BEHAV3D Tumor Profiler to map heterogeneous cancer cell behavior in the tumor microenvironment
Curation statements for this article:-
Curated by eLife

eLife Assessment
This is a useful tool for code-less analysis of patterns in cell migratory behaviours in vivo using intravital microscopy data and allows correlation with spatial features of the tumour microenvironment. There is a clear need for these tools to make quantitative analysis, comparison and interpretation of complex cell tracking data more accessible and solid evidence is provided of its applicability to tracks generated by both proprietary and open tracking software.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Intravital microscopy (IVM) enables live imaging of animals at single-cell level, offering essential insights into cancer progression. This technique allows for the observation of single-cell behaviors within their natural 3D tissue environments, shedding light on how genetic and microenvironmental changes influence the complex dynamics of tumors. IVM generates highly complex datasets that often exceed the analytical capacity of traditional uni-parametric approaches, which can neglect single-cell heterogeneous in vivo behavior and limit insights into microenvironmental influences on cellular behavior. To overcome these limitations, we present BEHAV3D Tumor Profiler (BEHAV3D-TP), a computational framework that enables unbiased single-cell classification based on a range of morphological, environmental, and dynamic single-cell features. BEHAV3D-TP integrates with widely used 2D and 3D image processing pipelines, enabling researchers without advanced computational expertise to profile cancer and healthy cell dynamics in IVM data from mouse models. Here, we apply BEHAV3D-TP to study diffuse midline glioma (DMG), a highly aggressive pediatric brain tumor characterized by invasive progression. By extending BEHAV3D-TP to incorporate tumor microenvironment (TME) data from IVM or fixed correlative imaging, we demonstrate that distinct migratory behaviors of DMG cells are associated with specific TME components, including tumor-associated macrophages and vasculature. BEHAV3D-TP enhances the accessibility of computational tools for analyzing the complex behaviors of cancer cells and their interactions with the TME in IVM data.
Article activity feed
-

-
-

eLife Assessment
This is a useful tool for code-less analysis of patterns in cell migratory behaviours in vivo using intravital microscopy data and allows correlation with spatial features of the tumour microenvironment. There is a clear need for these tools to make quantitative analysis, comparison and interpretation of complex cell tracking data more accessible and solid evidence is provided of its applicability to tracks generated by both proprietary and open tracking software.
-
Reviewer #1 (Public review):
In this work, Rios-Jimenez and Zomer et al have developed a 'zero-code' accessible computational framework (BEHAV3D-Tumour Profiler) designed to facilitate unbiased analysis of Intravital imaging (IVM) data to investigate tumour cell dynamics (via the tool's central 'heterogeneity module' ) and their interactions with the tumour microenvironment (via the 'large-scale phenotyping' and 'small-scale phenotyping' modules). A key strength is that it is designed as an open-source modular Jupyter Notebook with a user-friendly graphical user interface and can be implemented with Google Colab, facilitating efficient, cloud-based computational analysis at no cost. In addition, demo datasets are available on the authors GitHub repository to aid user training and enhance the usability of the developed pipeline.
To …
Reviewer #1 (Public review):
In this work, Rios-Jimenez and Zomer et al have developed a 'zero-code' accessible computational framework (BEHAV3D-Tumour Profiler) designed to facilitate unbiased analysis of Intravital imaging (IVM) data to investigate tumour cell dynamics (via the tool's central 'heterogeneity module' ) and their interactions with the tumour microenvironment (via the 'large-scale phenotyping' and 'small-scale phenotyping' modules). A key strength is that it is designed as an open-source modular Jupyter Notebook with a user-friendly graphical user interface and can be implemented with Google Colab, facilitating efficient, cloud-based computational analysis at no cost. In addition, demo datasets are available on the authors GitHub repository to aid user training and enhance the usability of the developed pipeline.
To demonstrate the utility of BEHAV3D-TP, they apply the pipeline to timelapse IVM imaging datasets to investigate the in vivo migratory behaviour of fluorescently labelled DMG cells in tumour bearing mice. Using the tool's 'heterogeneity module' they were able to identify distinct single-cell behavioural patterns (based on multiple parameters such as directionality, speed, displacement, distance from tumour edge) which was used to group cells into distinct categories (e.g. retreating, invasive, static, erratic). They next applied the framework's 'large-scale phenotyping' and 'small-scale phenotyping' modules to investigate whether the tumour microenvironment (TME) may influence the distinct migratory behaviours identified. To achieve this, they combine TME visualisation in vivo during IVM (using fluorescent probes to label distinct TME components) or ex vivo after IVM (by large-scale imaging of harvested, immunostained tumours) to correlate different tumour behavioural patterns with the composition of the TME. They conclude that this tool has helped reveal links between TME composition (e.g. degree of vascularisation, presence of tumour-associated macrophages) and the invasiveness and directionality of tumour cells, which would have been challenging to identify when analysing single kinetic parameters in isolation.
While the analysis provides only preliminary evidence in support of the authors conclusions on DMG cell migratory behaviours and their relationship with components of the tumour microenvironment, conclusions are appropriately tempered in the absence of additional experiments and controls.The authors also evaluated the BEHAV3D TP heterogeneity module using available IVM datasets of distinct breast cancer cell lines transplanted in vivo, as well as healthy mammary epithelial cells to test its usability in non-tumour contexts where the migratory phenotypes of cells may be more subtle. This generated data is consistent with that produced during the original studies, as well as providing some additional (albeit preliminary) insights above that previously reported. Collectively, this provides some confidence in BEHAV3D TP's ability to uncover complex, multi-parametric cellular behaviours that may be missed using traditional approaches.
While the tool does not facilitate the extraction of quantitative kinetic cellular parameters (e.g. speed, directionality, persistence and displacement) from intravital images, the authors have developed their tool to facilitate the integration of other data formats generated by open-source Fiji plugins (e.g. TrackMate, MTrackJ, ManualTracking) which will help ensure its accessibility to a broader range of researchers. Overall, this computational framework appears to represent a useful and comparatively user-friendly tool to analyse dynamic multi-parametric data to help identify patterns in cell migratory behaviours, and to assess whether these behaviours might be influenced by neighbouring cells and structures in their microenvironment.
When combined with other methods, it therefore has the potential to be a valuable addition to a researcher's IVM analysis 'tool-box'.
-
Reviewer #2 (Public review):
Summary:
The authors produce a new tool, BEHAV3D to analyse tracking data and to integrate these analyses with large and small scale architectural features of the tissue. This is similar to several other published methods to analyse spatio-temporal data, however, the connection to tissue features is a nice addition, as is the lack of requirement for coding. The tool is then used to analyse tracking data of tumour cells in diffuse midline glioma. They suggest 7 clusters exist within these tracks and that they differ spatially. They ultimately suggest that these behaviours occur in distinct spatial areas as determined by CytoMAP.
Strengths:
The tool appears relatively user-friendly and is open source. The combination with CytoMAP represents a nice option for researchers.
The identification of associations …
Reviewer #2 (Public review):
Summary:
The authors produce a new tool, BEHAV3D to analyse tracking data and to integrate these analyses with large and small scale architectural features of the tissue. This is similar to several other published methods to analyse spatio-temporal data, however, the connection to tissue features is a nice addition, as is the lack of requirement for coding. The tool is then used to analyse tracking data of tumour cells in diffuse midline glioma. They suggest 7 clusters exist within these tracks and that they differ spatially. They ultimately suggest that these behaviours occur in distinct spatial areas as determined by CytoMAP.
Strengths:
The tool appears relatively user-friendly and is open source. The combination with CytoMAP represents a nice option for researchers.
The identification of associations between cell track phenotype and spatial features is exciting and the diffuse midline glioma data nicely demonstrates how this could be used.
-
Reviewer #3 (Public review):
The manuscript by Rios-Jimenez developed a software tool, BEHAV3D Tumor Profiler, to analyze 3D intravital imaging data and identify distinctive tumor cell migratory phenotypes based on the quantified 3D image data. Moreover, the heterogeneity module in this software tool can correlate the different cell migration phenotypes with variable features of the tumor microenvironment. Overall, this is a useful tool for intravital imaging data analysis and its open-source nature makes it accessible to all interested users.
Strengths:
An open-source software tool that can quantify cell migratory dynamics from intravital imaging data and identify distinctive migratory phenotypes that correlate with variable features of the tumor microenvironment.
Weaknesses:
Motility is the main tumor cell feature analyzed in the …
Reviewer #3 (Public review):
The manuscript by Rios-Jimenez developed a software tool, BEHAV3D Tumor Profiler, to analyze 3D intravital imaging data and identify distinctive tumor cell migratory phenotypes based on the quantified 3D image data. Moreover, the heterogeneity module in this software tool can correlate the different cell migration phenotypes with variable features of the tumor microenvironment. Overall, this is a useful tool for intravital imaging data analysis and its open-source nature makes it accessible to all interested users.
Strengths:
An open-source software tool that can quantify cell migratory dynamics from intravital imaging data and identify distinctive migratory phenotypes that correlate with variable features of the tumor microenvironment.
Weaknesses:
Motility is the main tumor cell feature analyzed in the study together with some other tumor-intrinsic features, such as morphology. However, these features are insufficient to characterize and identify the heterogeneity of the tumor cell population that impacts their behaviors in the complex tumor microenvironment (TME). For instance, there are important non-tumor cell types in the TME, and the interaction dynamics of tumor cells with other cell types, e.g., fibroblasts and distinct immune cells, play a crucial role in regulating tumor behaviors. BEHAV3D-TP focuses on analysis of tumor-alone features, and cannot be applied to analyze important cell-cell interaction dynamics in 3D.
-
Author response:
The following is the authors’ response to the current reviews
Reviewer #1 (Public review):
In this work, Rios-Jimenez and Zomer et al have developed a 'zero-code' accessible computational framework (BEHAV3D-Tumour Profiler) designed to facilitate unbiased analysis of Intravital imaging (IVM) data to investigate tumour cell dynamics (via the tool's central 'heterogeneity module' ) and their interactions with the tumour microenvironment (via the 'large-scale phenotyping' and 'small-scale phenotyping' modules). A key strength is that it is designed as an open-source modular Jupyter Notebook with a user-friendly graphical user interface and can be implemented with Google Colab, facilitating efficient, cloud-based computational analysis at no cost. In addition, demo datasets are available on the authors GitHub repository to …
Author response:
The following is the authors’ response to the current reviews
Reviewer #1 (Public review):
In this work, Rios-Jimenez and Zomer et al have developed a 'zero-code' accessible computational framework (BEHAV3D-Tumour Profiler) designed to facilitate unbiased analysis of Intravital imaging (IVM) data to investigate tumour cell dynamics (via the tool's central 'heterogeneity module' ) and their interactions with the tumour microenvironment (via the 'large-scale phenotyping' and 'small-scale phenotyping' modules). A key strength is that it is designed as an open-source modular Jupyter Notebook with a user-friendly graphical user interface and can be implemented with Google Colab, facilitating efficient, cloud-based computational analysis at no cost. In addition, demo datasets are available on the authors GitHub repository to aid user training and enhance the usability of the developed pipeline.
To demonstrate the utility of BEHAV3D-TP, they apply the pipeline to timelapse IVM imaging datasets to investigate the in vivo migratory behaviour of fluorescently labelled DMG cells in tumour bearing mice. Using the tool's 'heterogeneity module' they were able to identify distinct single-cell behavioural patterns (based on multiple parameters such as directionality, speed, displacement, distance from tumour edge) which was used to group cells into distinct categories (e.g. retreating, invasive, static, erratic). They next applied the framework's 'large-scale phenotyping' and 'small-scale phenotyping' modules to investigate whether the tumour microenvironment (TME) may influence the distinct migratory behaviours identified. To achieve this, they combine TME visualisation in vivo during IVM (using fluorescent probes to label distinct TME components) or ex vivo after IVM (by large-scale imaging of harvested, immunostained tumours) to correlate different tumour behavioural patterns with the composition of the TME. They conclude that this tool has helped reveal links between TME composition (e.g. degree of vascularisation, presence of tumour-associated macrophages) and the invasiveness and directionality of tumour cells, which would have been challenging to identify when analysing single kinetic parameters in isolation.
While the analysis provides only preliminary evidence in support of the authors conclusions on DMG cell migratory behaviours and their relationship with components of the tumour microenvironment, conclusions are appropriately tempered in the absence of additional experiments and controls.
The authors also evaluated the BEHAV3D TP heterogeneity module using available IVM datasets of distinct breast cancer cell lines transplanted in vivo, as well as healthy mammary epithelial cells to test its usability in non-tumour contexts where the migratory phenotypes of cells may be more subtle. This generated data is consistent with that produced during the original studies, as well as providing some additional (albeit preliminary) insights above that previously reported. Collectively, this provides some confidence in BEHAV3D TP's ability to uncover complex, multi-parametric cellular behaviours that may be missed using traditional approaches.
While the tool does not facilitate the extraction of quantitative kinetic cellular parameters (e.g. speed, directionality, persistence and displacement) from intravital images, the authors have developed their tool to facilitate the integration of other data formats generated by open-source Fiji plugins (e.g. TrackMate, MTrackJ, ManualTracking) which will help ensure its accessibility to a broader range of researchers. Overall, this computational framework appears to represent a useful and comparatively user-friendly tool to analyse dynamic multi-parametric data to help identify patterns in cell migratory behaviours, and to assess whether these behaviours might be influenced by neighbouring cells and structures in their microenvironment.
When combined with other methods, it therefore has the potential to be a valuable addition to a researcher's IVM analysis 'tool-box'.
