Orai-mediated calcium entry determines activity of central dopaminergic neurons by regulation of gene expression
Curation statements for this article:-
Curated by eLife

eLife assessment
In Drosophila melanogaster, the SOCE channel Orai is required for the development of flight promoting dopaminergic neurons. The Hasan laboratory has previously shown that disabling Orai function impairs Drosophila flight due to aberrant neuronal development at the pupal stage. In this fundamental study, Mitra et al show that SOCE drives a transcriptional feedback loop via the homeobox transcription factor, 'Trithorax-like' (Trl), and histone modifiers, Set2 and E(z), to regulate the expression of key genes required for the function of dopaminergic flight neurons, including the muscarinic acetylcholine receptor and the inositol 1,4,5-trisphosphate receptor. This solid study is carefully performed with validated methodology and most of the analyses are rigorous.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Maturation and fine-tuning of neural circuits frequently require neuromodulatory signals that set the excitability threshold, neuronal connectivity, and synaptic strength. Here, we present a mechanistic study of how neuromodulator-stimulated intracellular Ca 2+ signals, through the store-operated Ca 2+ channel Orai, regulate intrinsic neuronal properties by control of developmental gene expression in flight-promoting central dopaminergic neurons (fpDANs). The fpDANs receive cholinergic inputs for release of dopamine at a central brain tripartite synapse that sustains flight (Sharma and Hasan, 2020). Cholinergic inputs act on the muscarinic acetylcholine receptor to stimulate intracellular Ca 2+ release through the endoplasmic reticulum (ER) localised inositol 1,4,5-trisphosphate receptor followed by ER-store depletion and Orai-mediated store-operated Ca 2+ entry (SOCE). Analysis of gene expression in fpDANs followed by genetic, cellular, and molecular studies identified Orai-mediated Ca 2+ entry as a key regulator of excitability in fpDANs during circuit maturation. SOCE activates the transcription factor trithorax-like (Trl), which in turn drives expression of a set of genes, including Set2 , that encodes a histone 3 lysine 36 methyltransferase (H3K36me3). Set2 function establishes a positive feedback loop, essential for receiving neuromodulatory cholinergic inputs and sustaining SOCE. Chromatin-modifying activity of Set2 changes the epigenetic status of fpDANs and drives expression of key ion channel and signalling genes that determine fpDAN activity. Loss of activity reduces the axonal arborisation of fpDANs within the MB lobe and prevents dopamine release required for the maintenance of long flight.
Article activity feed
-

