Virus adaptation to heparan sulfate comes with capsid stability tradeoff
Curation statements for this article:-
Curated by eLife

eLife Assessment
This is an important work and it correlates capsid stability with mutations that promote heparan sulfate binding. The data is solid, but there is a need for further analysis and experiments to support the claims and to propose a more detailed mechanism that could explain how these mutations altered capsid stability.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Because of high mutation rates, viruses constantly adapt to new environments. When propagated in cell lines, certain viruses acquire positively charged amino acids on their surface proteins, enabling them to utilize negatively charged heparan sulfate (HS) as an attachment receptor. In this study, we used enterovirus A71 (EV-A71) as the model and demonstrated that, unlike the parental MP4 variant, the cell-adapted strong HS-binder MP4-97R/167 G does not require acidification for uncoating and releases its genome in the neutral or weakly acidic environment of early endosomes. We experimentally confirmed that this pH-independent entry is not associated with the use of HS as an attachment receptor but rather with compromised capsid stability. We then extended these findings to another HS-dependent strain. In summary, our data indicate that the acquisition of capsid mutations conferring affinity for HS comes together with decreased capsid stability and allows EV-A71 to enter the cell via a pH-independent pathway. This pH-independent entry mechanism boosts viral replication in cell lines but may prove deleterious in vivo , especially for enteric viruses crossing the acidic gastric environment before reaching their primary replication site, the intestine. Our study thus provides new insight into the mechanisms underlying the in vivo attenuation of HS-binding EV-A71 strains. Not only are these viruses hindered in tissues rich in HS due to viral trapping, as generally accepted, but our research reveals that their diminished capsid stability further contributes to attenuation in vivo . This underscores the complex relationship between HS-binding, capsid stability, and viral fitness, where increased replication in cell lines coincides with attenuation in harsh in vivo environments like the gastrointestinal tract.
Article activity feed
-

-

Author Response:
Reviewer #4 (Public Review):
In this work, Tee et al. study the implications of Heparan Sulfate (HS) binding mutations observed on the Enterovirus A71 (EV-A71) capsid. HS-binding mutations are observed for several virus infections and are often presumed to be a cell culture adaptation. However, in the case of EV-A71, the presence of HS-binding mutations in clinical samples and the contradictory findings in animal studies have made the clinical relevance of HS-binding a subject of debate. Therefore, to better understand the role of HS-binding in EV-A71, the authors use a mouse-adapted EV-A71 variant (MP4) and compare it to a cell-adapted strong HS-binder (MP4-97R/167G). Using these two variants, the authors show that the strong HS-binder does not require acidification for uncoating and genome release. Furthermore, it is …
Author Response:
Reviewer #4 (Public Review):
In this work, Tee et al. study the implications of Heparan Sulfate (HS) binding mutations observed on the Enterovirus A71 (EV-A71) capsid. HS-binding mutations are observed for several virus infections and are often presumed to be a cell culture adaptation. However, in the case of EV-A71, the presence of HS-binding mutations in clinical samples and the contradictory findings in animal studies have made the clinical relevance of HS-binding a subject of debate. Therefore, to better understand the role of HS-binding in EV-A71, the authors use a mouse-adapted EV-A71 variant (MP4) and compare it to a cell-adapted strong HS-binder (MP4-97R/167G). Using these two variants, the authors show that the strong HS-binder does not require acidification for uncoating and genome release. Furthermore, it is demonstrated that the capsid stability of the HS-binding variant is compromised, resulting in pH-independent uncoating. Overall, this study provides new insights demonstrating that seemingly beneficial mutations increasing viral replication may be counterbalanced by other unintended consequences.
Strengths:
The thoroughness of the experiments performed to demonstrate that the HS-binding phenotype results in pH-independent entry and capsid destabilisation is worth highlighting. In this regard, the authors have explored viral entry using a range of approaches involving lysosomotropic drugs, viral binding assays, and neutral red-labelled viruses coupled with diverse techniques such as FISH, RNAscope, and transient expression of constitutively active molecules to inhibit parts of the viral cycle. In my opinion, this is necessary to rule out the other downstream effects of the lysomotropic drugs and to confirm the role of the HS-binding mutation in the entry phase. The use of in silico analysis coupled with negative staining electron microscopy and environmental challenge assays is notable. Finally, the demonstration of some of the work using a human-relevant strain is commendable.