We thank the reviewer for carefully considering our manuscript and providing constructive comments. We appreciate the recognition of BEHAV3D-TP’s user-friendliness, modular design, and ability to link cell behavior with the tumor microenvironment. In the future, we plan to extend the tool to incorporate segmentation and tracking modules, once we have approaches that are broadly applicable or allow for personalized model training, further enhancing its utility for the community.
Reviewer #2 (Public review):
Summary:
The authors produce a new tool, BEHAV3D to analyse tracking data and to integrate these analyses with large and small scale architectural features of the tissue. This is similar to several other published methods to analyse spatio-temporal data, however, the connection to tissue features is a nice addition, as is the lack of requirement for coding. The tool is then used to analyse tracking data of tumour cells in diffuse midline glioma. They suggest 7 clusters exist within these tracks and that they differ spatially. They ultimately suggest that these behaviours occur in distinct spatial areas as determined by CytoMAP.
Strengths:
- The tool appears relatively user-friendly and is open source. The combination with CytoMAP represents a nice option for researchers.
- The identification of associations between cell track phenotype and spatial features is exciting and the diffuse midline glioma data nicely demonstrates how this could be used.
We thank the reviewer for their careful reading and thoughtful comments. Feedback from all revision rounds has helped us clarify key points and improve the manuscript, and we are grateful for the positive remarks regarding our application to diffuse midline glioma and the potential of the tool to enable new biological insights.
Reviewer #3 (Public review):
The manuscript by Rios-Jimenez developed a software tool, BEHAV3D Tumor Profiler, to analyze 3D intravital imaging data and identify distinctive tumor cell migratory phenotypes based on the quantified 3D image data. Moreover, the heterogeneity module in this software tool can correlate the different cell migration phenotypes with variable features of the tumor microenvironment. Overall, this is a useful tool for intravital imaging data analysis and its open-source nature makes it accessible to all interested users.
Strengths:
An open-source software tool that can quantify cell migratory dynamics from intravital imaging data and identify distinctive migratory phenotypes that correlate with variable features of the tumor microenvironment.
Weaknesses:
Motility is the main tumor cell feature analyzed in the study together with some other tumor-intrinsic features, such as morphology. However, these features are insufficient to characterize and identify the heterogeneity of the tumor cell population that impacts their behaviors in the complex tumor microenvironment (TME). For instance, there are important non-tumor cell types in the TME, and the interaction dynamics of tumor cells with other cell types, e.g., fibroblasts and distinct immune cells, play a crucial role in regulating tumor behaviors. BEHAV3D-TP focuses on analysis of tumor-alone features, and cannot be applied to analyze important cell-cell interaction dynamics in 3D.
We thank the reviewer for their careful assessment and encouraging remarks regarding BEHAV3D-TP.
Regarding the concern about the tool’s current focus on motility features, we would like to clarify again that BEHAV3D-TP is designed to be highly flexible and extensible. Users can incorporate a wide range of features—including dynamic, morphological, and spatial parameters—into their analyses. In the latest revision, we have make this even more explicit by explaining that the feature selection interface allows users to either (i) directly select them for clustering or (ii) select features for correlation with clusters (See Small scale phenotyping module section in Methods).
Importantly, while our current analysis emphasizes clustering based on dynamic behaviors, Figure 4 demonstrates that these behavioral clusters are associated at the single-cell level with distinct proximities to key TME components, such as TAMMs and blood vessels. These spatial interaction features could also have been included in the clustering itself—creating dynamic-spatial clusters—but we deliberately chose not to do so. This decision was guided by established principles of feature selection: including features with unknown or potentially irrelevant variability can introduce noise and obscure biologically meaningful patterns, ultimately reducing the clarity and interpretability of the resulting clusters. Instead, we adopted a two-step approach—first identifying clusters based on core dynamic features, then examining their relationships with spatial and interaction metrics. This allowed us to reveal meaningful associations of particular cell behavior such as the invading cluster in proximity of TAMMs without overfitting or complicating the clustering model.
To address the reviewer’s point in the latest revision round, we have updated the Small-scale phenotyping module to highlight the possibility of including spatial interaction features with various TME cell types. We also revised the manuscript text and Figure 1 to clarify that these environmental features can be used both upstream as clustering input (Option 1) and for downstream analysis (Option 2), depending on the user’s experimental goals. Attached to this rebuttal letter, we also provide an additional figure illustrating these options in the feature selection panels of the Colab notebook.
In summary, while the clustering presented in this study is based on dynamic parameters, BEHAV3D-TP fully supports the integration of interaction features and other non-motility descriptors. This modularity enables users to customize their analysis pipelines according to specific biological questions, including those involving cell–cell interactions and spatial dynamics within the TME.
The following is the authors’ response to the original reviews.
Reviewer #1 (Public review):
Summary:
Intravital microscopy (IVM) is a powerful tool that facilitates live imaging of individual cells over time in vivo in their native 3D tissue environment. Extracting and analysing multi-parametric data from IVM images however is challenging, particularly for researchers with limited programming and image analysis skills. In this work, RiosJimenez and Zomer et al have developed a 'zero-code' accessible computational framework (BEHAV3D-Tumour Profiler) designed to facilitate unbiased analysis of IVM data to investigate tumour cell dynamics (via the tool's central 'heterogeneity module' ) and their interactions with the tumour microenvironment (via the 'large-scale phenotyping' and 'small-scale phenotyping' modules). It is designed as an open-source modular Jupyter Notebook with a user-friendly graphical user interface and can be implemented with Google Colab, facilitating efficient, cloud-based computational analysis at no cost. Demo datasets are also available on the authors GitHub repository to aid user training and enhance the usability of the developed pipeline.
To demonstrate the utility of BEHAV3D-TP, they apply the pipeline to timelapse IVM imaging datasets to investigate the in vivo migratory behaviour of fluorescently labelled DMG cells in tumour bearing mice. Using the tool's 'heterogeneity module' they were able to identify distinct single-cell behavioural patterns (based on multiple parameters such as directionality, speed, displacement, distance from tumour edge) which was used to group cells into distinct categories (e.g. retreating, invasive, static, erratic). They next applied the framework's 'large-scale phenotyping' and 'small-scale phenotyping' modules to investigate whether the tumour microenvironment (TME) may influence the distinct migratory behaviours identified. To achieve this, they combine TME visualisation in vivo during IVM (using fluorescent probes to label distinct TME components) or ex vivo after IVM (by large-scale imaging of harvested, immunostained tumours) to correlate different tumour behavioural patterns with the composition of the TME. They conclude that this tool has helped reveal links between TME composition (e.g. degree of vascularisation, presence of tumour-associated macrophages) and the invasiveness and directionality of tumour cells, which would have been challenging to identify when analysing single kinetic parameters in isolation.
The authors also evaluated the BEHAV3D TP heterogeneity module using available IVM datasets of distinct breast cancer cell lines transplanted in vivo, as well as healthy mammary epithelial cells to test its usability in non-tumour contexts where the migratory phenotypes of cells may be more subtle. This generated data is consistent with that produced during the original studies, as well as providing some additional (albeit preliminary) insights above that previously reported. Collectively, this provides some confidence in BEHAV3D TP's ability to uncover complex, multi-parametric cellular behaviours that may be missed using traditional approaches.
Overall, this computational framework appears to represent a useful and comparatively user-friendly tool to analyse dynamic multi-parametric data to help identify patterns in cell migratory behaviours, and to assess whether these behaviours might be influenced by neighbouring cells and structures in their microenvironment. When combined with other methods, it therefore has the potential to be a valuable addition to a researcher's IVM analysis 'tool-box'.
Strengths:
• Figures are clearly presented, and the manuscript is easy to follow.
• The pipeline appears to be intuitive and user-friendly for researchers with limited computational expertise. A detailed step-by-step video and demo datasets are also included to support its uptake.
• The different computational modules have been tested using relevant datasets, including imaging data of normal and tumour cells in vivo.
• All code is open source, and the pipeline can be implemented with Google Colab.
• The tool combines multiple dynamic parameters extracted from timelapse IVM images to identify single-cell behavioural patterns and to cluster cells into distinct groups sharing similar behaviours, and provides avenues to map these onto in vivo or ex vivo imaging data of the tumour microenvironment
Weaknesses:
• The tool does not facilitate the extraction of quantitative kinetic cellular parameters (e.g. speed, directionality, persistence and displacement) from intravital images. To use the tool researchers must first extract dynamic cellular parameters from their IVM datasets using other software including Imaris, which is expensive and therefore not available to all. Nonetheless, the authors have developed their tool to facilitate the integration of other data formats generated by open-source Fiji plugins (e.g. TrackMate, MTrackJ, ManualTracking) which will help ensure its accessibility to a broader range of researchers.
• The analysis provides only preliminary evidence in support of the authors conclusions on DMG cell migratory behaviours and their relationship with components of the tumour microenvironment. The authors acknowledge this however, and conclusions are appropriately tempered in the absence of additional experiments and controls.
We thank the reviewer for their thorough and constructive assessment of our work and are pleased that the accessibility, functionality, and potential impact of BEHAV3DTumour Profiler were well received. We particularly appreciate the acknowledgment of the tool’s ease of use for researchers with limited computational expertise, the clarity of the manuscript, and the relevance of our approach for identifying multi-parametric migratory behaviours and their correlation with the tumour microenvironment.
Regarding the weaknesses raised:
(1) Lack of built-in tracking and kinetic parameter extraction – As noted in our initial revision, while we agree that integrating open-source tracking and segmentation functionality could be valuable, it is beyond the scope of the current work. Our tool is designed to focus specifically on downstream analysis of already extracted kinetic data, addressing a gap in post-processing tools for exploring complex migratory behaviour and spatial correlations. Since different experimental systems often require tailored imaging and segmentation pipelines, we believe that decoupling tracking from the downstream analysis can actually be a strength, offering greater versatility. Researchers can use their preferred or most appropriate tracking software—whether proprietary or opensource—and then analyze the resulting data with BEHAV3D-TP. To support this, we ensured compatibility with widely used tools including open-source Fiji plugins (e.g., TrackMate, MTrackJ, ManualTracking), and we also cited several relevant studies and that address the upstream processing steps. Importantly, the main aim of our tool is to fill the gap in post-tracking analysis, enabling quantitative interpretation and pattern recognition that has until now required substantial coding effort or custom solutions.
(2) Preliminary nature of the biological conclusions – We fully agree with this assessment and have explicitly acknowledged this limitation in the manuscript. Our aim was to demonstrate the utility of BEHAV3D-TP in uncovering heterogeneity and spatial associations in vivo, while encouraging further hypothesis-driven studies using complementary biological approaches. We are grateful that the reviewer recognizes the cautious interpretation of our results and their added value beyond single-parameter analysis.
Reviewer #2 (Public review):
Summary:
The authors produce a new tool, BEHAV3D to analyse tracking data and to integrate these analyses with large and small scale architectural features of the tissue. This is similar to several other published methods to analyse spatio-temporal data, however, the connection to tissue features is a nice addition, as is the lack of requirement for coding. The tool is then used to analyse tracking data of tumour cells in diffuse midline glioma. They suggest 7 clusters exist within these tracks and that they differ spatially. They ultimately suggest that there these behaviours occur in distinct spatial areas as determined by CytoMAP.
Strengths:
- The tool appears relatively user-friendly and is open source. The combination with CytoMAP represents a nice option for researchers.
- The identification of associations between cell track phenotype and spatial features is exciting and the diffuse midline glioma data nicely demonstrates how this could be used.
Weaknesses:
The revision has dealt with many concerns, however, the statistics generated by the process are still flawed. While the statistics have been clarified within the legends and this is a great improvement in terms of clarity the underlying assumptions of the tests used are violated. The problem is that individual imaging positions or tracks are treated as independent and then analysed by ANOVA. As separate imaging positions within the same mouse are not independent, nor are individual cells within a single mouse, this makes the statistical analyses inappropriate. For a deeper analysis of this that is feasible within a review please see Lord, Samuel J., et al. "SuperPlots: Communicating reproducibility and variability in cell biology." The Journal of cell biology 219.6 (2020): e202001064. Ultimately, while this is a neat piece of software facilitating the analysis of complex data, the fact that it will produce flawed statistical analysis is a major problem. This problem is compounded by the fact that much imaging analysis has been analysed in this inappropriate manner in the past, leading to issues of interpretation and ultimately reproducibility.
We thank the reviewer for their careful reading and thoughtful feedback. We are encouraged by the recognition of BEHAV3D-TP’s ease of use, open-source accessibility, and the value of integrating cell behaviour with spatial features of the tissue. We appreciate the positive remarks regarding our application to diffuse midline glioma (DMG) and the potential for the tool to enable new biological insights.
We also appreciate the reviewer’s continued concern regarding the statistical treatment of the data. While we agree with the broader principle that care must be taken to avoid violating assumptions of independence, we respectfully disagree that all instances where individual tracks or imaging positions are used constitute flawed analysis. Importantly, our work is centered on characterizing heterogeneity at the single-cell level in distinct TME regions. Therefore, in certain cases—especially when comparing distinct behavioral subtypes across varying TME environments and multiple mice—it is appropriate to treat individual imaging positions as independent units. This approach is particularly relevant given our findings that large-scale TME regions differ across positions. When analyzing features such as the percentage of DMG cells in proximity to TAMMs, averaging per mouse would obscure these regional differences and reduce the resolution of biologically meaningful variation.
To address this concern further, we have revised the figure legends, main text, and documentation, carefully considering the appropriate statistical unit for each analysis. As detailed below, we used mouse-level aggregation where the experimental question required inter-mouse reproducibility, and a position-based approach where the aim was to explore intra-tumoral heterogeneity.
