Unfolding and identification of membrane proteins in situ
Curation statements for this article:-
Curated by eLife

Evaluation Summary:
This paper presents a method to identify membrane proteins in native cell membranes based on a combination of single molecule AFM and an unsupervised clustering procedure to identify clusters of single-protein curves. This original approach represents a definitive step forward for AFM technology and methodology, which can generally only be used to characterize purified biomolecules of known identity. The work will be of interest to all students of membrane biology and especially membrane proteins.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #2 and Reviewer #3 agreed to share their names with the authors.)
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Single-molecule force spectroscopy (SMFS) uses the cantilever tip of an atomic force microscope (AFM) to apply a force able to unfold a single protein. The obtained force-distance curve encodes the unfolding pathway, and from its analysis it is possible to characterize the folded domains. SMFS has been mostly used to study the unfolding of purified proteins, in solution or reconstituted in a lipid bilayer. Here, we describe a pipeline for analyzing membrane proteins based on SMFS, which involves the isolation of the plasma membrane of single cells and the harvesting of force-distance curves directly from it. We characterized and identified the embedded membrane proteins combining, within a Bayesian framework, the information of the shape of the obtained curves, with the information from mass spectrometry and proteomic databases. The pipeline was tested with purified/reconstituted proteins and applied to five cell types where we classified the unfolding of their most abundant membrane proteins. We validated our pipeline by overexpressing four constructs, and this allowed us to gather structural insights of the identified proteins, revealing variable elements in the loop regions. Our results set the basis for the investigation of the unfolding of membrane proteins in situ, and for performing proteomics from a membrane fragment.
Article activity feed
-

