called Niemann Pick disease type A/B. The lipid profile of ASM-deficient cells is massively altered18, which will result in severe side effects. Short-term inhibition by small molecules therefore poses a clear benefit when compared to the usage of ASM K.O. cells.
As to the treatment with a bacterial sphingomyelinase:
Treatment with the bacterial SMase (bSMase, here: β-toxin) was performed in two different ways:
i) Pretreatment of host cells with β-toxin to remove SM from the host cell surface before infection. This removes the substrate of ASM from the cell surface prior to addition of the bacteria (Figure 2e, Figure 4d-f). Since SM is not present on the extracellular plasma membrane leaflet after treatment, a release of ASM cannot cause localized ceramide formation at the sites of lysosomal exocytosis. Similar observations were made by others.19
ii) Addition of bSMase to host cells together with the bacteria to complement for the absence of ASM (Figure 2f).
Removal of the ASM substrate before infection (i) prevents localized ASM-mediated conversion of SM to Cer during infection and resulted in a decreased invasion, while addition of the SMase during infection resulted in an increased invasion in TPC1 and SYT7 ablated cells. Thus, both experiments are consistent with each other and in line with our other observations.
Removal of SM from the plasma membrane by β-toxin was indirectly demonstrated by the absence of Lysenin recruitment to phagosomes/escaped bacteria when host cells were pretreatment with the toxin before infection (Figure4F). In another publication, we recently quantified the effectiveness of β-toxin treatment, even though with slightly longer treatment times (75 min vs. 3h).20 We will repeat the measurements also for shorter treatment times.
To clarify our experimental approaches to the readership we will add an explanatory section to the revised manuscript.
As to the general conclusions regarding the role of ASM: ASM and lysosomal exocytosis has been shown to be involved in uptake of a variety of pathogens19, 21-25 supporting its role in the process.
The use of fluorescent analogs of sphingomyelin and ceramide is not well justified and it is unclear what conclusions can be derived from these observations. Despite the low resolution of the images provided, it appears as if the labeled lipids are largely in endomembrane compartments, where they would presumably be inaccessible to the secreted ASM. Moreover, considering the location of the BODIPY probe, the authors would be unable to distinguish intact sphingomyelin from its breakdown product, ceramide. What can be concluded from these experiments? Incidentally, the authors report only 10% of BODIPY-positive events after 10 min. What are the implications of this finding? That 90% of the invasion events are unrelated to sphingomyelin, ASM, and ceramide?
During the experiments with fluorescent SM analogues (Figure 3a,b), S. aureus was added to the samples immediately before start of video recording. Hence, bacteria are slowly trickling onto the host cells and we thus can image the initial contact between them and the bacteria, for instance, the bacteria depicted in Figure 3a contact the host cell about 9 min before becoming BODIPY-FL-positive (see Supp. Video 1, 55 min). Hence, we think that in these cases we see the formation of phagosomes around bacteria rather than bacteria in endomembrane compartments. Since generation of phagosomes happens at the plasma membrane, SM is accessible to secreted ASM.
The “trickling” approach for infection is an experimental difference to our invasion measurements, in which we synchronized the infection by a very slow centrifugation. This ensures that all bacteria have contact to host cells and are not just floating in the culture medium. However, live cell imaging of initial bacterial-host contact and synchronization of infection is technically not combinable.
In our invasion measurements -with synchronization-, we typically see internalization of ~20% of all added bacteria after 30 min. Hence, most bacteria that are visible in our videos likely are still extracellular and only a small proportion was internalized. This explains why only 10% of total bacteria are positive for BODIPY-FL-SM after 10 min. The proportion of internalized bacteria that are positive for BODIPY-FL-SM should be way higher but cannot be determined with this method.