Figure 3d and Supplementary Figure 5d: In this analysis, we treated imaging positions as independent units because our data specifically demonstrate that, within individual mice, different positions correspond to distinct large-scale tumor microenvironment phenotypes. Therefore, averaging across the whole mouse would obscure these important spatial differences and not accurately reflect the heterogeneity we aim to characterize.
Figure 4c-e; Supplementary Figure 6d: While our initial aim was to highlight single-cell variability, we acknowledge that the original presentation may have been misleading. In the revised manuscript, we have updated the graphs for greater clarity. To quantify how often tumor cells of each behavioral type are located near TAMMs (Fig. 4c) or blood vessels (Fig. 4e), we now calculate the percentage of tumor cells "close" to environmental feature per behavioral cluster within each imaging position. This classification is based on the distance to the TME feature of interest and is detailed in the “Large-scale phenotyping” section of the Methods. For the number of SR101 objects in a 30um radius we averaged per position.
We treated individual imaging positions as the units of analysis rather than averaging per mouse, as our data (see Figure 2) show that positions vary in their TME phenotypes—such as Void, TAMM/Oligo, and TAMM/Vascularized—as well as in the number of TAMMs, SR101 cells or blood vessels per position. These differences are biologically meaningful and relevant to the quantification that we performed – percentage of tumor cell in close proximity to distinct TME features.
To account for inter-mouse and TME region variability, we applied a linear mixedeffects model with both mouse and TME class included as random effects.
Supplementary Figure 3d: Following the reviewer’s suggestion, we have averaged the distance to the 3 closest GBM neighbours per mouse, treating each mouse as an independent unit for comparison across distinct GBM morphodynamic clusters. To account for inter-mouse variability when assessing statistical significance, we employed a linear mixed model with mouse included as a random effect.
Distance to 3 neighbours is a feature not used in the clustering, thus variability between mice can be more pronounced—for example, due to differences in tumor compactness or microenvironment structure across individual mice. To appropriately account for this, mouse was included as a random effect in the model.
Supplementary Figure 4c: Following the reviewer’s suggestion, we averaged cell speed per mouse, treating each mouse as an independent unit for comparison across distinct DMG behavioral clusters. Statistical significance was assessed using ANOVA followed by Tukey’s post hoc test. When comparing cell speed, which is a feature used in the clustering process, inter-mouse variability was already addressed during clustering itself. Therefore, in the downstream analysis of this cluster-derived feature, it is appropriate to treat each mouse as an independent unit without including mouse as a random effect.
Supplementary Figure 5e-g: Following the reviewer’s suggestion, we averaged cell speed per mouse, treating each mouse as an independent unit for comparison across distinct DMG behavioral clusters. Statistical significance was assessed using ANOVA followed by Tukey’s post hoc test.
Supplementary Figure 6c: Following the reviewer’s suggestion, we averaged cell distance to the 10 closest DMG neighbours per mouse, treating each mouse as an independent unit for comparison across distinct DMG behavioral clusters. To account for inter-mouse variability, we used a linear mixed model with mouse included as a random effect.
Reviewer #3 (Public review):
The manuscript by Rios-Jimenez developed a software tool, BEHAV3D Tumor Profiler, to analyze 3D intravital imaging data and identify distinctive tumor cell migratory phenotypes based on the quantified 3D image data. Moreover, the heterogeneity module in this software tool can correlate the different cell migration phenotypes with variable features of the tumor microenvironment. Overall, this is a useful tool for intravital imaging data analysis and its open-source nature makes it accessible to all interested users.
Strengths:
An open-source software tool that can quantify cell migratory dynamics from intravital imaging data and identify distinctive migratory phenotypes that correlate with variable features of the tumor microenvironment.
Weaknesses:
Motility is only one tumor cell feature and is probably not sufficient to characterize and identify the heterogeneity of the tumor cell population that impacts their behaviors in the complex tumor microenvironment (TME). For instance, there are important nontumor cell types in the TME, and the interaction dynamics of tumor cells with other cell types, e.g., fibroblasts and distinct immune cells, play a crucial role in regulating tumor behaviors. BEHAV3D-TP focuses on only motility feature analysis, and cannot be applied to analyze other tumor cell dynamic features or cell-cell interaction dynamics.
Regarding the concern about the tool’s current focus on motility features, we would like to clarify that BEHAV3D-TP is designed to be highly flexible and extensible. As described in our first revision, users can incorporate a wide range of features—including dynamic, morphological, and spatial parameters—into their analyses. In the current revision, we have make this even more explicit by explaining that the feature selection interface allows users to either (i) directly select them for clustering or (ii) select features for correlation with clusters (See Small scale phenotyping module section in Methods and Rebuttal Figure).
Importantly, while our current analysis emphasizes clustering based on dynamic behaviors, Figure 4 demonstrates that these behavioral clusters are associated at the single-cell level with distinct proximities to key TME components, such as TAMMs and blood vessels. These spatial interaction features could also have been included in the clustering itself—creating dynamic-spatial clusters—but we deliberately chose not to do so. This decision was guided by established principles of feature selection: including features with unknown or potentially irrelevant variability can introduce noise and obscure biologically meaningful patterns, ultimately reducing the clarity and interpretability of the resulting clusters. Instead, we adopted a two-step approach—first identifying clusters based on core dynamic features, then examining their relationships with spatial and interaction metrics. This allowed us to reveal meaningful associations of particular cell behavior such as the invading cluster in proximity of TAMMs without overfitting or complicating the clustering model.
To further address the reviewer’s point, we have updated the Small-scale phenotyping module to highlight the possibility of including spatial interaction features with various TME cell types. We also revised the manuscript text and Figure 1 to clarify that these environmental features can be used both upstream as clustering input (Option 1) and for downstream analysis (Option 2), depending on the user’s experimental goals. Author response image 1 illustrates these options in the feature selection panels of the Colab notebook.
Author response image 1.
(a) In the small-scale phenotyping module, microenvironmental factors (MEFs) detected in the segmented IVM movies are identified and their coordinates imported. From here, there are two options: (b) include the relationship to these MEFs as a feature for clustering, or (c) exclude this relationship and instead correlate MEFs with cell behavior to assess potential spatial associations.
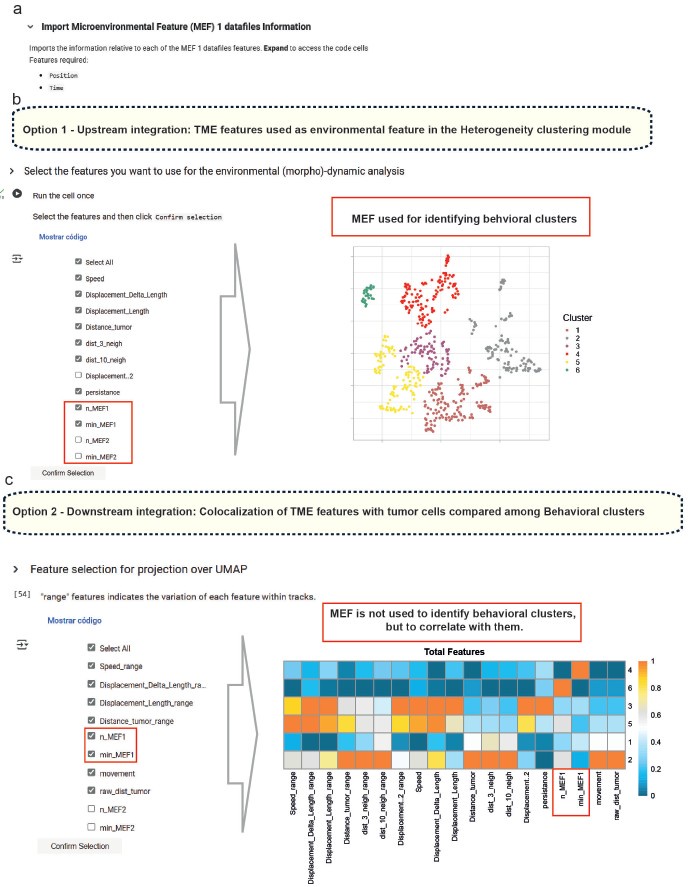
In summary, while the clustering presented in this study is based on dynamic parameters, BEHAV3D-TP fully supports the integration of interaction features and other non-motility descriptors. This modularity enables users to customize their analysis pipelines according to specific biological questions, including those involving cell–cell interactions and spatial dynamics within the TME.
Reviewer #2 (Recommendations for the authors):
If the software were adjusted to produce analyses following best practices in the field as outlined in Lord, Samuel J., et al. "SuperPlots: Communicating reproducibility and variability in cell biology." The Journal of cell biology 219.6 (2020): e202001064. this could be a helpful piece of software. The major current issue would be that it democratises the ability to analyse complex imaging data, allowing non-experts to carry out these analyses but misleads them and encourages poor statistical practice.
We appreciate the reviewer’s suggestion and the reference to best practices outlined in Lord et al., 2020. As discussed in detail in our point-by-point response to Reviewer #2, we have revised several figures to enhance clarity and statistical rigor, including Figure 4c,e; Supplementary Figures 3d, 4c, 5e–g, and 6c–d. Specifically, we adjusted how data are summarized and displayed—averaging per mouse where appropriate and clarifying the statistical methods used. Where imaging positions were retained as the unit of analysis, this decision was grounded in the biological relevance of intra-mouse spatial heterogeneity (as demonstrated in Figure 2). Additionally, we applied linear mixed-effects models in cases where inter-mouse or inter-Large scale TME regions variability needed to be accounted for. We believe these changes address the core concern about reproducibility and statistical interpretation while preserving the biological insights captured by our approach.
-
-
-
eLife Assessment
This is a useful tool for code-less analysis of patterns in cell migratory behaviours in vivo using intravital microscopy data and allows correlation with spatial features of the tumour microenvironment. There is a need for these tools to make quantitative analysis, comparison and interpretation of complex cell tracking data more accessible and evidence is provided of its applicability to tracks generated by both proprietary and open tracking software. However, it is incomplete due to limitations imposed by the assumptions that apply to the statistical tests employed.
-
Reviewer #1 (Public review):
Summary:
Intravital microscopy (IVM) is a powerful tool that facilitates live imaging of individual cells over time in vivo in their native 3D tissue environment. Extracting and analysing multi-parametric data from IVM images however is challenging, particularly for researchers with limited programming and image analysis skills. In this work, Rios-Jimenez and Zomer et al have developed a 'zero-code' accessible computational framework (BEHAV3D-Tumour Profiler) designed to facilitate unbiased analysis of IVM data to investigate tumour cell dynamics (via the tool's central 'heterogeneity module' ) and their interactions with the tumour microenvironment (via the 'large-scale phenotyping' and 'small-scale phenotyping' modules). It is designed as an open-source modular Jupyter Notebook with a user-friendly …
Reviewer #1 (Public review):
Summary:
Intravital microscopy (IVM) is a powerful tool that facilitates live imaging of individual cells over time in vivo in their native 3D tissue environment. Extracting and analysing multi-parametric data from IVM images however is challenging, particularly for researchers with limited programming and image analysis skills. In this work, Rios-Jimenez and Zomer et al have developed a 'zero-code' accessible computational framework (BEHAV3D-Tumour Profiler) designed to facilitate unbiased analysis of IVM data to investigate tumour cell dynamics (via the tool's central 'heterogeneity module' ) and their interactions with the tumour microenvironment (via the 'large-scale phenotyping' and 'small-scale phenotyping' modules). It is designed as an open-source modular Jupyter Notebook with a user-friendly graphical user interface and can be implemented with Google Colab, facilitating efficient, cloud-based computational analysis at no cost. Demo datasets are also available on the authors GitHub repository to aid user training and enhance the usability of the developed pipeline.
To demonstrate the utility of BEHAV3D-TP, they apply the pipeline to timelapse IVM imaging datasets to investigate the in vivo migratory behaviour of fluorescently labelled DMG cells in tumour bearing mice. Using the tool's 'heterogeneity module' they were able to identify distinct single-cell behavioural patterns (based on multiple parameters such as directionality, speed, displacement, distance from tumour edge) which was used to group cells into distinct categories (e.g. retreating, invasive, static, erratic). They next applied the framework's 'large-scale phenotyping' and 'small-scale phenotyping' modules to investigate whether the tumour microenvironment (TME) may influence the distinct migratory behaviours identified. To achieve this, they combine TME visualisation in vivo during IVM (using fluorescent probes to label distinct TME components) or ex vivo after IVM (by large-scale imaging of harvested, immunostained tumours) to correlate different tumour behavioural patterns with the composition of the TME. They conclude that this tool has helped reveal links between TME composition (e.g. degree of vascularisation, presence of tumour-associated macrophages) and the invasiveness and directionality of tumour cells, which would have been challenging to identify when analysing single kinetic parameters in isolation.
The authors also evaluated the BEHAV3D TP heterogeneity module using available IVM datasets of distinct breast cancer cell lines transplanted in vivo, as well as healthy mammary epithelial cells to test its usability in non-tumour contexts where the migratory phenotypes of cells may be more subtle. This generated data is consistent with that produced during the original studies, as well as providing some additional (albeit preliminary) insights above that previously reported. Collectively, this provides some confidence in BEHAV3D TP's ability to uncover complex, multi-parametric cellular behaviours that may be missed using traditional approaches.
Overall, this computational framework appears to represent a useful and comparatively user-friendly tool to analyse dynamic multi-parametric data to help identify patterns in cell migratory behaviours, and to assess whether these behaviours might be influenced by neighbouring cells and structures in their microenvironment. When combined with other methods, it therefore has the potential to be a valuable addition to a researcher's IVM analysis 'tool-box'.
Strengths:
- Figures are clearly presented, and the manuscript is easy to follow.