-

Author Response
Reviewer #3 (Public Review):
Gavanetto et al. propose an interesting method to identify membrane proteins based on the analysis of single-molecule AFM (smAFM) force-extension traces obtained from native plasma membranes. In the proposed pipeline, the authors use smAFM to non-specifically probe isolated plasma membranes by recording a large number (millions) of force-extension traces. While, as expected, most of them lack any binding or represent spurious events, the authors use an unsupervised clustering algorithm to identify groups of force-extension curves with a similar mechanical pattern, suggesting that each cluster corresponds to a unique protein species that can be fingerprinted by its specific force-extension pattern. By implementing a Bayesian framework, the authors contrast the identified groups with …
Author Response
Reviewer #3 (Public Review):
Gavanetto et al. propose an interesting method to identify membrane proteins based on the analysis of single-molecule AFM (smAFM) force-extension traces obtained from native plasma membranes. In the proposed pipeline, the authors use smAFM to non-specifically probe isolated plasma membranes by recording a large number (millions) of force-extension traces. While, as expected, most of them lack any binding or represent spurious events, the authors use an unsupervised clustering algorithm to identify groups of force-extension curves with a similar mechanical pattern, suggesting that each cluster corresponds to a unique protein species that can be fingerprinted by its specific force-extension pattern. By implementing a Bayesian framework, the authors contrast the identified groups with proteomics databases, which provide the most likely proteins that correspond to the identified force-extension clusters. A set of control experiments complements the manuscript to validate the proposed methodology, such as the application of their pipeline using purified samples or overexpressing a specific protein species to enrich its population.
The primary strength of the manuscript is its originality, as it proposes a novel application of smAFM as a protein-detection method that can be applied in native samples. This methodology combines ingredients from conventional mass spectrometry and cryoEM; the contour length released upon extending a protein is a direct measure of its sequence extension (related to its mass), but the force pattern contains insightful information about the protein's structure. In this sense, the authors' proposal is very smart. However, the relationship between protein structure and mechanics is far from straightforward, and here perhaps lies one of the main limitations of the proposed method. This is particularly true for the case of membrane proteins, where we cannot talk about protein unfolding in its classical sense but rather about pullout events which is likely what each peak corresponds to (indeed, the authors speak throughout the paper about unfolding events, which I believe is not the correct term).
We fully agree with the semantics concern of reviewer #3 about the term unfolding. A membrane protein when pulled with the tip of the AFM is pulled out of the membrane (see 2 in the image below) and, simultaneously, the segment that is pulled out unfolds (see 3). To our knowledge, force peaks corresponding to a contour length equal to 2 where not consistently observed or reported (when e.g. a transmembrane alpha helix is out of the membrane but folded).
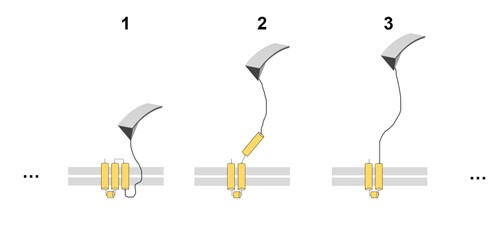
Since the field evolved with the practice of using the term ‘unfolding’ even for membrane proteins (see for instance (Kessler and Gaub, 2006; Oesterhelt et al., 2000; Yu et al., 2017) and many others), we would prefer to stick with this term.
In the context of membrane proteins the term unfolding therefore refers to at least the tertiary structure of the protein, because it is not clear when and at which timescale the secondary structures really unfolds.
We pointed this out in Line 131 (and following Lines).
-
Evaluation Summary:
This paper presents a method to identify membrane proteins in native cell membranes based on a combination of single molecule AFM and an unsupervised clustering procedure to identify clusters of single-protein curves. This original approach represents a definitive step forward for AFM technology and methodology, which can generally only be used to characterize purified biomolecules of known identity. The work will be of interest to all students of membrane biology and especially membrane proteins.
(This preprint has been reviewed by eLife. We include the public reviews from the reviewers here; the authors also receive private feedback with suggested changes to the manuscript. Reviewer #2 and Reviewer #3 agreed to share their names with the authors.)
-
Reviewer #1 (Public Review):
The work aims to develop a new data analysis pipeline that allows them to identify individual membrane proteins from native membranes using analysis of AFM force-extension curves. They introduce a method to isolate patches of cell membrane for AFM force spectroscopy analysis. To validate their approach, they first demonstrate the ability to measure unfolding traces of known membrane proteins transfected into cells. They develop a custom data analysis pipeline that clusters repeated patterns in the unfolding traces. My understanding is that the data analysis procedure is not completely automated, but may require some user intervention to select useable regions of the curves. The data processing is therefore likely a combination of manual and automated steps. Following the demonstration to cluster the curves, …
Reviewer #1 (Public Review):
The work aims to develop a new data analysis pipeline that allows them to identify individual membrane proteins from native membranes using analysis of AFM force-extension curves. They introduce a method to isolate patches of cell membrane for AFM force spectroscopy analysis. To validate their approach, they first demonstrate the ability to measure unfolding traces of known membrane proteins transfected into cells. They develop a custom data analysis pipeline that clusters repeated patterns in the unfolding traces. My understanding is that the data analysis procedure is not completely automated, but may require some user intervention to select useable regions of the curves. The data processing is therefore likely a combination of manual and automated steps. Following the demonstration to cluster the curves, they use mass spectrometry databases along with protein structure databases to make Bayesian predictions about which membrane proteins are most likely observed. This provides a new bioanalytical technique for membranes.
-
Reviewer #2 (Public Review):
The authors present experimental and computational advances towards the development of their approach, as well as very nice experiments validating their technique. The discussion of the different artifacts, and features of their method that address these artifacts, is very nice. The discussion of the method's limitations in the discussion section is also nice.
However, I do not feel that this manuscript has taken the critical step of clearly illustrating the benefits that this approach brings to the broader scientific community. The authors should look through their dataset for useful insights to better illustrate the utility of their technique. Again, I think the technique is very interesting and important but additional analyses that clearly illustrate the utility of the technique to scientific research …Reviewer #2 (Public Review):
The authors present experimental and computational advances towards the development of their approach, as well as very nice experiments validating their technique. The discussion of the different artifacts, and features of their method that address these artifacts, is very nice. The discussion of the method's limitations in the discussion section is also nice.
However, I do not feel that this manuscript has taken the critical step of clearly illustrating the benefits that this approach brings to the broader scientific community. The authors should look through their dataset for useful insights to better illustrate the utility of their technique. Again, I think the technique is very interesting and important but additional analyses that clearly illustrate the utility of the technique to scientific research will be necessary. -
Reviewer #3 (Public Review):
Gavanetto et al. propose an interesting method to identify membrane proteins based on the analysis of single-molecule AFM (smAFM) force-extension traces obtained from native plasma membranes. In the proposed pipeline, the authors use smAFM to non-specifically probe isolated plasma membranes by recording a large number (millions) of force-extension traces. While, as expected, most of them lack any binding or represent spurious events, the authors use an unsupervised clustering algorithm to identify groups of force-extension curves with a similar mechanical pattern, suggesting that each cluster corresponds to a unique protein species that can be fingerprinted by its specific force-extension pattern. By implementing a Bayesian framework, the authors contrast the identified groups with proteomics databases, …
Reviewer #3 (Public Review):
Gavanetto et al. propose an interesting method to identify membrane proteins based on the analysis of single-molecule AFM (smAFM) force-extension traces obtained from native plasma membranes. In the proposed pipeline, the authors use smAFM to non-specifically probe isolated plasma membranes by recording a large number (millions) of force-extension traces. While, as expected, most of them lack any binding or represent spurious events, the authors use an unsupervised clustering algorithm to identify groups of force-extension curves with a similar mechanical pattern, suggesting that each cluster corresponds to a unique protein species that can be fingerprinted by its specific force-extension pattern. By implementing a Bayesian framework, the authors contrast the identified groups with proteomics databases, which provide the most likely proteins that correspond to the identified force-extension clusters. A set of control experiments complements the manuscript to validate the proposed methodology, such as the application of their pipeline using purified samples or overexpressing a specific protein species to enrich its population.
The primary strength of the manuscript is its originality, as it proposes a novel application of smAFM as a protein-detection method that can be applied in native samples. This methodology combines ingredients from conventional mass spectrometry and cryoEM; the contour length released upon extending a protein is a direct measure of its sequence extension (related to its mass), but the force pattern contains insightful information about the protein's structure. In this sense, the authors' proposal is very smart. However, the relationship between protein structure and mechanics is far from straightforward, and here perhaps lies one of the main limitations of the proposed method. This is particularly true for the case of membrane proteins, where we cannot talk about protein unfolding in its classical sense but rather about pullout events which is likely what each peak corresponds to (indeed, the authors speak throughout the paper about unfolding events, which I believe is not the correct term). Unlike most proteins-which unfold along a unique pathway, especially at high forces-the removal of membrane proteins is typically rather heterogeneous, with multiple parallel pathways. In this sense, the authors are likely to identify only those proteins with a unique or dominant pathway and miss potential proteins that exhibit heterogeneity. A second limitation recognized by the authors is the low yield of their method since only a meagre percentage of traces (~2%) correspond to identifiable protein patterns. This implies that only very abundant proteins can be identified, severely limiting the proposed method's practical applicability.
Overall, I believe that recognizing its natural limitations; the present work is a remarkable contribution to protein science due to the originality of the approach and the careful control experiments that validate the method.
-