We agree with the reviewer that we cannot observe conversion of BODIPY-FL-SM by ASM. In order to do that, we attempted to visualize the conversion of a visible-range SM FRET probe (Supp. Figure 3), but the structure of the probe is not compatible with measurement of conversion on the plasma membrane, since the FITC fluorophore released into the culture medium by the ASM activity thereby gets lost for imaging. In general, the visualization of SM conversion with subcellular resolution is challenging and even with novel tools developed in our lab26 visualization of SM on the plasma membrane is difficult.
The conclusion we draw from these experiments are that i.) S. aureus invasion is associated with SM and ii.) SM-associated invasion can be very fast, since bacteria are rapidly engulfed by BODIPY-FL-SM containing membranes.
It is also unclear how the authors can distinguish lysenin entry into ruptured vacuoles from the entry of RFP-CWT, used as a criterion of bacterial escape. Surely the molecular weights of the probes are not sufficiently different to prevent the latter one from traversing the permeabilized membrane until such time that the bacteria escape from the vacuole.
We here want to clarify that both, the Lysenin as well as the CWT reporter have access to rupture vacuoles (Figure 4b). We used the Lysenin reporter in these experiments for estimation of SM content of phagosomal membranes. If a vacuole is ruptured, both the bacteria and the luminal leaflet of the phagosomal membrane remnants get in contact with the cytosol and hence with the cytosolically expressed reporters YFP-Lysenin as well as RFP-CWT resulting in “Lysenin-positive escape” when phagosomes contained SM (see Figure 4f). By contrast, either β-toxin expression by S. aureus or pre-treatment with the bSMase resulted in absence of Lysenin recruitment suggesting that the phagosomal SM levels were decreased/undetectable (Figure 4f, Supp Figure 5f, g, i, j).
This approach does not enable a quantitative measurement of phagosomal SM and rather gives a “yes or no” answer. However, we think this method is sufficient to show that β-toxin expression and pretreatment markedly decreased phagosomal SM levels in the host cells.
The approach we used here to analyze “Lysenin-positive escape” can clearly be distinguished from Lysenin-based methods that were used by others.27 There Lysenin was used to show trans-bilayer movement of SM before rupture of bacteria-containing phagosomes.
To clarify the function of Lysenin in our approach we will add an additional figure to the revised manuscript.
Both SMase inhibitors (Figure 4C) and SMase pretreatment increased bacterial escape from the vacuole. The former should prevent SM hydrolysis and formation of ceramide, while the latter treatment should have the exact opposite effects, yet the end result is the same. What can one conclude regarding the need and role of the SMase products in the escape process?
As pointed out above, pretreatment of host cells with SMase removes SM from the plasma membrane and hence, ASM does not have access to its substrate. Hence, both treatment with either ASM inhibitors or pretreatment with bacterial SMase prevent ASM from being active on the plasma membrane and hence block the ASM-dependent uptake (Figure 2 c, e). Although overall less bacteria were internalized by host cells under these conditions, the bacteria that invaded host cells did so in an ASM-independent manner.
Since blockage of the ASM-dependent internalization pathway (with ASM inhibitor [Figure 4c], SMase pretreatment [Figure 4e] and Vacuolin-1[Supp. Fig.4f]) always resulted in enhanced phagosomal escape, we conclude that bacteria that were internalized in an ASM-independent fashion cause enhanced escape. Vice versa, bacteria that enter host cells in an ASM-dependent manner demonstrate lower escape rates.
This is supported by comparing the escape rates of “early” and “late” invaders [Figure 4g/h], which in our opinion is a key experiment that supports this hypothesis. The “early” invaders are predominantly ASM-dependent (see e.g. Figure 3e) and thus, bacteria that entered host cell in the first 10 min of infection should have been internalized predominantly in an ASM-dependent fashion, while slower entry pathways are active later during infection. The early ASM dependent invaders possessed lower escape rates, which is in line with the data obtained with inhibitors (e.g. Figure 4c and Supp. Fig. 4f).
We hypothesize that the activity of ASM on the plasma membrane during invasion mediates the recruitment of a specific subset of receptors, which then influence downstream phagosomal maturation and escape. This hypothesis is supported by the fact that the subset of receptors interacting with S. aureus is altered upon inhibition of the ASM-dependent uptake pathway. We describe this in another study that is currently under evaluation elsewhere.