- The pipeline appears to be intuitive and user-friendly for researchers with limited computational expertise. A detailed step-by-step video and demo datasets are also included to support its uptake.
- The different computational modules have been tested using relevant datasets, including imaging data of normal and tumour cells in vivo.
- All code is open source, and the pipeline can be implemented with Google Colab.
- The tool combines multiple dynamic parameters extracted from timelapse IVM images to identify single-cell behavioural patterns and to cluster cells into distinct groups sharing similar behaviours, and provides avenues to map these onto in vivo or ex vivo imaging data of the tumour microenvironmentWeaknesses:
- The tool does not facilitate the extraction of quantitative kinetic cellular parameters (e.g. speed, directionality, persistence and displacement) from intravital images. To use the tool researchers must first extract dynamic cellular parameters from their IVM datasets using other software including Imaris, which is expensive and therefore not available to all. Nonetheless, the authors have developed their tool to facilitate the integration of other data formats generated by open-source Fiji plugins (e.g. TrackMate, MTrackJ, ManualTracking) which will help ensure its accessibility to a broader range of researchers.
- The analysis provides only preliminary evidence in support of the authors conclusions on DMG cell migratory behaviours and their relationship with components of the tumour microenvironment. The authors acknowledge this however, and conclusions are appropriately tempered in the absence of additional experiments and controls. -
Reviewer #2 (Public review):
Summary:
The authors produce a new tool, BEHAV3D to analyse tracking data and to integrate these analyses with large and small scale architectural features of the tissue. This is similar to several other published methods to analyse spatio-temporal data, however, the connection to tissue features is a nice addition, as is the lack of requirement for coding. The tool is then used to analyse tracking data of tumour cells in diffuse midline glioma. They suggest 7 clusters exist within these tracks and that they differ spatially. They ultimately suggest that there these behaviours occur in distinct spatial areas as determined by CytoMAP.
Strengths:
- The tool appears relatively user-friendly and is open source. The combination with CytoMAP represents a nice option for researchers.
- The identification of …
Reviewer #2 (Public review):
Summary:
The authors produce a new tool, BEHAV3D to analyse tracking data and to integrate these analyses with large and small scale architectural features of the tissue. This is similar to several other published methods to analyse spatio-temporal data, however, the connection to tissue features is a nice addition, as is the lack of requirement for coding. The tool is then used to analyse tracking data of tumour cells in diffuse midline glioma. They suggest 7 clusters exist within these tracks and that they differ spatially. They ultimately suggest that there these behaviours occur in distinct spatial areas as determined by CytoMAP.
Strengths:
- The tool appears relatively user-friendly and is open source. The combination with CytoMAP represents a nice option for researchers.
- The identification of associations between cell track phenotype and spatial features is exciting and the diffuse midline glioma data nicely demonstrates how this could be used.
Weaknesses:
- The revision has dealt with many concerns, however, the statistics generated by the process are still flawed. While the statistics have been clarified within the legends and this is a great improvement in terms of clarity the underlying assumptions of the tests used are violated. The problem is that individual imaging positions or tracks are treated as independent and then analysed by ANOVA. As separate imaging positions within the same mouse are not independent, nor are individual cells within a single mouse, this makes the statistical analyses inappropriate. For a deeper analysis of this that is feasible within a review please see Lord, Samuel J., et al. "SuperPlots: Communicating reproducibility and variability in cell biology." The Journal of cell biology 219.6 (2020): e202001064. Ultimately, while this is a neat piece of software facilitating the analysis of complex data, the fact that it will produce flawed statistical analysis is a major problem. This problem is compounded by the fact that much imaging analysis has been analysed in this inappropriate manner in the past, leading to issues of interpretation and ultimately reproducibility.
-
Reviewer #3 (Public review):
The manuscript by Rios-Jimenez developed a software tool, BEHAV3D Tumor Profiler, to analyze 3D intravital imaging data and identify distinctive tumor cell migratory phenotypes based on the quantified 3D image data. Moreover, the heterogeneity module in this software tool can correlate the different cell migration phenotypes with variable features of the tumor microenvironment. Overall, this is a useful tool for intravital imaging data analysis and its open-source nature makes it accessible to all interested users.
Strengths:
An open-source software tool that can quantify cell migratory dynamics from intravital imaging data and identify distinctive migratory phenotypes that correlate with variable features of the tumor microenvironment.
Weaknesses:
Motility is only one tumor cell feature and is probably not …
Reviewer #3 (Public review):
The manuscript by Rios-Jimenez developed a software tool, BEHAV3D Tumor Profiler, to analyze 3D intravital imaging data and identify distinctive tumor cell migratory phenotypes based on the quantified 3D image data. Moreover, the heterogeneity module in this software tool can correlate the different cell migration phenotypes with variable features of the tumor microenvironment. Overall, this is a useful tool for intravital imaging data analysis and its open-source nature makes it accessible to all interested users.
Strengths:
An open-source software tool that can quantify cell migratory dynamics from intravital imaging data and identify distinctive migratory phenotypes that correlate with variable features of the tumor microenvironment.
Weaknesses:
Motility is only one tumor cell feature and is probably not sufficient to characterize and identify the heterogeneity of the tumor cell population that impacts their behaviors in the complex tumor microenvironment (TME). For instance, there are important non-tumor cell types in the TME, and the interaction dynamics of tumor cells with other cell types, e.g., fibroblasts and distinct immune cells, play a crucial role in regulating tumor behaviors. BEHAV3D-TP focuses on only motility feature analysis, and cannot be applied to analyze other tumor cell dynamic features or cell-cell interaction dynamics.
-
Author response:
The following is the authors’ response to the original reviews
We thank the reviewers for their positive and constructive comments on the manuscript. In the revised manuscript we addressed these comments, which we believe have improved the quality of our work.
In summary:
(1) We acknowledge the reviewer's suggestion to incorporate open-source segmentation and tracking functionalities, increasing its accessibility to a wider user base; however, these additions fall outside the primary scope of our current work, which is to provide an analytical framework for IVM data after segmentation and tracking. Developing open-source segmentation and tracking tools represents a substantial undertaking in its own right, which has been comprehensively explored in other studies (e.g. https://doi.org/10.4049/jimmunol.2100811; https://doi…
Author response:
The following is the authors’ response to the original reviews
We thank the reviewers for their positive and constructive comments on the manuscript. In the revised manuscript we addressed these comments, which we believe have improved the quality of our work.
In summary:
(1) We acknowledge the reviewer's suggestion to incorporate open-source segmentation and tracking functionalities, increasing its accessibility to a wider user base; however, these additions fall outside the primary scope of our current work, which is to provide an analytical framework for IVM data after segmentation and tracking. Developing open-source segmentation and tracking tools represents a substantial undertaking in its own right, which has been comprehensively explored in other studies (e.g. https://doi.org/10.4049/jimmunol.2100811; https://doi.org/10.7554/eLife.60547; https://doi.org/10.1016/j.media.2022.102358; https://doi.org/10.1038/s41592024-02295-6 - now cited in our revised manuscript).
In our analyses, we used data processed with Imaris, a commercial software that, despite its limitations, is widely used by the intravital microscopy community due to its user-friendly platform for 3D image visualization and analysis. Nevertheless, recognizing the need for compatibility with tracking data from various pipelines, we have modified our tool to accept other data formats, such as those generated by open-source Fiji plugins like TrackMate, MTrackJ, ManualTracking (https://github.com/imAIgene-Dream3D/BEHAV3D_Tumor_Profiler?tab=readme-ov-file#data-input). These updates are available in our GitHub repository and are described in the revised manuscript.
(2) We appreciate the reviewer #3 suggestion to incorporate additional features into our analytical pipeline. In response, we have already updated the GitHub repository to allow users to input and select which features (dynamic, morphological, or spatial) they wish to include in the analysis (https://github.com/imAIgene-Dream3D/BEHAV3D_Tumor_Profiler?tab=readmeov-file#feature-selection ). In the revised manuscript, we highlighted this new functionality and provided examples using alternative datasets to demonstrate the application of these features.
(3) We appreciate the constructive feedback of reviewers #1 and #2 regarding the statistical analysis and interpretation of the data presented in Figures 3 and 4. We understand the importance of clarity and rigor in data analysis and presentation, and we addressed the concerns raised in the revised version of the manuscript.
(4) We appreciate reviewer #1's suggestion regarding the inclusion of demo data, as we believe it would greatly enhance the usability of our pipeline. We acknowledge that this was an oversight on our part. To address this, we have now added demos to our GitHub repository (https://github.com/imAIgene-
Dream3D/BEHAV3D_Tumor_Profiler/tree/BEHAV3D_TP-v2.0/demo_datasets). In the revised manuscript, we referenced this addition and present new figures with examples of these demo’s processing different IVM dataset (2D/3D, different tumors and healthy tissues). Additionally, we have provided processed DMG IVM movie samples in an imaging repository.
(5) Finally, we made some small changes to the manuscript based on the reviewers’ feedback.
Below we provide a point-by-point response to the reviewers’ comments
Reviewer #1 (Public review):
Comment #1: A key limitation of the pipeline is that it does not overcome the main challenges and bottlenecks associated with processing and extracting quantitative cellular data from timelapse and longitudinal intravital images. This includes correcting breathing-induced movement artifacts, automated registration of longitudinal images taken over days/weeks, and accurate, automated segmentation and tracking of individual cells over time. Indeed, there are currently no standardised computational methods available for IVM data processing and analysis, with most laboratories relying on custom-built solutions or manual methods. This isn't made explicit in the manuscript early on (described below), and the researchers rely on expensive software packages such as IMARIS for image processing and data extraction to feed the required parameters into their pipeline. This limitation unfortunately reduces the likely impact of BEHAV3D-TP on the IVM field.
As highlighted above, the tool does not facilitate the extraction of quantitative kinetic cellular parameters (e.g. speed, directionality, persistence, and displacement) from intravital images. Indeed, to use the tool researchers must first extract dynamic cellular parameters from their IVM datasets, requiring access to expensive software (e.g. IMARIS as used here) and/or above-average computational expertise to develop and use custom-made open-source solutions. This limitation is not made explicit or discussed in the text.
We acknowledge the reviewer's suggestion to incorporate open-source segmentation and tracking functionalities, increasing its accessibility to a wider user base; however, these additions fall outside the primary scope of our current work and represent a substantial undertaking in their own right. Several studies (e.g., Diego Ulisse Pizzagalli et al., J Immunol (2022); Aby Joseph et al., eLife (2020); Molina-Moreno et al., Medical Image Analysis (2022); Hidalgo-Cenalmor et al., Nat Methods (2024); Ershov et al., Nat Methods (2022)) have comprehensively addressed these topics, and we now reference them in the revised manuscript to provide readers with relevant background.
The objective of our manuscript is not to develop a complete segmentation or tracking pipeline but rather to introduce an analytical framework capable of extracting enhanced insights from the data generated by existing tools. This goal arises from our observations of the field: despite significant investment in image processing, researchers often rely on simplistic approaches, such as averaging single parameters across conditions, which can obscure tumor heterogeneity and spatial behavioral dynamics within the tumor microenvironment.
Our current tool focuses on providing this much-needed analytical capability. For our analysis we used Imaris, a widely utilized software in the intravital microscopy (IVM) community, known for its intuitive 3D visualization and analysis platform despite certain limitations.
In our own literature search of recent IVM studies published by leading laboratories in high-impact journals, we found that close to half used Imaris, while the remainder primarily relied on manual workflows with Fiji plugins. Thus, we consider it valuable to offer a pipeline compatible with such commonly used software, given its prevalence in the field.
However, following the suggestion of the reviewer, and to enhance the tool’s flexibility and compatibility, we have expanded the pipeline to accept data formats generated by open-source Fiji plugins, such as TrackMate, MTrackJ, and ManualTracking. These updates are detailed in the revised manuscript and are implemented in our GitHub repository (https://github.com/imAIgene-Dream3D/BEHAV3D_Tumor_Profiler?tab=readme-ov-file#data-input ), where we also provide several demos using TrackMate and Imaris processed data. This addition demonstrates our tool's capability to integrate with segmented and tracked datasets from diverse platforms, increasing its applicability to a broader range of researchers using both commercial and open-source pipelines.
Comment #2: The number of cells (e.g. per behavioural cluster), and the number of independent mice, represented in each result figure, is not included in the figure legends and are difficult to ascertain from the methods.
We appreciate the reviewer's constructive feedback regarding the clarity of the number and type of replicates used in our analyses. In the revised manuscript, we have included detailed information in the figure legends and the number of independent mice represented in each figure legend to ensure transparency. Regarding the number
of cells, we have indicated the total number of processed cells in Figure 2b legend (953 cells). Additionally, we have now included figures (Sup Fig 4c, Sup Fig 5e-g, Fig 5c,e, Sup Fig 6 c,d) for each cluster, where individual dots represent the individual cell tracks with color indicating the position and the shape indicating individual mice.
Comment #3: The data used to test the pipeline in this manuscript is currently not available, making it difficult to assess its usability. It would be important to include this for researchers to use as a 'training dataset'.
As stated above we acknowledge that this was an oversight on our part and thank the reviewer for pointing this out. To address this, we have now added demo data to our GitHub repository (BEHAV3D_Tumor_Profiler/demo_datasets at main · imAIgeneDream3D/BEHAV3D_Tumor_Profiler · GitHub). In the revised manuscript we have referenced this addition in the Data availability section. Since we included now processing with Fiji as well, we provide 4 demo datasets (https://github.com/imAIgeneDream3D/BEHAV3D_Tumor_Profiler/tree/main/demo_datasets), one processed with Imaris in 3D; and one with CellPose2.0 and Trackmate in 2D; one processed with µSAM and Trackmate in 3D and one manually processed with MtrackJ in 2D . Moreover, we now provide Imaris-processed DMG IVM movie samples in an open-source repository.