We appreciate the reviewer recognition of the significance of our study and the precious advises.
Weaknesses:
A major weakness in this study is the focus on using a mouse-adapted EV-A71 strain (MP4). In the introduction, it is argued that HS-binding mutations are controversial due to their occurrence in cell culture. However, due to host limitations, mice are not the natural hosts for EV-A71 and thus, the same argument can be made for a mouse-adapted strain. It is not clear how different this strain is from circulating EV-A71 strains and the relevance of these findings to the human situation is questionable. This is particularly made evident in the discussion where it is highlighted that HS-binding variants (VP1-145G/Q mutants) have been associated with severe neurological cases while the same variants show attenuated phenotypes in mice and monkeys. This contrast between clinical data and animal studies should be highlighted in the introduction, rather than later in the discussion, as currently the in vivo animal studies are presented as the optimal situation and may lead to misconstrued conclusions from the results.
As requested by the reviewer, we included new experiments performed with a clinical strain isolated in an immunosuppressed patient (Cordey et al., 2012). We compared the sensitivity of this human strain harboring or not the VP1 L97R and E167G mutations to HCQ and confirmed that the similar differential sensitivity to HCQ was observed as with the MP4 variant. This result is presented as a new supplementary figure (Figure 6-figure supplement 1) and is described in the result section of the revised manuscript (Page 7, lines 251).
Page 7, lines 251: To determine if our observations are applicable to human strains, we examined the sensitivity of a closely related clinical strain. This strain was isolated from the respiratory tract of an immunosuppressed patient with a disseminated EV-A71 infection27. Additionally, we tested a strong HS-binding derivative that harbors the same VP1-L97R and E167G mutations as our MP4 double mutant. Notably, this human clinical strain shares 98.3% amino acid similarity with the MP4 variant used in this study and exhibits similar HS-binding phenotypes28. As shown in Figure 6-figure supplement 1, the original human strain was inhibited by HCQ, whereas the double mutant exhibited insensitivity to the drug.
We also added the comment about discrepancy between clinical data and animal studies in the introduction as requested (page 2, lines 69-76): However, epidemiological surveillance of human EV-A71 infections19-21 and experimental evidence from 2D human fetal intestinal models22, human airway organoids23 and air-liquid interface cultures24 suggest that HS binding may enhance viral replication and virulence in humans. In addition, recent research has shown that EV-A71 can be released and transmitted via cellular extrusions25 or exosomes26, potentially preventing viral trapping of HS-binding strains in the circulation. Further studies are required to evaluate the true impact of HS-binding mutations on the spread and virulence of EV-A71 in both animal models and humans.
An important consideration is that the results are based primarily on image analysis. The inclusion of RT-qPCR and/or plaque assays as supplementary data will help strengthen the findings.
We have performed RT-qPCR to confirm the immunostaining data and included them in the supplementary data (Figure 1-figure supplement 1E). Reference to these data is made in the result section [Page 4, lines 114-116: These results were confirmed by viral load quantification with real-time RT-PCR (Figure 1-figure supplement 1E).]
Moreover, there are suggestions of an intermediate binder having a different phenotype. As this intermediate binder is the clinical phenotype, data on the entry of this intermediate binder will be valuable.