Reviewer #2 (Public review):
Summary:
In this manuscript, Ruhling et al propose a rapid uptake pathway that is dependent on lysosomal exocytosis, lysosomal Ca2+ and acid sphingomyelinase, and further suggest that the intracellular trafficking and fate of the pathogen is dictated by the mode of entry.
The evidence provided is solid, methods used are appropriate and results largely support their conclusions, but can be substantiated further as detailed below. The weakness is a reliance on chemical inhibitors that can be non-specific to delineate critical steps.
Specific comments:
A large number of experiments rely on treatment with chemical inhibitors. While this approach is reasonable, many of the inhibitors employed such as amitriptyline and vacuolin1 have other or non-defined cellular targets and pleiotropic effects cannot be ruled out. Given the centrality of ASM for the manuscript, it will be important to replicate some key results with ASM KO cells.
We thank the reviewer for the critical evaluation of our manuscript and plenty of constructive comments.
We agree with the reviewer, that ASM inhibitors such as functional inhibitors of ASM (FIASMA) like amitriptyline used in our study have unspecific side effects given their mode-of-action. FIASMAs induce the detachment of ASM from lysosomal membranes resulting in degradation of the enzyme.15 However, we want to emphasize that we also used the competitive inhibitor ARC39 in our study16, 17 which acts on the enzyme by a completely different mechanism. All phenotypes (reduced invasion [Figure 2c, d], effect on invasion dynamics [Figure 3d], enhanced escape [Figure 4c and Supp Figure 4e] and differential recruitment of Rab7 [Supp. Figure 4b]) were observed with both inhibitors thereby supporting the role of ASM in the process.
We further agree that experiments with genetic evidence usually support and improve scientific findings. However, ASM is a cellular key player for SM degradation and recycling. In a clinical context, deficiency in ASM results in a so-called Niemann Pick disease type A/B. The lipid profile of ASM-deficient cells is massively altered18, which in itself will result in severe side effects. Thus, the usage of inhibitors provides a clear benefit when compared to ASM K.O. cells, since ASM activity can be targeted in a short-term fashion thereby preventing larger alterations in cellular lipid composition.
Most experiments are done in HeLa cells. Given the pathway is projected as generic, it will be important to further characterize cell type specificity for the process. Some evidence for a similar mechanism in other cell types S. aureus infects, perhaps phagocytic cell type, might be good.
Whenever possible we performed the experiments not only in HeLa but also in HuLECs. For example, we refer to experiments concerning the role of Ca2+ (Figure 1c/Supp.Figure1e), lysosomal Ca2+/Ned19 (Figure1d/Supp Figure 1g), lysosomal exocytosis/Vacuolin-1 (Figure 2a/Supp. Figure2a), ASM/ARC39 and amitriptyline (Figure 2c), surface SM/β-toxin (Figure 2e/Supp. Figure 2g), analysis of invasion dynamics (complete Figure 3) and measurement of cell death during infection (Figure 5c-e, Supp. Figure 6a+b).
HuLECs, however, are not really genetically amenable and hence we were not able to generate gene deletions in these cells and upon introduction of the fluorescence escape reporter the cells are not readily growing.
As to ASM involvement in phagocytic cells: a role for ASM during the uptake of S. aureus by macrophages was previously reported by others.23 However, in professional phagocytes S. aureus does not escape from the phagosome and replicates within the vacuole.28
I'm a little confused about the role of ASM on the surface. Presumably, it converts SM to ceramide, as the final model suggests. Overexpression of b-toxin results in the near complete absence of SM on phagosomes (having representative images will help appreciate this), but why is phagosomal SM detected at high levels in untreated conditions? If bacteria are engulfed by SM-containing membrane compartments, what role does ASM play on the surface? If surface SM is necessary for phagosomal escape within the cell, do the authors imply that ASM is tuning the surface SM levels to a certain optimal range? Alternatively, can there be additional roles for ASM on the cell surface? Can surface SM levels be visualized (for example, in Figure 4 E, F)?