Comment #4: Precisely how the BEHAV3D-TP large-scale phenotyping module can map large-scale spatial phenotyping data generated using LSR-3D imaging data and Cytomap to 3D intravital imaging movies is unclear. Further details in the text and methods would be beneficial to aid understanding.
We appreciate the reviewer’s comment and in the revised manuscript we have now provided details in the methods section “Tumor large-scale spatial phenotyping with Cytomap” to clarify how the BEHAV3D-TP module maps LSR-3D and Cytomap data to 3D intravital imaging movies:
“To map the assigned regions onto IVM movies, a 3D image of the cluster distribution within the tumor was generated and exported for each sample (Figure Supplement 5a). Next, regions within the IVM movies were visually matched to the corresponding regions identified by the Large-Scale Phenotyping module of Cytomap (Figure 3c). For each mouse, at least one or two representative positions per matched region type were selected, cropped, and analyzed to assess tumor cell behavior, following the previously described cell tracking methodology (Imaris Cell tracking).”
Moreover, we updated Figure 3 c to further clarify these steps.
Comment #5: The analysis provides only preliminary evidence in support of the authors' conclusions on DMG cell migratory behaviours and their relationship with components of the tumour microenvironment. Conclusions should therefore be tempered in the absence of additional experiments and controls.
We appreciate the reviewer’s comment and acknowledge that our conclusions should be tempered due to the preliminary nature of our evidence. In the revised version of the manuscript we have revised our conclusions accordingly and emphasize the necessity for additional experiments and controls to further validate our findings on DMG cell migratory behaviors and their relationship with the tumor microenvironment.
In discussion: “While our findings suggest that microenvironmental factors may influence tumor cell migration, further studies will be necessary to establish causal relationships. Additional experimental validation, such as macrophage ablation experiments, could help clarify the specific contributions of these factors.”
Reviewer #1 (Recommendations for the authors):
(1) To test the ability of the pipeline to identify relevant patterns of migratory behaviours additional 'control' experiments would be helpful e.g. comparing non-invasive vs invasive tumour cell lines, artificially controlling migratory behaviours of cells such as implanting beads soaked in factors that would attract/repel cells?
(2) Does the pipeline work well for a variety of cell types/contexts? e.g. can it identify and cluster more subtle migratory behaviours such as non-tumour cells during tissue development or regeneration conditions?
We appreciate the reviewer’s valuable suggestions. In the revised manuscript, we have included additional examples demonstrating the capability of our pipeline to investigate heterogeneous cell behavior across two additional experimental setups:
(1) We have now evaluated our BEHAV3D TP heterogeneity module using IVM data from breast cancer cell lines with varying migratory capacities (DOI: 10.1016/j.yexcr.2019.04.009). In these datasets, our pipeline extends beyond predefined characteristics based solely on speed, enabling the identification of distinct cell populations. Notably, our analysis reveals that the breast cancer lines exhibit different proportions of different migratory behaviors such as Fast, Intermediate, Very slow and Static (Supplementary Fig 1).
(2) We have now evaluated our BEHAV3D TP heterogeneity module using IVM data from healthy breast epithelial cells (DOI: 10.1016/j.celrep.2024.115073), where we identify distinct morhophynamic epithelial cell populations in the terminal end but of the mammary gland that have a distinct distribution among Hormone receptor (HR) + and HR- terminal end but cells.
(3) To support biological conclusions could the authors show that ablating tumourassociated macrophages or vasculature alters the migratory patterns of nearby tumour cells?
We appreciate the reviewer's suggestion regarding the potential effects of ablating tumor-associated macrophages or vasculature on the migratory patterns of nearby tumor cells. While these experiments would functionally validate the observations made by our method, we would like to clarify that the primary focus of our study was on the development and application of computational tools for behavioral analysis and thus we consider that delving deeper in understanding the biology behind our observation is out of the scope of the current study. However, as mentioned previously, we have carefully tempered our conclusions to acknowledge the limitations of our current study. In the revised manuscript, we explicitly highlight that experiments involving the ablation of tumor-associated macrophages or vasculature would be crucial for further understanding the biological relevance of our findings.
Minor corrections to text:
(4) Line 63 - are references formatted correctly?
Thank you for pointing out this error. We have corrected it in the revised manuscript.
(5) Lines 161 -162 - 'intravitally imaged' used twice in a sentence.
Thank you for pointing out the typo. We have corrected it in the revised manuscript.
Reviewer #2 (Public review):
Comment#1: The strength of democratizing this kind of analysis is undercut by the reliance upon Imaris for segmentation, so it would be nice if this was changed to an open-source option for track generation.
As noted in our previous response to Reviewer #1, we would like to point out that although Imaris is a commercial software, it is widely used in the intravital microscopy community due to its user-friendly interface. We conducted a literature review to evaluate this aspect and below we include references from leading laboratories in the IVM field that utilize Imaris. One of its key advantages, which we also utilized, is semi-automated data tracking that allows for manual corrections in 3D—a process that can be more challenging in other open-source software with less effective data visualization.
However, we recognize that enhancing our pipeline's compatibility with open-source options is important. To this end, we have updated our tool to support 2D and 3D data formats generated by open-source Fiji plugins like TrackMate, MTrackJ, and ManualTracking, improving compatibility with various segmentation and tracking pipelines (https://github.com/imAIgene-Dream3D/BEHAV3D_Tumor_Profiler?tab=readme-ov-file#data-input ). In the revised manuscript, we describe the new functionality and demonstrate the operation of the BEHAV3D-TP heterogeneity module across various IVM datasets, processed in both 2D and 3D with different processing pipelines (Supplementary Fig 1-3). This includes CellPose 2.0 and the novel 'Segment Anything' model, followed by TrackMate tracking, applied to both tumor and healthy IVM data. Moreover we have developed a new web application that integrates morphological and tracking information from Segment Anything segmentation and Trackmate tracking, depicted in Supplementary Fig 3 a (https://morphotrack-merger.streamlit.app/ ). Additionally, we have updated the introduction to better clarify the scope of our study and include references to existing image processing solutions.
Comment#2: The main issue is with the interpretation of the biological data in Figure 3 where ANOVA was used to analyse the proportional distribution of different clusters. Firstly the n is not listed so it is unclear if this represents an n of 3 where each mouse is an individual or whether each track is being treated as a test unit. If the latter this is seriously flawed as these tracks can't be treated as independent. Also, a more appropriate test would be something like a Chi-squared test or Fisher's exact test. Also, no error bars are included on the stacked bar graphs making interpretation impossible. Ultimately this is severely flawed and also appears to show very small differences which may be statistically different but may not represent biologically important findings. This would need further study.
We appreciate the reviewer’s insightful comments regarding the interpretation of the biological data in Figure 3.
To clarify, each imaged position is considered an independent biological replicate (n = 18 from a total of 6 mice). We acknowledge that the description of the statistical methods and the experimental units was not sufficiently clear in the previous version. In our original submission, we used an ANOVA to test whether the proportion of each behavioral cluster differed across the tumor microenvironment regions. Post hoc pairwise comparisons were performed using Tukey’s test, with the results shown in Supplementary Figure 2d (currently Fig 3d). However, we agree with the reviewer that this approach may be misleading when paired with stacked bar plots that lack error bars, as it can obscure individual variability and does not explicitly represent statistical uncertainty.
In the revised manuscript, we present the data as boxplots with individual data points, where each dot represents an imaged position, and the shape corresponds to a specific mouse. In Figure 3 d the y-axis displays the normalized percentage of each cluster across TME regions, expressed as z-scores. This normalization corrects for inter-mouse variability and facilitates a comparison of the relative distribution of clusters across TME regions, independent of the overall abundance differences between mice. We performed an ANOVA with Tukey's post hoc test for each individual behavioral cluster to assess differences across TME regions. Additionally, for transparency, in Supplementary Figure 5 d we provide the raw percentage values. The legends provide the number of positions and mice included in the analysis.
Comment#3: Figure 4 has similar statistical issues in that the n is not listed and, again, it is unclear whether they are treating each cell track as independent which, again, would be inappropriate. The best practice for this type of data would be the use of super plots as outlined in Lord et al. (2020) JCI - SuperPlots: Communicating reproducibility and variability in cell biology.
We appreciate the reviewer’s comments and suggestions regarding Figure 4. In this case as we are comparing overall the behavioral clusters features, each individual cell is treated as a unit. In the revised manuscript, we have clarified this point in the figure legend and incorporated plots in Figure 4c and 4e, indicating the mouse and imaging position each data point originates from. This enhances the visualization of reproducibility and variability in our data, demonstrating that the results are consistent across multiple mice and positions and are not driven by a single mouse or imaging position.
Comment#4: The main issue that this raises is that the large-scale phenotyping module and the heterogeneity module appear designed to produce these statistical analyses that are used in these figures and, if they are based on the assumption that each track is independent, then this will produce inappropriate analyses as a default.
We appreciate the reviewer’s comment, although we are unclear about the specific concern being raised. To clarify, in our large-scale phenotyping analysis, each position is assigned to a TME niche based on the CytoMAP analysis and the workflow outlined in Figure 3c. Multiple positions are imaged per mouse. For each position, we measure the proportion of tumor cells exhibiting a specific behavioral phenotype, and these proportions are subsequently used for statistical analysis (Figure 3 d).
In contrast, in Supplementary Fig. 5e-g, we treat each cell track as an individual unit, grouping them by their assigned large-scale region. Here, we assess whether differences between regions can be detected using a conventional single-feature analysis—a more traditional approach. However, we find that this method loses important behavioral patterns and distinctions that BEHAV3D-TP captures.
We hope that this explanation, along with the modifications made to the figures and figure legends, provides greater clarity.
Reviewer #3 (Public review):
Comment #1: The most challenging task of analyzing 3D time-lapse imaging data is to accurately segment and track the individual cells in 3D over a long time duration. BEHAV3D Tumor Profiler did not provide any new advancement in this regard, and instead relies on commercial software, Imaris, for this critical step. Imaris is known to have a very high error rate when used for analyzing 3D time-lapse data. In the Methods section, the authors themselves stated that "Tumor cell tracks were manually corrected to ensure accurate tracking". Based on our own experience of using Imaris, such manual correction is tedious and often required for every time step of the movie. Therefore, Imaris is not a satisfactory tool for analyzing 3D time-lapse data. Moreover, Imaris is expensive and many research labs probably can't afford to buy it. The fact that BEHAV3D Tumor Profiler critically depends on the faulty ImarisTrack module makes it unclear whether the BEHAV3D tool or the results are reliable.
If the authors want to "democratize the analysis of heterogeneous cancer cell behaviors", they should perform image segmentation and tracking using open-source codes (e.g., Cellpose, Stardisk & 3DCellTracker) and not rely on the expensive and inaccurate ImarisTrack Module for the image analysis step of BEHAV3D.
We appreciate the reviewer’s comments on the challenges of segmenting and tracking individual cells in 3D time-lapse imaging data. As mentioned previously (please refer to comment #1 to reviewer #1), our primary focus is to develop an analytical tool for comprehensive data analysis rather than developing tools for image processing. However to enhance accessibility, we have updated our tool to support data formats from open-source Fiji plugins, such as TrackMate, which will benefit users without access to commercial software (https://github.com/imAIgeneDream3D/BEHAV3D_Tumor_Profiler?tab=readme-ov-file#data-input ). In Supplementary Figures 1, 2, and 3, we present IVM data from different sources, processed using three distinct methods: MTrackJ (Supplementary Fig. 1), Cellpose + TrackMate (Supplementary Fig. 2), and µSAM + TrackMate (Supplementary Fig. 3). The latter two represent state-of-the-art deep learning approaches.
On the other hand, while we recognize the limitations of Imaris, it remains widely used in the intravital microscopy community due to its user-friendly interface for 3D visualization and semi-automated segmentation capabilities. Since no perfect tracking method currently exists, we initially utilized Imaris for its ability to allow manual correction of faulty tracks, ensuring the reliability of our results. This approach, not only widely used (see above) but was the best available option when we began our analysis, allowing us to obtain accurate results efficiently.
In the revised manuscript, we clarify the scope of our study and provide information on both Imaris and alternative processing options to strengthen the reliability of our findings:
In introduction: “While significant efforts have been made to develop opensource segmentation and tracking tools for live imaging data, including IVM22–27 fewer tools exist for the unbiased analysis of tumor dynamics. One major barrier is that implementing such analytical methods often requires substantial computational expertise, limiting accessibility for many biomedical researchers conducting IVM experiments. To bridge this gap, we present BEHAV3D Tumor Profiler (BEHAV3D-TP) by providing a robust, user-friendly tool that allows researchers to extract meaningful insights from dynamic cellular behaviors without requiring advanced programming skills.”
In the Methods, we describe now describe not only Imaris processing pipeline, but also the µSAM segmentation pipelines and reference to CellPose IVM processing, which are combined with TrackMate for tracking. Additionally, to integrate morphological information from µSAM with tracking data from TrackMate, we developed a web tool to merge the outputs from both processing steps: https://morphotrack-merger.streamlit.app/
Comment #2: The authors developed a "Heterogeneity module" to extract distinctive tumor migratory phenotypes from the cell tracks quantified by Imaris. The cell tracks of the individual tumor cells are all quite short, indicating relatively low motility of the tumor cells. It's unclear whether such short migratory tracks are sufficient to warrant the PCA analysis to identify the 7 distinctive migratory phenotypes shown in Figure 2d. It's also unclear whether these 7 migratory phenotypes correspond to unique functional phenotypes.