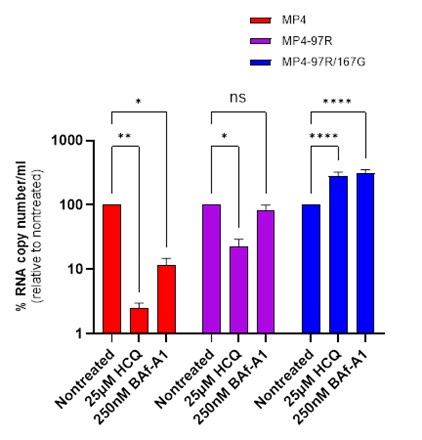
While we agree with reviewer that the single mutant is an intermediate binder and exhibits a clinical phenotype, we made the decision to work with variants that display clear phenotypes, selecting MP4 and the double mutant, as the latter is fully attenuated in both immunocompetent and immunosuppressed mice (Weng et al., 2023). Additionally, we performed an experiment using HCQ, where we observed an intermediate effect with the single mutant. This further confirmed our decision to proceed with MP4 and the double mutant for all experiments. The data supporting this are shown in Author response image 1, which we are sharing exclusively with the reviewer.
Author response image 1.
Differential sensitivity of MP4, MP4-97R and MP4-97R167G to Lysosomotropic drugs
Another weakness in the study is the lack of contextualization of the results to current EV-A71 literature. For instance, SCARB2 is referred to as the internalization receptor but a recent study has shown that SCARB2 is not required for internalization (https://doi.org/10.1128%2Fjvi.02042-21). The findings from this study are consistent with the localization of SCARB2 in the lysosomal membranes. Furthermore, the same study has highlighted host sulfation as a key factor in EV-A71 entry. Post-translational sulfation introduces negatively charged residues on host proteins including HS and SCARB2. This increases the binding of HS-binding strains to these proteins. In this regard, the reduced infectivity upon soluble SCARB2 treatment may simply be due to enhanced binding rather than capsid opening as suggested in the results. Therefore, additional experiments (e.g. nSEM following soluble SCARB2 treatment) must be performed to support the conclusion of capsid opening, due to inherent instability, upon SCARB2 binding.
We apologize for not citing this relevant literature excluding the role of SCARB2 in viral attachment. We have now included these references in the revised version of the manuscript. (Page 2, lines 54-56: “Since SCARB2 is mostly localized on endosomal and lysosomal membrane and sparsely on plasma membrane3,5, it seems to play only a minor role in EV-A71 cell attachment6,7.
We thank the reviewer for mentioning the possibility that the sulfation of SCARB2 may enhance its binding to the mutated virus compared to the wild-type virus, potentially explaining the selective competitive inhibition of this variant by soluble SCARB2 produced in mammalian cells. To investigate this hypothesis, we performed nsEM imaging of the double mutant incubated with soluble SCARB2 and we observed an increase in the proportion of empty capsids in the presence of soluble SCARB2 (4% versus 0.7%), supporting our original findings that the inactivation is indeed associated with capsid opening. The results are included in the revised manuscript in Figure 5-figure supplement 4 and described on Page 7, lines 243-245: “However, the double mutant exhibited a ~5-fold increase in empty capsid percentage after treatment with sSCARB2 (Figure 5-figure supplement 4), consistent with the functional data above.”
In addition to the above, other existing literature on EV-A71 pathogenesis using organoids contradicts some of the explanations of differential phenotype in clinical observations versus mice models. In the introduction, it is suggested that reduced neurovirulence of HS-binding strains is due to binding to the vascular endothelia. However, the correlation of clinical severity to viremia (https://doi.org/10.1186/1471-2334-14-417) and the association of HS-binding mutants to clinical disease counteract this suggestion. Similarly, viral infection in human organoids with EV-A71 results in as low as 0.4% of the cells being infected (https://doi.org/10.1038/s41564-023-01339-5). In this case, if viral binding to (ubiquitously expressed) HS results in viral trapping then the HS-binding mutants should show lowered infectivity in organoid models rather than the observed higher infectivity (https://doi.org/10.3389/fmicb.2023.1045587, https://doi.org/10.1038/s41426-018-0077-2). Finally, EV-A71 release has also been shown to occur in exosomes (https://doi.org/10.1093%2Finfdis%2Fjiaa174) which effectively provides a protective lipid membrane. These recent findings must be incorporated into the article and will help better contextualize their findings.