We initially hypothesized that we would detect higher phagosomal SM levels upon inhibition of ASM, since our model suggests SM cleavage by ASM on the host cell surface during bacterial cell entry. However, we did not detect any changes in our experiments (Supp. Figure 4d). We currently favor the following explanation: SM is the most abundant sphingolipid in human cells.29 If peripheral lysosomes are exocytosed and thereby release ASM, only a localized and relative small proportion of SM may get converted to Cer, which most likely is below our detection limit. In addition, the detection of cytosolically exposed phagosomal SM by YFP-Lysenin is not quantitative and provides a “Yes or No” measurement. Hence, we think that the rather limited SM to Cer conversion in combination with the high abundance of SM in cellular membranes does not visibly affect the recruitment of the Lysenin reporter.
In our experiments that employ BODIPY-FL-SM (Figure 3a+b), we cannot distinguish between native SM and downstream metabolites such as Cer. Hence, again we cannot make any assumptions on the extent to which SM is converted on the surface during bacterial internalization. Although our laboratory recently used trifunctional sphingolipid analogs to analyze the SM to Cer conversion20, the visualization of this process on the plasma membrane is currently still challenging.
Overall, we hypothesize that the localized generation of Cer on the surface by released ASM leads to generation of Cer-enriched platforms. Subsequently, a certain subset of receptors may be recruited to these platforms and influence the uptake process. These platforms are supposed to be very small, which also would explain that we did not detect changes in Lysenin recruitment.
Related to that, why is ASM activity on the cell surface important? Its role in non-infectious or other contexts can be discussed.
ASM release by lysosomal exocytosis is implied in plasma membrane repair upon injury. We will this discuss this in the revised version of the manuscript.
If SM removal is so crucial for uptake, can exocytosis of lysosomes alone provide sufficient ASM for SM removal? How much or to what extent is lysosomal exocytosis enhanced by initial signaling events? Do the authors envisage the early events in their model happening in localized confines of the PM, this can be discussed.
Ionomycin treatment led to a release of ~10 % of all lysosomes and also increased extracellular ASM activity.7, 8 However, it is currently unclear– to our knowledge -to which extent the released ASM affects surface SM levels. Also, it is unknown which percentage of the lysosomes is released during infection with S. aureus. However, one has to speculate that this will be only a fraction of the “releasable lysosomes” as we assume that the effects (lysosomal Ca2+ liberation, lysosomal exocytosis and ASM activity) are very localized and take place only at host-pathogen contact sites (see also above). In initial experimentation we attempted to visualize the local ASM activity on the cell surface by using a visible range FRET probe (Supp. Fig. 3). Cleavage of the probe by ASM on the surface leads to release of FITC into the cell culture medium which does not contribute a measurable signal at the surface.
How are inhibitor doses determined? How efficient is the removal of extracellular bacteria at 10 min? It will be good to substantiate the cfu experiments for infectivity with imaging-based methods. Are the roles of TPC1 and TPC2 redundant? If so, why does silencing TPC1 alone result in a decrease in infectivity? For these and other assays, it would be better to show raw values for infectivity. Please show alterations in lysosomal Ca2+ at the doses of inhibitors indicated. Is lysosomal Ca2+ released upon S. aureus binding to the cell surface? Will be good to directly visualize this.
Concerning the inhibitor concentrations, we either used values established in published studies or recommendations of the suppliers (e.g. 2-APB, Ned19, Vacuolin-1). For ASM inhibitors, we determined proper inhibition of ASM by activity assays. Concentrations of ionomycin resulting in Ca2+ influx and lysosomal exocytosis was determined in earlier studies of our lab.8, 30
As to the removal of bacteria at 10 min p.i.: Lysostaphin is very efficient for removal of extracellular S. aureus and sterilizes the tissue culture supernatant. It significantly lyses bacteria within a few minutes, as determined by turbidity assays.31
As to imaging-based infectivity assays: We will add an analysis of imaging-based invasion assays in the revised manuscript.