For the 7 distinctive motility clusters, the authors should provide a more detailed analysis of the differences between them. It's unclear whether the difference in retreating, slow retreating, erratic, static, slow, slow invading, and invading correspond to functional difference of the tumor cells.
While some tumor cells exhibit limited motility, indicated by short tracks, others demonstrate significant migratory capabilities (Figure 2 Invading and Retreating cells). This variability in tumor cell behavior is a central focus of our analysis, and our tool is specifically designed to identify and distinguish these differences. Our PCA analysis effectively captures this variability, as illustrated in Figure 2 d-f. It differentiates between cells exhibiting varying degrees of migratory behavior, including both highly and less migratory phenotypes, as well as their directionality relative to the tumor core and the persistence of their movements. Thus, we believe that our approach provides valuable insights into the distinct migratory phenotypes within the tumor microenvironment.
While our current manuscript does not provide explicit evidence linking each motility cluster to functional differences among the tumor cells, it is important to note that the state of the field supports the idea that cell dynamics can predict cell states and phenotypes. Research conducted by ourselves (Dekkers, Alieva et al., Nat Biotech, 2023) and others, such as Craiciuc et al. (Nature, 2022) and Freckmann et al. (Nat Comm, 2022) has shown that variations in cell motility patterns are indicative of underlying functional characteristics. For instance, cell morphodynamic features have been shown to reflect differences in cell types, T cell targeting states (Dekkers, Alieva et al., Nat Biotech, 2023), immune cell types (Crainiciuc et al. (Nature, 2022)), tumor metastatic potential, and drug resistance states (Freckmann et al. (Nat Comm, 2022)). In the revised manuscript, we have referenced relevant studies to underscore the biological significance of these behaviors. By doing so, we hope to clarify the potential implications of our findings and strengthen the overall narrative of our research:
In discussion: “While our current study does not provide direct functional validation of the distinct motility clusters identified, existing literature strongly supports the notion that cell dynamics can serve as a proxy for functional states and phenotypic heterogeneity. Prior work, including studies by our group[19,66] as well as Crainiciuc et al.[35] and Freckmann et al.[20], has demonstrated that variations in cell motility patterns can reflect underlying functional characteristics. Specifically, cell morpho-dynamic features have been shown to correlate with differences in cell type identity, T-cell engagement, metastatic potential, and drug resistance states. This growing body of evidence suggests that tumor cell behavior, as captured by BEHAV3D-TP, may serve as a predictive tool for deciphering functional tumor heterogeneity. Future studies integrating transcriptomic or proteomic profiling of motility-defined subpopulations could further elucidate the biological significance of these behavioral phenotypes.”
Comment #3: Using only motility to classify tumor cell behaviours in the tumor microenvironment (TME) is probably not sufficient to capture the tumor cell difference. There are also other non-tumor cell types in the TME. If the authors aim to develop a computational tool that can elucidate tumor cell behaviors in the TME, they should consider other tumor cell features, e.g., morphology, proliferation state, and tumor cell interaction with other cell types, e.g., fibroblasts and distinct immune cells.
The authors should expand the scale of tumor behavior features to classify the tumor phenotype clusters, e.g., to include tumor morphology, proliferation state, and tumor cell interaction with other TME cell types.
We believe that using dynamic features alone is sufficient to capture differences in tumor behavior, as demonstrated by our results in Figure 2. However, we appreciate the reviewer’s suggestion to consider additional features, such as cell morphology, to finetune our analyses. To this end, we have adapted our pipeline to be compatible with any dynamic, morphologic or spatial features present in the data. In the revised manuscript we showcase this new addition with the analyses of two new dataset: 2D IVM data from healthy epithelial breast cells (Supplementary Fig 2) and 3D IVM data from adult gliomas (Supplementary Fig 3). These analyses identified cells with specific morphodynamic characteristics, which exhibited distinct kinetic behaviors or spatial distributions.
However, we would like to point out that not all features may provide informative insights and that a wide range of features can instead introduce biologically irrelevant noise, making interpretation more challenging. For instance, in 3D microscopy, the zaxis resolution is typically lower, which can lead to artifacts like elongation in that direction. Adding morphological features that capture this may skew the analysis. Therefore, we believe that incorporating additional features should be approached with caution. We clarify these considerations in the revised manuscript to better guide users in utilizing our computational tool effectively:
In discussion: “In addition to motility-based classification, features such as tumor cell morphology, proliferation state, and interactions with the tumor microenvironment can further refine tumor phenotyping. BEHAV3D-TP allows for the selection of diverse feature types, supporting datasets that include both dynamic, morphological and spatial parameters. However, we recognize that expanding the feature set may introduce biologically irrelevant noise, particularly in 3D microscopy data where limited z-axis resolution can lead to morphological artifacts. This highlights the potential need in the future to include unbiased feature selection strategies, such as bootstrapping methods67, to ensure the identification of meaningful and biologically relevant parameters. Careful consideration of these aspects is key to maximizing the interpretability and predictive value of analyses performed with BEHAV3D-TP.”
Comment #4: The authors have already published two papers on BEHAV3D [Alieva M et al. Nat Protoc. 2024 Jul;19(7): 2052-2084; Dekkers JF, et al. Nat Biotechnol. 2023 Jan;41(1):60-69]. Although the previous two papers used BEHAV3D to analyze T cells, the basic pipeline and computational steps are similar, in particular regarding cell segmentation and tracking. The addition of a "Heterogeneity module" based on PCA analysis does not make a significant advancement in terms of image analysis and quantification.
We want to emphasize that we have no intention of duplicating our previous publications. In this manuscript, we have consistently cited our foundational papers, where BEHAV3D was first developed for T cell migratory analysis in in vitro settings. In the introduction, we clearly state that our earlier work inspired us to adopt a similar approach for analyzing cell behavior in intravital microscopy (IVM) data, addressing the specific needs and complexities of analyzing tumor cell behaviors in the tumor microenvironment.
Importantly, our new work provides several key advancements: 1) a pipeline specifically adapted for intravital microscopy (IVM) data; 2) integration of spatial characteristics from both large-scale and small-scale phenotyping; and 3) a zero-code approach designed to empower researchers without coding skills to effectively utilize the tool. We believe that these enhancements represent meaningful progress in the analysis of cell behaviors within the tumor microenvironment which will be valuable for the IVM community. We ensure that these points are clearly articulated in the revised manuscript:
In introduction: “In line with this concept of characterizing cellular dynamic properties for cell classification, we have previously developed an analytical platform termed BEHAV3D 19,21 allowing to perform behavioral phenotyping of engineered T cells targeting cancer. While BEHAV3D was initially developed to analyze T cell migratory behavior under controlled in vitro conditions, we sought to expand its application to investigate tumor cell behaviors in IVM data, where the complexity of the TME presents distinct analytical challenges. This manuscript builds on our foundational work but represents a significant advancement by adapting the pipeline specifically for IVM datasets.”
Reviewer #3 (Recommendations for the authors):
(1) If the authors want to "democratize the analysis of heterogeneous cancer cell behaviors", they should perform image segmentation and tracking using open-source codes (e.g., Cellpose, Stardisk & 3DCellTracker) and not rely on the expensive and inaccurate ImarisTrack Module for the image analysis step of BEHAV3D.
We thank the reviewer for this recommendation and as stated above we recognize that enhancing our pipeline's compatibility with open-source options is important. To this end, we have updated our tool to support data formats generated by open-source Fiji plugins like TrackMate, MTrackJ, and ManualTracking, improving compatibility with various segmentation and tracking pipelines (https://github.com/imAIgeneDream3D/BEHAV3D_Tumor_Profiler?tab=readme-ov-file#data-input ). In the revised manuscript, we detail this new functionality and demonstrate the operation of the BEHAV3D-TP heterogeneity module using an example dataset of glioma tumors.
Additionally, we have updated the introduction to better clarify the scope of our study (See comment #1 from Review #3) and include references to existing image processing solutions.
(2) For the 7 distinctive motility clusters, the authors should provide a more detailed analysis of the differences between them. It's unclear whether the difference in retreating, slow retreating, erratic, static, slow, slow invading, and invading correspond to functional difference of the tumor cells.
As noted in the comment above, the revised manuscript now incorporates references to relevant literature that support our understanding that behavioral differences among cells are driven by their underlying functional differences (See comment #2 from Reviewer #3). Additionally, we would like to point to Figure 2d and Supplementary Fig 4 c that provide evidence of the functional distinctions between the identified clusters.
(3) The authors should expand the scale of tumor behavior features to classify the tumor phenotype clusters, e.g., to include tumor morphology, proliferation state, and tumor cell interaction with other TME cell types.
We thank the reviewer for this valuable suggestion. In the revised manuscript, we have added the flexibility to incorporate a wide range of features, including morphological ones, and enabled users to select the specific features they wish to include in their analysis. To illustrate this functionality, we have included 2 example dataset analyzed using this approach (See comment #3 from Reviewer #3). Additionally, as indicated above we emphasize the importance of careful selection and interpretation of features, as improper choices may lead to biologically irrelevant results. This clarification is intended to ensure that users apply the tool thoughtfully and derive meaningful insights.
-
-
eLife Assessment
This study represents a potentially useful tool for extracting quantitative data from intravital microscopy directed at in vivo cancer models. In general, this is an area of interest as accessible non-proprietary tools are needed and some evidence of the tool's utility is provided. However, the work in its current form is incomplete as it is heavily reliant on proprietary software to segment, track, and correct the data. In addition, there are significant reservations regarding the methods used to produce statistics in the software, limiting its applicability and the potential advance over other approaches.
-
Reviewer #1 (Public review):
Summary:
Intravital microscopy (IVM) is a powerful tool that facilitates live imaging of individual cells over time in vivo in their native 3D tissue environment. Extracting and analysing multi-parametric data from IVM images however is challenging, particularly for researchers with limited programming and image analysis skills. In this work, Rios-Jimenez and Zomer et al have developed a 'zero-code' accessible computational framework (BEHAV3D-Tumour Profiler) designed to facilitate unbiased analysis of IVM data to investigate tumour cell dynamics (via the tool's central 'heterogeneity module') and their interactions with the tumour microenvironment (via the 'large-scale phenotyping' and 'small-scale phenotyping' modules). It is designed as an open-source modular Jupyter Notebook with a user-friendly …
Reviewer #1 (Public review):
Summary:
Intravital microscopy (IVM) is a powerful tool that facilitates live imaging of individual cells over time in vivo in their native 3D tissue environment. Extracting and analysing multi-parametric data from IVM images however is challenging, particularly for researchers with limited programming and image analysis skills. In this work, Rios-Jimenez and Zomer et al have developed a 'zero-code' accessible computational framework (BEHAV3D-Tumour Profiler) designed to facilitate unbiased analysis of IVM data to investigate tumour cell dynamics (via the tool's central 'heterogeneity module') and their interactions with the tumour microenvironment (via the 'large-scale phenotyping' and 'small-scale phenotyping' modules). It is designed as an open-source modular Jupyter Notebook with a user-friendly graphical user interface and can be implemented with Google Colab, facilitating efficient, cloud-based computational analysis at no cost.
To demonstrate the utility of BEHAV3D-TP, they apply the pipeline to timelapse IVM imaging datasets to investigate the in vivo migratory behaviour of fluorescently labelled DMG cells in tumour-bearing mice. Using the tool's 'heterogeneity module' they were able to identify distinct single-cell behavioural patterns (based on multiple parameters such as directionality, speed, displacement, and distance from tumour edge) which was used to group cells into distinct categories (e.g. retreating, invasive, static, erratic). They next applied the framework's 'large-scale phenotyping' and 'small-scale phenotyping' modules to investigate whether the tumour microenvironment (TME) may influence the distinct migratory behaviours identified. To achieve this, they combine TME visualisation in vivo during IVM (using fluorescent probes to label distinct TME components) or ex vivo after IVM (by large-scale imaging of harvested, immunostained tumours) to correlate different tumour behavioural patterns with the composition of the TME. They conclude that this tool has helped reveal links between TME composition (e.g. degree of vascularisation, presence of tumour-associated macrophages) and the invasiveness and directionality of tumour cells, which would have been challenging to identify when analysing single kinetic parameters in isolation.
A key limitation of the pipeline is that it does not overcome the main challenges and bottlenecks associated with processing and extracting quantitative cellular data from timelapse and longitudinal intravital images. This includes correcting breathing-induced movement artifacts, automated registration of longitudinal images taken over days/weeks, and accurate, automated segmentation and tracking of individual cells over time. Indeed, there are currently no standardised computational methods available for IVM data processing and analysis, with most laboratories relying on custom-built solutions or manual methods. This isn't made explicit in the manuscript early on (described below), and the researchers rely on expensive software packages such as IMARIS for image processing and data extraction to feed the required parameters into their pipeline. This limitation unfortunately reduces the likely impact of BEHAV3D-TP on the IVM field.
Nonetheless, this computational framework appears to represent a useful and comparatively user-friendly tool to analyse dynamic multi-parametric data to help identify patterns in cell migratory behaviours, and to assess whether these behaviours might be influenced by neighbouring cells and structures in their microenvironment. When combined with other methods, it, therefore, has the potential to be a valuable addition to a researcher's IVM analysis 'tool-box'.
Strengths:
(1) The figures are clearly presented, and the manuscript is easy to follow.
(2) The pipeline appears to be intuitive and user-friendly for researchers with limited computational expertise. A detailed step-by-step video is also included to support its uptake.
(3) The different computational modules have been tested using a relevant dataset.
(4) All code is open source, and the pipeline can be implemented with Google Colab.