We appreciate the reviewer thoughtful comments. We do not believe that the correlation between clinical severity and viremia contradicts the viral trapping hypothesis. For strains that do not bind to HS, the absence of viral trapping could indeed lead to higher viral concentrations in the bloodstream, potentially increasing neurovirulence. However, we agree with the reviewer that other observations in humans, along with experimental data from more relevant models such as organoids, challenge the trapping hypothesis. We are grateful for the suggested citations and have incorporated these references in the introduction, where we discuss this point in more detail
Page 2, lines 69-76: “However, epidemiological surveillance of human EV-A71 infections19-21 and experimental evidence from 2D human fetal intestinal models22, human airway organoids23 and air-liquid interface cultures24 suggest that HS binding may enhance viral replication and virulence in humans. In addition, recent research has shown that EV-A71 can be released and transmitted via cellular extrusions25 or exosomes26, potentially preventing viral trapping of HS-binding strains in the circulation. Further studies are required to evaluate the true impact of HS-binding mutations on the spread and virulence of EV-A71 in both animal models and humans.”
Overall, the authors present new findings with convincing methodology. The manuscript can be improved in the contextualization of the findings and highlighting the weakness in translating these findings to resolve the debate surrounding the relevance of HS-binding phenotype. The inclusion of additional experiments and data recommended to the authors will also help strengthen the manuscript.
-
eLife Assessment
This is an important work and it correlates capsid stability with mutations that promote heparan sulfate binding. The data is solid, but there is a need for further analysis and experiments to support the claims and to propose a more detailed mechanism that could explain how these mutations altered capsid stability.
-
Reviewer #1 (Public Review):
This article is interesting because the phenotype of the virus with mutations that alter the affinity of HS has been associated with how the viral particle interacts with HS and, thus, with binding and entry. However, the data in this manuscript is compelling and strongly suggests that the mutation that increases the affinity of HS alters capsid stability. To my knowledge, this is the first evidence that such mutation causes capsid destabilization. Furthermore, the idea that this mutation increases infectivity in cell lines by also using a pH-independent route and that, in vivo, this mutation attenuates the virus is very novel. Last year Wa-Chu's lab proposed that encephalopathic Alphaviruses produce capsids with different sizes and that this helps to attenuate highly pathogenic viruses (which might not be …
Reviewer #1 (Public Review):
This article is interesting because the phenotype of the virus with mutations that alter the affinity of HS has been associated with how the viral particle interacts with HS and, thus, with binding and entry. However, the data in this manuscript is compelling and strongly suggests that the mutation that increases the affinity of HS alters capsid stability. To my knowledge, this is the first evidence that such mutation causes capsid destabilization. Furthermore, the idea that this mutation increases infectivity in cell lines by also using a pH-independent route and that, in vivo, this mutation attenuates the virus is very novel. Last year Wa-Chu's lab proposed that encephalopathic Alphaviruses produce capsids with different sizes and that this helps to attenuate highly pathogenic viruses (which might not be the case for non encepahlopatic Alphavirsues). However, they did not demonstrate whether these alterations attenuate the virus and if the altered morphology affects capsid stability. Therefore, this manuscript is fundamental as it contributes to understanding how the assembly/disassembly mechanism can be used to attenuate a virus. Furthermore, it is possible this mechanism could not be restricted to viruses that belong to the Picornaviridae family and opens a new door to understanding viral attenuation in other icosahedral viruses.