Regarding the roles of TPC1 and TPC2: from our data we cannot conclude whether the roles of TPC1 and TPC2 are redundant. One could speculate that since blockage of TPC1 alone is sufficient to reduce internalization of bacteria, that both channels may have distinct roles. On the other hand, there might be a Ca2+ threshold in order to initiate lysosomal exocytosis that can only be attained if TPC1 and TPC2 are activated in parallel. Thus, our observations are in line with another study that shows reduced Ebola virus infection in absence of either TPC1 or TPC2.32
As to raw CFU counts: whereas the observed effects upon blocking the invasion of S. aureus are stable, the number of internalized bacteria varies between individual biological replicates, for instance, by differences in host cell fitness or growth differences in bacterial cultures, which are prepared freshly for each experiment.
With respect to visualization of lysosomal Ca2+ release: we agree with the reviewer that direct visual demonstration of lysosomal Ca2+ release upon infection will improve the manuscript. We therefore will perform additional experimentation to show alterations of Ca2+ at the lysosomes during infection.
The precise identification of cytosolic vs phagosomal bacteria is not very easy to appreciate. The methods section indicates how this distinction is made, but how do the authors deal with partial overlaps and ambiguities generally associated with such analyses? Please show respective images. The number of events (individual bacteria) for the live cell imaging data should be clearly mentioned.
We apologize for not having sufficiently explained the technology to detect escaped S. aureus. The cytosolic location of S. aureus is indicated by recruitment of RFP-CWT.33 CWT is the cell wall targeting domain of lysostaphin, which efficiently binds to the pentaglycine cross bridge in the peptidoglycan of S. aureus. This reporter is exclusively and homogenously expressed in the host cytosol. Only upon rupture of phagoendosomal membranes the reporter can be recruited to the cell wall of now cytosolically located bacteria. S. aureus mutants, for instance in the agr quorum sensing system, cannot break down the phagosomal membrane in non-professional phagocytes and thus stay unlabeled by the CWT-reporter.33 We will include respective images/movies of escape events and the bacteria numbers for live cell experiments in the revised version of the manuscript.
In the phagosome maturation experiments, what is the proportion of bacteria in Rab5 or Rab7 compartments at each time point? Will the decreased Rab7 association be accompanied by increased Rab5? Showing raw values and images will help appreciate such differences. Given the expertise and tools available in live cell imaging, can the authors trace Rab5 and Rab7 positive compartment times for the same bacteria?
We will include the proportion of Rab7-associated bacteria in the revised manuscript. Usually, we observe that Rab5 is only transiently (for a few minutes) present on phagosomes and only afterwards the phagosomes become positive for Rab7. We do not think that a decrease in Rab7-positive phagosomes would increase the proportion of Rab5-positive phagosomes. However, we cannot exclude this hypothesis with our data.
We can achieve tracing of individual bacteria for recruitment of Rab5/Rab7 only manually, which impedes a quantitative evaluation. However, we will include information that illustrates the consecutive recruitment of the GTPases.
The results with longer-term infection are interesting. Live cell imaging suggests that ASM-inhibited cells show accelerated phagosomal escape that reduces by 6 hpi. Where are the bacteria at this time point ? Presumably, they should have reached lysosomes. The relationship between cytosolic escape, replication, and host cell death is interesting, but the evidence, as presented is correlative for the populations. Given the use of live cell imaging, can the authors show these events in the same cell?
We think that most bacteria-containing phagoendosomes should have fused with lysosomes 6 h p.i. as we have previously shown by acidification to pH of 5 and LAMP1 decoration.34
We will provide images/videos to show the correlation between escape and replication in the revised manuscript.