(5) The tool combines multiple dynamic parameters extracted from time-lapse IVM images to identify single-cell behavioural patterns and to cluster cells into distinct groups sharing similar behaviours, and provides avenues to map these onto in vivo or ex vivo imaging data of the tumour microenvironment.
Weaknesses:
(1) As highlighted above, the tool does not facilitate the extraction of quantitative kinetic cellular parameters (e.g. speed, directionality, persistence, and displacement) from intravital images. Indeed, to use the tool researchers must first extract dynamic cellular parameters from their IVM datasets, requiring access to expensive software (e.g. IMARIS as used here) and/or above-average computational expertise to develop and use custom-made open-source solutions. This limitation is not made explicit or discussed in the text.
(2) The number of cells (e.g. per behavioural cluster), and the number of independent mice, represented in each result figure, is not included in the figure legends and are difficult to ascertain from the methods.
(3) The data used to test the pipeline in this manuscript is currently not available, making it difficult to assess its usability. It would be important to include this for researchers to use as a 'training dataset'.
(4) Precisely how the BEHAV3D-TP large-scale phenotyping module can map large-scale spatial phenotyping data generated using LSR-3D imaging data and Cytomap to 3D intravital imaging movies is unclear. Further details in the text and methods would be beneficial to aid understanding.
(5) The analysis provides only preliminary evidence in support of the authors' conclusions on DMG cell migratory behaviours and their relationship with components of the tumour microenvironment. Conclusions should therefore be tempered in the absence of additional experiments and controls.
-
Reviewer #2 (Public review):
Summary:
The authors produce a new tool, BEHAV3D to analyse tracking data and to integrate these analyses with large and small-scale architectural features of the tissue. This is similar to several other published methods to analyse spatiotemporal data, however, the connection to tissue features is a nice addition, as is the lack of requirement for coding. The tool is then used to analyse tracking data of tumour cells in diffuse midline glioma. They suggest that 7 clusters exist within these tracks and that they differ spatially. They ultimately suggest that these behaviours occur in distinct spatial areas as determined by CytoMAP.Strengths:
(1) The tool appears relatively user-friendly and is open source. The combination with CytoMAP represents a nice option for researchers.
- The identification of …
Reviewer #2 (Public review):
Summary:
The authors produce a new tool, BEHAV3D to analyse tracking data and to integrate these analyses with large and small-scale architectural features of the tissue. This is similar to several other published methods to analyse spatiotemporal data, however, the connection to tissue features is a nice addition, as is the lack of requirement for coding. The tool is then used to analyse tracking data of tumour cells in diffuse midline glioma. They suggest that 7 clusters exist within these tracks and that they differ spatially. They ultimately suggest that these behaviours occur in distinct spatial areas as determined by CytoMAP.Strengths:
(1) The tool appears relatively user-friendly and is open source. The combination with CytoMAP represents a nice option for researchers.
- The identification of associations between cell track phenotype and spatial features is exciting and the diffuse midline glioma data nicely demonstrates how this could be used.
Weaknesses:
(1) The strength of democratizing this kind of analysis is undercut by the reliance upon Imaris for segmentation, so it would be nice if this was changed to an open-source option for track generation.
(2) The main issue is with the interpretation of the biological data in Figure 3 where ANOVA was used to analyse the proportional distribution of different clusters. Firstly the n is not listed so it is unclear if this represents an n of 3 where each mouse is an individual or whether each track is being treated as a test unit. If the latter this is seriously flawed as these tracks can't be treated as independent. Also, a more appropriate test would be something like a Chi-squared test or Fisher's exact test. Also, no error bars are included on the stacked bar graphs making interpretation impossible. Ultimately this is severely flawed and also appears to show very small differences which may be statistically different but may not represent biologically important findings. This would need further study.
(3) Figure 4 has similar statistical issues in that the n is not listed and, again, it is unclear whether they are treating each cell track as independent which, again, would be inappropriate. The best practice for this type of data would be the use of super plots as outlined in Lord et al. (2020) JCI - SuperPlots: Communicating reproducibility and variability in cell biology.
(4) The main issue that this raises is that the large-scale phenotyping module and the heterogeneity module appear designed to produce these statistical analyses that are used in these figures and, if they are based on the assumption that each track is independent, then this will produce inappropriate analyses as a default.
-
Reviewer #3 (Public review):
Summary:
The manuscript by Rios-Jimenez developed a computational tool, BEHAV3D Tumor Profiler, to analyze intravital imaging data and extract distinctive tumor cell migratory phenotypes based on the quantified 3D image data.
Weaknesses:
(1) The most challenging task of analyzing 3D time-lapse imaging data is to accurately segment and track the individual cells in 3D over a long time duration. BEHAV3D Tumor Profiler did not provide any new advancement in this regard, and instead relies on commercial software, Imaris, for this critical step. Imaris is known to have a very high error rate when used for analyzing 3D time-lapse data. In the Methods section, the authors themselves stated that "Tumor cell tracks were manually corrected to ensure accurate tracking". Based on our own experience of using Imaris, such …
Reviewer #3 (Public review):
Summary:
The manuscript by Rios-Jimenez developed a computational tool, BEHAV3D Tumor Profiler, to analyze intravital imaging data and extract distinctive tumor cell migratory phenotypes based on the quantified 3D image data.
Weaknesses:
(1) The most challenging task of analyzing 3D time-lapse imaging data is to accurately segment and track the individual cells in 3D over a long time duration. BEHAV3D Tumor Profiler did not provide any new advancement in this regard, and instead relies on commercial software, Imaris, for this critical step. Imaris is known to have a very high error rate when used for analyzing 3D time-lapse data. In the Methods section, the authors themselves stated that "Tumor cell tracks were manually corrected to ensure accurate tracking". Based on our own experience of using Imaris, such manual correction is tedious and often required for every time step of the movie. Therefore, Imaris is not a satisfactory tool for analyzing 3D time-lapse data. Moreover, Imaris is expensive and many research labs probably can't afford to buy it. The fact that BEHAV3D Tumor Profiler critically depends on the faulty ImarisTrack module makes it unclear whether the BEHAV3D tool or the results are reliable.
(2) The authors developed a "Heterogeneity module" to extract distinctive tumor migratory phenotypes from the cell tracks quantified by Imaris. The cell tracks of the individual tumor cells are all quite short, indicating relatively low motility of the tumor cells. It's unclear whether such short migratory tracks are sufficient to warrant the PCA analysis to identify the 7 distinctive migratory phenotypes shown in Figure 2d. It's also unclear whether these 7 migratory phenotypes correspond to unique functional phenotypes.
(3) Using only motility to classify tumor cell behaviours in the tumor microenvironment (TME) is probably not sufficient to capture the tumor cell difference. There are also other non-tumor cell types in the TME. If the authors aim to develop a computational tool that can elucidate tumor cell behaviors in the TME, they should consider other tumor cell features, e.g., morphology, proliferation state, and tumor cell interaction with other cell types, e.g., fibroblasts and distinct immune cells.
(4) The authors have already published two papers on BEHAV3D [Alieva M et al. Nat Protoc. 2024 Jul;19(7): 2052-2084; Dekkers JF, et al. Nat Biotechnol. 2023 Jan;41(1):60-69]. Although the previous two papers used BEHAV3D to analyze T cells, the basic pipeline and computational steps are similar, in particular regarding cell segmentation and tracking. The addition of a "Heterogeneity module" based on PCA analysis does not make a significant advancement in terms of image analysis and quantification.
-
Author response:
We want to thank the reviewers for their positive and constructive comments on the manuscript. We already addressed some of their concerns and are planning the following revisions to both BEHAV3D-TP and the corresponding manuscript to address the reviewers’ comments. Below, we provide a response to the most significant comments, followed by a detailed, point-by-point response:
(1) We acknowledge the reviewer's suggestion to incorporate open-source segmentation and tracking functionalities, increasing its accessibility to a wider user base; however, these additions fall outside the primary scope of our current work and represent a substantial undertaking in their own right. This topic has been comprehensively explored in other studies (e.g. https://doi.org/10.4049/jimmunol.2100811 ; https://doi.org/10.7554/eLife.60547 ; h…
Author response:
We want to thank the reviewers for their positive and constructive comments on the manuscript. We already addressed some of their concerns and are planning the following revisions to both BEHAV3D-TP and the corresponding manuscript to address the reviewers’ comments. Below, we provide a response to the most significant comments, followed by a detailed, point-by-point response:
(1) We acknowledge the reviewer's suggestion to incorporate open-source segmentation and tracking functionalities, increasing its accessibility to a wider user base; however, these additions fall outside the primary scope of our current work and represent a substantial undertaking in their own right. This topic has been comprehensively explored in other studies (e.g. https://doi.org/10.4049/jimmunol.2100811 ; https://doi.org/10.7554/eLife.60547 ; https://doi.org/10.1016/j.media.2022.102358 ; https://doi.org/10.1038/s41592-024-02295-6), which we will cite in our revised manuscript as indicated in our responses to the reviewers’ comments. Instead, the goal of our manuscript is to provide an analytical framework for processing data generated by existing segmentation and tracking pipelines. In our analyses, we used data processed with Imaris, a commercial software that, despite its limitations, is widely used by the intravital microscopy community due to its user-friendly platform for 3D image visualization and analysis. Nevertheless, to enhance compatibility with tracking data from various pipelines, we have modified our tool to accept data formats, such as those generated by open-source Fiji plugins like TrackMate (https://github.com/imAIgene-Dream3D/BEHAV3D_Tumor_Profiler?tab=readme-ov-file#data-input ). These updates are available in our GitHub repository, and we will describe this feature in the revised manuscript to emphasize compatibility with segmented and tracked data from diverse open-source platforms.
(2) We appreciate the reviewer’s suggestion to incorporate additional features into our analytical pipeline. In response, we have already updated the GitHub repository to allow users to input and select which features (dynamic, morphological, or spatial) they wish to include in the analysis (https://github.com/imAIgene-Dream3D/BEHAV3D_Tumor_Profiler?tab=readme-ov-file#feature-selection ) . In the revised manuscript, we will highlight this new functionality and provide examples using alternative datasets to demonstrate the application of these features.
(3) We appreciate the constructive feedback of reviewers #1 and #2 regarding the statistical analysis and interpretation of the data presented in Figures 3 and 4. We understand the importance of clarity and rigor in data analysis and presentation, and we are committed to addressing the concerns raised in the revised version of the manuscript.
(4) We appreciate Reviewer #1's suggestion regarding the inclusion of demo data, as we believe it would greatly enhance the usability of our pipeline. We acknowledge that this was an oversight on our part. To address this, we have now added demo data to our GitHub repository (https://github.com/imAIgene-Dream3D/BEHAV3D_Tumor_Profiler/tree/BEHAV3D_TP-v2.0/demo_datasets). In the upcoming revised manuscript, we will also ensure to reference this addition. Additionally, we will provide both original and processed IVM movie samples to support users in navigating the complete pipeline effectively.
(5) Finally, we agree with the reviewers to make some small changes to the manuscript based on their feedback.
Below we provide a point-by-point response to the reviewers’ comments, along with proposed revisions.
Reviewer #1:
Comment: A key limitation of the pipeline is that it does not overcome the main challenges and bottlenecks associated with processing and extracting quantitative cellular data from timelapse and longitudinal intravital images. This includes correcting breathing-induced movement artifacts, automated registration of longitudinal images taken over days/weeks, and accurate, automated segmentation and tracking of individual cells over time. Indeed, there are currently no standardised computational methods available for IVM data processing and analysis, with most laboratories relying on custom-built solutions or manual methods. This isn't made explicit in the manuscript early on (described below), and the researchers rely on expensive software packages such as IMARIS for image processing and data extraction to feed the required parameters into their pipeline. This limitation unfortunately reduces the likely impact of BEHAV3D-TP on the IVM field.
As highlighted above, the tool does not facilitate the extraction of quantitative kinetic cellular parameters (e.g. speed, directionality, persistence, and displacement) from intravital images. Indeed, to use the tool researchers must first extract dynamic cellular parameters from their IVM datasets, requiring access to expensive software (e.g. IMARIS as used here) and/or above-average computational expertise to develop and use custom-made open-source solutions. This limitation is not made explicit or discussed in the text.
As mentioned previously, we agree with the reviewer that image processing steps, such as segmentation, tracking, and motion correction, present significant challenges in intravital microscopy (IVM) data processing. While these aspects are being addressed by other researchers, our publication centers on the analysis of acquired data rather than on the image processing itself. Our motivation, as outlined in the manuscript, arises from our own experience: despite the substantial effort invested in image processing, researchers often rely on simplistic analytical approaches, such as averaging single parameters and comparing them across conditions. These approaches tend to overlook potential tumor heterogeneity.
Our work aimed to develop an analytical tool that provides a comprehensive framework for extracting more insights from processed IVM data, with a focus on two key aspects: capturing the heterogeneity of tumor behavior and examining the spatial distribution of these behaviors within the tumor microenvironment. In the revised manuscript, we will clarify the scope of our study, emphasizing its limitations as an analytical tool rather than an image-processing solution. Additionally, we will provide references to relevant literature on available (open-source) software options for image processing (e.g. Diego Ulisse Pizzagalli et al J Immunol (2022); Aby Joseph et al eLife (2020) ;Molina-Moreno M et al Medical Image Analysis (2022); Hidalgo-Cenalmor, I et al, Nat Methods (2024); Ershov. D et al Nat Methods (2022)).
Regarding the reviewer’s comment on our use of Imaris, we acknowledge that Imaris is a costly commercial software. However, based on our experience, it is widely used by the intravital microscopy community due to its user-friendly interface for 3D image visualization and analysis. Despite its limitations in accuracy and the fact that it is not open-source, we believe that including data processed with Imaris will be valuable to the IVM community.