-
Reviewer #3 (Public Review):
Heparan Sulphate is a general association factor in the extracellular matrix which assists in host cell entry for a multitude of viral and bacterial pathogens by concentrating them in the vicinity of cellular membranes. The neurotropic picornavirus, EV-71 utilizes a protein receptor SCARB-2, in conjunction with Heparan Sulfate, in order to enter cells through the endo-lysosomal pathway. The uncoating and release of viral genome requires both receptor binding and late endosomal pH conditions. The authors have attempted to address a seeming contradiction in the in vitro and in vivo infectivity of strain MP4 variants of EV-71. One of the cell culture adapted strains MP4-L97R/E167G has stronger association with HS, which translates to higher infectivity in cell culture models; however, viral virulence is …
Reviewer #3 (Public Review):
Heparan Sulphate is a general association factor in the extracellular matrix which assists in host cell entry for a multitude of viral and bacterial pathogens by concentrating them in the vicinity of cellular membranes. The neurotropic picornavirus, EV-71 utilizes a protein receptor SCARB-2, in conjunction with Heparan Sulfate, in order to enter cells through the endo-lysosomal pathway. The uncoating and release of viral genome requires both receptor binding and late endosomal pH conditions. The authors have attempted to address a seeming contradiction in the in vitro and in vivo infectivity of strain MP4 variants of EV-71. One of the cell culture adapted strains MP4-L97R/E167G has stronger association with HS, which translates to higher infectivity in cell culture models; however, viral virulence is significantly lower in animal models.
Using an elegant and methodical set of experiments, the authors have probed the steps in the cellular entry pathway of MP4 and its L97R/E167G variant. Their experiments strongly suggest a difference in capsid uncoating mechanisms in the variant, with the L97R/E167G variant being significantly less robust and prone to destabilize earlier in the pathway. While this confers an advantage in terms of cell culture based infectivity, it is posited that the particles will not survive the gastric pH intact, which compromises virulence in the animal model. While the cell culture based uncoating experiments somewhat support this hypothesis, the main weakness of this work is a lack of explanation for the mechanism(s) of capsid destabilization conferred by overall increased positive charge. The structural bioinformatics study in the supplementary section does not explain how receptor binding, pocket factor expulsion, subunit interactions and low pH based capsid dynamics may be influenced by the mutations. Capsid destabilization could be an outcome in alteration of any or all of these processes. It is also unclear whether it is suggested that all mutations enhancing the net positive charge of VP1, or any other structural protein, will cause capsid destabilization by similar pathways. A clearer analysis of the influence of overall charge alterations, or individual mutations, on subunit interaction or particle conformation is needed. The enhancement in cell culture infectivity of the L97R/E167G variant under elevated endosomal pH is also unclear and requires further experimentation.
It has been suggested earlier that increased HS binding in vivo results in virus "trapping" and decreased infectivity. This may still be a major reason for reduced infectivity in vivo, in addition to the capsid destabilization as proposed in this work.
-
Reviewer #4 (Public Review):
In this work, Tee et al. study the implications of Heparan Sulfate (HS) binding mutations observed on the Enterovirus A71 (EV-A71) capsid. HS-binding mutations are observed for several virus infections and are often presumed to be a cell culture adaptation. However, in the case of EV-A71, the presence of HS-binding mutations in clinical samples and the contradictory findings in animal studies have made the clinical relevance of HS-binding a subject of debate. Therefore, to better understand the role of HS-binding in EV-A71, the authors use a mouse-adapted EV-A71 variant (MP4) and compare it to a cell-adapted strong HS-binder (MP4-97R/167G). Using these two variants, the authors show that the strong HS-binder does not require acidification for uncoating and genome release. Furthermore, it is demonstrated that …
Reviewer #4 (Public Review):
In this work, Tee et al. study the implications of Heparan Sulfate (HS) binding mutations observed on the Enterovirus A71 (EV-A71) capsid. HS-binding mutations are observed for several virus infections and are often presumed to be a cell culture adaptation. However, in the case of EV-A71, the presence of HS-binding mutations in clinical samples and the contradictory findings in animal studies have made the clinical relevance of HS-binding a subject of debate. Therefore, to better understand the role of HS-binding in EV-A71, the authors use a mouse-adapted EV-A71 variant (MP4) and compare it to a cell-adapted strong HS-binder (MP4-97R/167G). Using these two variants, the authors show that the strong HS-binder does not require acidification for uncoating and genome release. Furthermore, it is demonstrated that the capsid stability of the HS-binding variant is compromised, resulting in pH-independent uncoating. Overall, this study provides new insights demonstrating that seemingly beneficial mutations increasing viral replication may be counterbalanced by other unintended consequences.