Given the inherent heterogeneity in uptake processes and the use of inhibitors in most experiments, the distinction between ASM-dependent and independent pathways might not be as clear-cut as the authors suggest. Some caution here will be good. Can the authors estimate what fraction of intracellular bacteria are taken up ASM-dependent?
We agree with the reviewer that an overlap between internalization pathways is likely. A clear distinction is therefore certainly non-trivial. Alternative to ASM-dependent and ASM-independent pathways, the ASM activity may also accelerate one or several internalization pathways. We will address this limitation in the revised manuscript.
Early in infection (~10 min after contact with the cells), the proportion of bacteria that enter host cells ASM-dependently is relatively high amounting to roughly 75% in HuLEC. After 30 min, this proportion is decreasing to about 50%. We will include this information in the revised version of the manuscript.
References
(1) Launay, P. et al. TRPM4 Is a Ca2+-Activated Nonselective Cation Channel Mediating Cell Membrane Depolarization. Cell 109, 397-407 (2002).
(2) Nilius, B. et al. The Ca2+‐activated cation channel TRPM4 is regulated by phosphatidylinositol 4,5‐biphosphate. The EMBO Journal 25, 467-478-478 (2006).
(3) Cáceres, M. et al. TRPM4 Is a Novel Component of the Adhesome Required for Focal Adhesion Disassembly, Migration and Contractility. PLoS One 10, e0130540 (2015).
(4) Silva, I., Brunett, M., Cáceres, M. & Cerda, O. TRPM4 modulates focal adhesion-associated calcium signals and dynamics. Biophysical Journal 123, 390a (2024).
(5) Schlesier, T., Siegmund, A., Rescher, U. & Heilmann, C. Characterization of the Atl-mediated staphylococcal internalization mechanism. International Journal of Medical Microbiology 310, 151463 (2020).
(6) Jevon, M. et al. Mechanisms of Internalization ofStaphylococcus aureus by Cultured Human Osteoblasts. Infection and Immunity 67, 2677-2681 (1999).
(7) Rodriguez, A., Webster, P., Ortego, J. & Andrews, N.W. Lysosomes behave as Ca2+-regulated exocytic vesicles in fibroblasts and epithelial cells. J Cell Biol 137, 93-104 (1997).
(8) Krones & Rühling et al. Staphylococcus aureus alpha-Toxin Induces Acid Sphingomyelinase Release From a Human Endothelial Cell Line. Front Microbiol 12, 694489 (2021).
(9) Sakurai, Y. et al. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347, 995-998 (2015).
(10) Aarhus, R., Graeff, R.M., Dickey, D.M., Walseth, T.F. & Lee, H.C. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J Biol Chem 270, 30327-30333 (1995).
(11) Schmid, F., Fliegert, R., Westphal, T., Bauche, A. & Guse, A.H. Nicotinic acid adenine dinucleotide phosphate (NAADP) degradation by alkaline phosphatase. J Biol Chem 287, 32525-32534 (2012).
(12) Angeletti, C. et al. SARM1 is a multi-functional NAD(P)ase with prominent base exchange activity, all regulated bymultiple physiologically relevant NAD metabolites. iScience 25, 103812 (2022).
(13) Gu, F. et al. Dual NADPH oxidases DUOX1 and DUOX2 synthesize NAADP and are necessary for Ca(2+) signaling during T cell activation. Sci Signal 14, eabe3800 (2021).
(14) Schonn, J.-S., Maximov, A., Lao, Y., Südhof, T.C. & Sørensen, J.B. Synaptotagmin-1 and -7 are functionally overlapping Ca2+ sensors for exocytosis in adrenal chromaffin cells. Proceedings of the National Academy of Sciences 105, 3998-4003 (2008).
(15) Kornhuber, J. et al. Functional Inhibitors of Acid Sphingomyelinase (FIASMAs): a novel pharmacological group of drugs with broad clinical applications. Cell Physiol Biochem 26, 9-20 (2010).