However, to improve compatibility with data from other segmentation and tracking pipelines, we have already updated our tool to support formats generated by open-source Fiji plugins like TrackMate. These updates are available in our GitHub repository, and we will describe this functionality in detail in the revised manuscript to ensure compatibility with segmented and tracked data from various open-source platforms.
Comment: The number of cells (e.g. per behavioural cluster), and the number of independent mice, represented in each result figure, is not included in the figure legends and are difficult to ascertain from the methods.
We appreciate the reviewer's constructive feedback regarding the clarity of the number and type of replicates used in our analyses. In the revised manuscript, we will include detailed information in the figure legends regarding the number of cells (e.g., per behavioral cluster) and the number of independent mice represented in each result figure to ensure transparency.
Comment: The data used to test the pipeline in this manuscript is currently not available, making it difficult to assess its usability. It would be important to include this for researchers to use as a 'training dataset'.
As stated above we acknowledge that this was an oversight on our part and thank the reviewer for pointing this out. To address this, we have now added demo data to our GitHub repository (https://github.com/imAIgene-Dream3D/BEHAV3D_Tumor_Profiler/tree/BEHAV3D_TP-v2.0/demo_datasets). In the upcoming revised manuscript, we will also make sure to reference this addition. Additionally, we intend to provide both original and processed IVM movie samples to support users in navigating the complete pipeline effectively.
Comment: Precisely how the BEHAV3D-TP large-scale phenotyping module can map large-scale spatial phenotyping data generated using LSR-3D imaging data and Cytomap to 3D intravital imaging movies is unclear. Further details in the text and methods would be beneficial to aid understanding.
We appreciate the reviewer’s comment and will provide additional details in the text and methods of the revised manuscript to clarify how the BEHAV3D-TP module maps LSR-3D and Cytomap data to 3D intravital imaging movies.
Comment: The analysis provides only preliminary evidence in support of the authors' conclusions on DMG cell migratory behaviours and their relationship with components of the tumour microenvironment. Conclusions should therefore be tempered in the absence of additional experiments and controls.
We appreciate the reviewer’s comment and acknowledge that our conclusions should be tempered due to the preliminary nature of our evidence. To be able to directly analyze the impact of the brain tumor microenvironment on cancer cell behavior, we will include a new set of analyses in the revised manuscript. Specifically, we will utilize BEHAV3D-TP to analyze existing IVM data from adult gliomas with and without macrophage depletion (Alieva et al, Scientific Reports, 2017; https://doi.org/10.1038/s41598-017-07660-4 ) to evaluate the differences in heterogeneous cell populations under these conditions. Since this analysis pertains to a different tumor type, we will revise our conclusions accordingly and emphasize the necessity for additional experiments and controls to further validate our findings on DMG cell migratory behaviors and their relationship with the tumor microenvironment.
Reviewer #2:
Comment: The strength of democratizing this kind of analysis is undercut by the reliance upon Imaris for segmentation, so it would be nice if this was changed to an open-source option for track generation.
As noted in our previous response to Reviewer #1, we would like to point out that although Imaris is a commercial software, it is widely used in the intravital microscopy (IVM) community due to its user-friendly interface. One of its key advantages, which we also utilized, is semi-automated data tracking that allows for manual corrections in 3D—a process that can be more challenging in other open-source software with less effective data visualization.
However, we recognize that enhancing our pipeline's compatibility with open-source options is important. To this end, we have already updated our tool to support data formats generated by open-source Fiji plugins like TrackMate, improving compatibility with various segmentation and tracking pipelines (https://github.com/imAIgene-Dream3D/BEHAV3D_Tumor_Profiler?tab=readme-ov-file#data-input ). We will describe these updates in the revised manuscript to clarify our study's scope and the available image processing options.
Comment: The main issue is with the interpretation of the biological data in Figure 3 where ANOVA was used to analyse the proportional distribution of different clusters. Firstly the n is not listed so it is unclear if this represents an n of 3 where each mouse is an individual or whether each track is being treated as a test unit. If the latter this is seriously flawed as these tracks can't be treated as independent. Also, a more appropriate test would be something like a Chi-squared test or Fisher's exact test. Also, no error bars are included on the stacked bar graphs making interpretation impossible. Ultimately this is severely flawed and also appears to show very small differences which may be statistically different but may not represent biologically important findings. This would need further study.
We appreciate the reviewer’s insightful comments regarding the interpretation of the biological data in Figure 3. To clarify, each mouse serves as an independent unit in this analysis. We believe that ANOVA is the appropriate test for comparing the proportions of different behavioral signatures across the tumor microenvironment (TME) regions identified by large-scale phenotyping. However, we acknowledge that using a stacked bar plot may have been misleading. While a Chi-squared test could show differences in the distribution of behavioral signatures, it would not indicate which specific signatures are responsible for those differences. Therefore, in the revised manuscript, we will retain the ANOVA analysis but will represent the proportions using a bar chart that clearly illustrates multiple conditions for each behavioral cluster. We also appreciate the reviewer’s concern regarding the transparency of our data. In the revised manuscript, we will include the number of replicates for all figures to enhance clarity and understanding.
Comment: Figure 4 has similar statistical issues in that the n is not listed and, again, it is unclear whether they are treating each cell track as independent which, again, would be inappropriate. The best practice for this type of data would be the use of super plots as outlined in Lord et al. (2020) JCI - SuperPlots: Communicating reproducibility and variability in cell biology.
We appreciate the reviewer’s comments and suggestions regarding Figure 4. In the revised manuscript, we will clarify the number of replicates used and our approach to treating cell tracks as independent units. We will implement super-plots where appropriate, to enhance the communication of reproducibility and variability in our data.
Comment: The main issue that this raises is that the large-scale phenotyping module and the heterogeneity module appear designed to produce these statistical analyses that are used in these figures and, if they are based on the assumption that each track is independent, then this will produce inappropriate analyses as a default.
We appreciate the reviewer’s comment, though we find ourselves unsure about the specific concern being raised. To clarify, each mouse is treated as an independent unit in our analyses. For each large-scale phenotyping region, we measure the proportion of tumor cells displaying a specific behavioral phenotype independently for each mouse. These proportions are then used for statistical analysis. We hope this explanation provides clarity, and we will adjust the manuscript to better convey this methodology.
Reviewer #3:
Comment: The most challenging task of analyzing 3D time-lapse imaging data is to accurately segment and track the individual cells in 3D over a long time duration. BEHAV3D Tumor Profiler did not provide any new advancement in this regard, and instead relies on commercial software, Imaris, for this critical step. Imaris is known to have a very high error rate when used for analyzing 3D time-lapse data. In the Methods section, the authors themselves stated that "Tumor cell tracks were manually corrected to ensure accurate tracking". Based on our own experience of using Imaris, such manual correction is tedious and often required for every time step of the movie. Therefore, Imaris is not a satisfactory tool for analyzing 3D time-lapse data. Moreover, Imaris is expensive and many research labs probably can't afford to buy it. The fact that BEHAV3D Tumor Profiler critically depends on the faulty ImarisTrack module makes it unclear whether the BEHAV3D tool or the results are reliable.
If the authors want to "democratize the analysis of heterogeneous cancer cell behaviors", they should perform image segmentation and tracking using open-source codes (e.g., Cellpose, Stardisk & 3DCellTracker) and not rely on the expensive and inaccurate ImarisTrack Module for the image analysis step of BEHAV3D.
We appreciate the reviewer’s comments on the challenges of segmenting and tracking individual cells in 3D time-lapse imaging data. As mentioned previously, our primary focus is to develop an analytical tool for comprehensive data analysis rather than developing tools for image processing. To enhance accessibility, we have updated our tool to support data formats from open-source Fiji plugins, such as TrackMate, which will benefit users without access to commercial software (https://github.com/imAIgene-Dream3D/BEHAV3D_Tumor_Profiler?tab=readme-ov-file#data-input ).
While we recognize the limitations of Imaris, it remains widely used in the intravital microscopy community due to its user-friendly interface for 3D visualization and semi-automated segmentation capabilities. Since no perfect tracking method currently exist, we utilized Imaris for its ability to allow manual corrections of faulty tracks, ensuring the reliability of our results. This approach was the best available option when we began our analysis, allowing us to obtain accurate results efficiently.
In the revised manuscript, we will clarify our methodology and provide information on both Imaris and alternative processing options to strengthen the reliability of our findings.
Comment: The authors developed a "Heterogeneity module" to extract distinctive tumor migratory phenotypes from the cell tracks quantified by Imaris. The cell tracks of the individual tumor cells are all quite short, indicating relatively low motility of the tumor cells. It's unclear whether such short migratory tracks are sufficient to warrant the PCA analysis to identify the 7 distinctive migratory phenotypes shown in Figure 2d. It's also unclear whether these 7 migratory phenotypes correspond to unique functional phenotypes.
For the 7 distinctive motility clusters, the authors should provide a more detailed analysis of the differences between them. It's unclear whether the difference in retreating, slow retreating, erratic, static, slow, slow invading, and invading correspond to functional difference of the tumor cells.
While some tumor cells exhibit limited motility, indicated by short tracks, others demonstrate significant migratory capabilities. This variability in tumor cell behavior is a central focus of our analysis, and our tool is specifically designed to identify and distinguish these differences. Our PCA analysis effectively captures this variability, as illustrated in Figure 2 d-f. It differentiates between cells exhibiting varying degrees of migratory behavior, including both highly migratory and less migratory phenotypes, as well as their directionality relative to the tumor core and the persistence of their movements. Thus, we believe that our approach provides valuable insights into the distinct migratory phenotypes within the tumor microenvironment. We will clarify these aspects further in the revised manuscript to enhance the reader's understanding of our findings.
While our current manuscript does not provide explicit evidence linking each motility cluster to functional differences among the tumor cells, it is important to note that the state of the field supports the idea that cell dynamics can predict cell states and phenotypes. Research conducted by ourselves (Dekkers, Alieva et al., Nat Biotech, 2023) and others, such as Craiciuc et al. (Nature, 2022) and Freckmann et al. (Nat Comm, 2022) has shown that variations in cell motility patterns are indicative of underlying functional characteristics. For instance, cell morphodynamic features have been shown to reflect differences in cell types, T cell targeting states, tumor metastatic potential, and drug resistance states. In the revised manuscript, we will reference relevant studies to underscore the biological significance of these behaviors. By doing so, we hope to clarify the potential implications of our findings and strengthen the overall narrative of our research.
Comment: Using only motility to classify tumor cell behaviours in the tumor microenvironment (TME) is probably not sufficient to capture the tumor cell difference. There are also other non-tumor cell types in the TME. If the authors aim to develop a computational tool that can elucidate tumor cell behaviors in the TME, they should consider other tumor cell features, e.g., morphology, proliferation state, and tumor cell interaction with other cell types, e.g., fibroblasts and distinct immune cells.
The authors should expand the scale of tumor behavior features to classify the tumor phenotype clusters, e.g., to include tumor morphology, proliferation state, and tumor cell interaction with other TME cell types.
We believe that using dynamic features alone is sufficient to capture differences in tumor behavior, as demonstrated by our results in Figure 2. However, we appreciate the reviewer’s suggestion to consider additional features, such as cell morphology and interactions with other cell types, to finetune our analyses. To this end, we have adapted our pipeline to be compatible with various features present in the data (https://github.com/imAIgene-Dream3D/BEHAV3D_Tumor_Profiler/tree/BEHAV3D_TP-v2.0?tab=readme-ov-file#feature-selection ). We will emphasize this in the revised manuscript. However, we would like to point out that not all features may provide informative insights and that a wide range of features can instead introduce biologically irrelevant noise, making interpretation more challenging. For instance, in 3D microscopy, the z-axis resolution is typically lower, which can lead to artifacts like elongation in that direction. Adding morphological features that capture this may skew the analysis. Therefore, we believe that incorporating additional features should be approached with caution. We will clarify these considerations in the revised manuscript to better guide users in utilizing our computational tool effectively. We will also reference the use of unbiased feature selection techniques, such as bootstrapping methods, to identify biologically relevant features based on the conditions provided (D.G. Aragones et al, Computers in Biology and Medicine (2024)).
Comment: The authors have already published two papers on BEHAV3D [Alieva M et al. Nat Protoc. 2024 Jul;19(7): 2052-2084; Dekkers JF, et al. Nat Biotechnol. 2023 Jan;41(1):60-69]. Although the previous two papers used BEHAV3D to analyze T cells, the basic pipeline and computational steps are similar, in particular regarding cell segmentation and tracking. The addition of a "Heterogeneity module" based on PCA analysis does not make a significant advancement in terms of image analysis and quantification.
We want to emphasize that we have no intention of duplicating our previous publications. In this manuscript, we have consistently cited our foundational papers, where BEHAV3D was first developed for T cell migratory analysis in in vitro settings. In the introduction, we clearly state that our earlier work inspired us to adopt a similar approach for analyzing cell behavior in intravital microscopy (IVM) data, addressing the specific needs and complexities of analyzing tumor cell behaviors in the tumor microenvironment.
Importantly, our new work provides several key advancements: 1) a pipeline specifically adapted for intravital microscopy (IVM) data; 2) integration of spatial characteristics from both large-scale and small-scale phenotyping; and 3) a zero-code approach designed to empower researchers without coding skills to effectively utilize the tool. We believe that these enhancements represent meaningful progress in the analysis of cell behaviors within the tumor microenvironment which will be valuable for the IVM community. We will ensure that these points are clearly articulated in the revised manuscript.
-