Strengths:
The thoroughness of the experiments performed to demonstrate that the HS-binding phenotype results in pH-independent entry and capsid destabilisation is worth highlighting. In this regard, the authors have explored viral entry using a range of approaches involving lysosomotropic drugs, viral binding assays, and neutral red-labelled viruses coupled with diverse techniques such as FISH, RNAscope, and transient expression of constitutively active molecules to inhibit parts of the viral cycle. In my opinion, this is necessary to rule out the other downstream effects of the lysomotropic drugs and to confirm the role of the HS-binding mutation in the entry phase. The use of in silico analysis coupled with negative staining electron microscopy and environmental challenge assays is notable. Finally, the demonstration of some of the work using a human-relevant strain is commendable.
Weaknesses:
A major weakness in this study is the focus on using a mouse-adapted EV-A71 strain (MP4). In the introduction, it is argued that HS-binding mutations are controversial due to their occurrence in cell culture. However, due to host limitations, mice are not the natural hosts for EV-A71 and thus, the same argument can be made for a mouse-adapted strain. It is not clear how different this strain is from circulating EV-A71 strains and the relevance of these findings to the human situation is questionable. This is particularly made evident in the discussion where it is highlighted that HS-binding variants (VP1-145G/Q mutants) have been associated with severe neurological cases while the same variants show attenuated phenotypes in mice and monkeys. This contrast between clinical data and animal studies should be highlighted in the introduction, rather than later in the discussion, as currently the in vivo animal studies are presented as the optimal situation and may lead to misconstrued conclusions from the results.
An important consideration is that the results are based primarily on image analysis. The inclusion of RT-qPCR and/or plaque assays as supplementary data will help strengthen the findings. Moreover, there are suggestions of an intermediate binder having a different phenotype. As this intermediate binder is the clinical phenotype, data on the entry of this intermediate binder will be valuable.
Another weakness in the study is the lack of contextualization of the results to current EV-A71 literature. For instance, SCARB2 is referred to as the internalization receptor but a recent study has shown that SCARB2 is not required for internalization (https://doi.org/10.1128%2Fjvi.02042-21). The findings from this study are consistent with the localization of SCARB2 in the lysosomal membranes. Furthermore, the same study has highlighted host sulfation as a key factor in EV-A71 entry. Post-translational sulfation introduces negatively charged residues on host proteins including HS and SCARB2. This increases the binding of HS-binding strains to these proteins. In this regard, the reduced infectivity upon soluble SCARB2 treatment may simply be due to enhanced binding rather than capsid opening as suggested in the results. Therefore, additional experiments (e.g. nSEM following soluble SCARB2 treatment) must be performed to support the conclusion of capsid opening, due to inherent instability, upon SCARB2 binding.
In addition to the above, other existing literature on EV-A71 pathogenesis using organoids contradicts some of the explanations of differential phenotype in clinical observations versus mice models. In the introduction, it is suggested that reduced neurovirulence of HS-binding strains is due to binding to the vascular endothelia. However, the correlation of clinical severity to viremia (https://doi.org/10.1186/1471-2334-14-417) and the association of HS-binding mutants to clinical disease counteract this suggestion. Similarly, viral infection in human organoids with EV-A71 results in as low as 0.4% of the cells being infected (https://doi.org/10.1038/s41564-023-01339-5). In this case, if viral binding to (ubiquitously expressed) HS results in viral trapping then the HS-binding mutants should show lowered infectivity in organoid models rather than the observed higher infectivity (https://doi.org/10.3389/fmicb.2023.1045587, https://doi.org/10.1038/s41426-018-0077-2). Finally, EV-A71 release has also been shown to occur in exosomes (https://doi.org/10.1093%2Finfdis%2Fjiaa174) which effectively provides a protective lipid membrane. These recent findings must be incorporated into the article and will help better contextualize their findings.
Overall, the authors present new findings with convincing methodology. The manuscript can be improved in the contextualization of the findings and highlighting the weakness in translating these findings to resolve the debate surrounding the relevance of HS-binding phenotype. The inclusion of additional experiments and data recommended to the authors will also help strengthen the manuscript.
-