(16) Naser, E. et al. Characterization of the small molecule ARC39, a direct and specific inhibitor of acid sphingomyelinase in vitro. J Lipid Res 61, 896-910 (2020).
(17) Roth, A.G. et al. Potent and selective inhibition of acid sphingomyelinase by bisphosphonates. Angew Chem Int Ed Engl 48, 7560-7563 (2009).
(18) Schuchman, E.H. & Desnick, R.J. Types A and B Niemann-Pick disease. Mol Genet Metab 120, 27-33 (2017).
(19) Miller, M.E., Adhikary, S., Kolokoltsov, A.A. & Davey, R.A. Ebolavirus Requires Acid Sphingomyelinase Activity and Plasma Membrane Sphingomyelin for Infection. Journal of Virology 86, 7473-7483 (2012).
(20) M. Rühling, L.K., F. Wagner, F. Schumacher, D. Wigger, D. A. Helmerich, T. Pfeuffer, R. Elflein, C. Kappe, M. Sauer, C. Arenz, B. Kleuser, T. Rudel, M. Fraunholz, J. Seibel Trifunctional sphingomyelin derivatives enable nanoscale resolution of sphingomyelin turnover in physiological and infection processes via expansion microscopy. Nat Commun accepted in principle (2024).
(21) Peters, S. et al. Neisseria meningitidis Type IV Pili Trigger Ca(2+)-Dependent Lysosomal Trafficking of the Acid Sphingomyelinase To Enhance Surface Ceramide Levels. Infect Immun 87 (2019).
(22) Grassmé, H. et al. Acidic sphingomyelinase mediates entry of N. gonorrhoeae into nonphagocytic cells. Cell 91, 605-615 (1997).
(23) Li, C. et al. Regulation of Staphylococcus aureus Infection of Macrophages by CD44, Reactive Oxygen Species, and Acid Sphingomyelinase. Antioxid Redox Signal 28, 916-934 (2018).
(24) Fernandes, M.C. et al. Trypanosoma cruzi subverts the sphingomyelinase-mediated plasma membrane repair pathway for cell invasion. J Exp Med 208, 909-921 (2011).
(25) Luisoni, S. et al. Co-option of Membrane Wounding Enables Virus Penetration into Cells. Cell Host & Microbe 18, 75-85 (2015).
(26) Rühling, M. et al. Trifunctional sphingomyelin derivatives enable nanoscale resolution of sphingomyelin turnover in physiological and infection processes via expansion microscopy. Nature Communications 15, 7456 (2024).
(27) Ellison, C.J., Kukulski, W., Boyle, K.B., Munro, S. & Randow, F. Transbilayer Movement of Sphingomyelin Precedes Catastrophic Breakage of Enterobacteria-Containing Vacuoles. Curr Biol 30, 2974-2983 e2976 (2020).
(28) Moldovan, A. & Fraunholz, M.J. In or out: Phagosomal escape of Staphylococcus aureus. Cell Microbiol 21, e12997 (2019).
(29) Slotte, J.P. Biological functions of sphingomyelins. Progress in Lipid Research 52, 424-437 (2013).
(30) Stelzner, K. et al. Intracellular Staphylococcus aureus Perturbs the Host Cell Ca(2+) Homeostasis To Promote Cell Death. mBio 11 (2020).
(31) Kunz, T.C. et al. The Expandables: Cracking the Staphylococcal Cell Wall for Expansion Microscopy. Front Cell Infect Microbiol 11, 644750 (2021).
(32) Sakurai, Y. et al. Ebola virus. Two-pore channels control Ebola virus host cell entry and are drug targets for disease treatment. Science 347, 995-998 (2015).
(33) Grosz, M. et al. Cytoplasmic replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin alpha. Cell Microbiol 16, 451-465 (2014).
(34) Giese, B. et al. Staphylococcal alpha-toxin is not sufficient to mediate escape from phagolysosomes in upper-airway epithelial cells. Infect Immun 77, 3611-3625 (2009).
-