-
-
-

-
Author Response
The following is the authors’ response to the previous reviews.
We thank the reviewers for collectively highlighting our study as “interesting and timely” and as making significant advances regarding the functional role of Orai in the activity of central dopaminergic neurons underlying the development of Drosophila flight behaviour. We hope that based on the revisions detailed below the data supporting our findings will be considered complete.
Reviewer 1:
- In this revision, the authors have addressed most points using text changes but there is still one important issue that continues to be inadequately addressed. This relates to point 1.
If Set2 is acting downstream of SOCE, it is not clear to me how STIM1 over expression rescues Set2-dependent downstream responses in flies that do not have Set2. It seems that if …
Author Response
The following is the authors’ response to the previous reviews.
We thank the reviewers for collectively highlighting our study as “interesting and timely” and as making significant advances regarding the functional role of Orai in the activity of central dopaminergic neurons underlying the development of Drosophila flight behaviour. We hope that based on the revisions detailed below the data supporting our findings will be considered complete.
Reviewer 1:
- In this revision, the authors have addressed most points using text changes but there is still one important issue that continues to be inadequately addressed. This relates to point 1.
If Set2 is acting downstream of SOCE, it is not clear to me how STIM1 over expression rescues Set2-dependent downstream responses in flies that do not have Set2. It seems that if STIM1 over-expression, which would presumably enhance SOCE, largely rescues Set2-dependent effector responses in the Set2RNAi flies, then the proposed pathway cannot be true (because if Set2 is downstream of SOCE, it shouldn't matter whether SOCE is boosted in flies that lack Set2). This discrepancy is not explained. Does STIM1 over-expression somehow restore Set2 expression in the Set2RNAi flies?
Ans: Based on the requirement of Orai-mediated Ca2+ entry for Set2 expression (THD’>OraiE180A neurons, Figure 2C) we had indeed proposed that rescue of flight in Set2RNAi flies by STIMOE is because Set2 expression in Set2RNAi flies is restored by STIMOE. However, we agree that this has not been tested experimentally. Since these data are supportive but not essential to our findings here, we have removed data demonstrating flight rescue of Set2RNAi by STIMOE from Figure 2 – supplement 5 and associated text from the revised manuscript. We plan to investigate the effect of STIMOE on Set2 in the context of Drosophila dopaminergic neurons in the future.
Reviewer 2:
The manuscript analyses the functional role of Orai in the excitability of central dopaminergic neurons in Drosophila. The authors answer the previous concerns, but several important issues have not been experimentally tested. Especially, the lack of characterization of SOCE or calcium release from the intracellular calcium stores limits considerably the impact of the study. They comment on a number of technical problems but, taking into account the nature of the study, based on Orai and SOCE, the lack of these experimental data reduces the relevance of the study. Below are some specific comments:
- The response to question 1 is unconvincing. The authors do not demonstrate experimentally that STIM over-expression enhances SOCE or how excess SOCE might overcome the loss of SET2.
Ans: The reason we have not performed experiments in this manuscript to investigate SOCE in STIM overexpression condition is two-fold. Firstly, extensive characterisation of SOCE by STIM overexpression in Drosophila pupal neurons forms part of an earlier publication (Chakraborty and Hasan, Front. Mol. Neurosci, 2017). A graph from Chakraborty and Hasan, 2017 where SOCE was measured in primary cultures of pupal neurons from an IP3R mutant (S224F/G1891S) of Drosophila. Reduced SOCE in IP3R mutant neurons (red trace) was restored by overexpression of STIM (black trace). The green trace is of wild-type neurons with STIM overexpression and the grey trace with STIMRNAi. Similar experiments were performed with Orai+STIM overexpression and the rescue in SOCE was compared with STIM overexpression in pupal neurons of wild type and IP3R mutant S224F/G1891S. See Chakraborty and Hasan, 2017 (Front. Mol. Neurosci. 10:111. doi: 10.3389/fnmol.2017.00111)
- Secondly, rescue by STIMOE is supportive but not essential to the findings of this manuscript which relate primarily to the analysis of an Orai-dependent transcriptional feed-back mechanism acting via Trl and Set2 in flight promoting dopaminergic neurons (See Fig 2C where we demonstrate that OraiE180A expression in THD’ neurons brings down Set2 expression).
We agree that we have not demonstrated how loss of Set2 can be compensated by STIM overexpression. Therefore, we have now removed the supplementary data relating to STIM rescue of Set2RNAi (THD’>Set2RNAi; STIMOE) flight phenotypes since as mentioned above it was supportive but not essential to the main theme of the manuscript. Consistent with this, we have also removed rescue of flight in TrlRNAi by STIMOE (Figure 4C).
- The authors do not present a characterization of SOCE in the cells investigated expressing native Orai or the dominant negative OraiE180A mutant yet. They comment on some technical problems for in situ determination or using culture cells but, apparently, in previous studies they have reported some results.
Ans: We respectfully submit that characterisation of SOCE in cells expressing native Orai and OraiE180A from primary cultures of Drosophila pupal dopaminergic neurons, form part of an earlier publication (Pathak, T., et al., (2015). The Journal of Neuroscience, 35, 13784–13799. https://doi.org/10.1523/jneurosci.1680-15.2015). As mentioned in lines 80-84 the dopaminergic neurons studied here (THD’) are a subset of the dopaminergic neurons studied in the Pathak et al., 2015 publication (TH). As evident in Figure 2 panels B-D expression of OraiE180A in dopaminergic neurons abrogates SOCE.
In this study we have focused on identifying the molecular mechanism by which OraiE180A expression and concomitant loss of cellular Ca2+ signals (Figure 3B, 3C) affects dopaminergic neuron function. In lines 270-274 (page 10) we have stated the technical reason why Ca2+ measurements made in this study from ex-vivo brain preps measure a composite of ER-Ca2+ release and SOCE. Our observation that the measured Ca2+ response is significantly attenuated in cells expressing OraiE180A leads us to the conclusion that we are indeed measuring an SOCE component in the ex-vivo brain preps. This is also explained in ‘Limitations of the study’.
- Concerning the question about the STIM:Orai stoichiometry the authors answer that "We agree that STIM-Orai stoichiometry is essential for SOCE, and propose that the rescue backgrounds possess sufficient WT Orai, which is recruited by the excess STIM to mediate the rescue"; however, again, this is not experimentally tested.
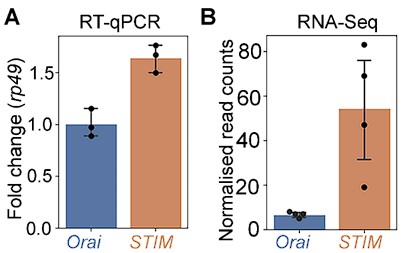
Ans: To address this point we have now measured relative stoichiometries of STIM and Orai mRNA by qPCR under WT conditions in Drosophila THD’ neurons at 72 hr APF. The observed stoichiometry as per these measurements is STIM:Orai =1.6:1 (~8:5). These data are in relative agreement with the normalised read counts of STIM and Orai in THD’ neurons in the RNAseq performed and described in Fig 1F. The qPCR (A) and RNAseq (B) measures of STIM and Orai are appended below.
Author response image 1.
In comparison to the numerous studies investigating structural, biophysical and cellular characterisation of Orai channels in heterologous systems, there are fewer studies which have traced systemic implications of Orai function through multiple tiers of investigation including organismal behaviour. Leveraging the wealth of genetic resources available in Drosophila, we have attempted this here. While we respectfully agree that questions pertaining to the stoichiometries of STIM/Orai proteins are indeed relevant to cellular regulation of SOCE, we submit they may be better suited for investigation in heterologous systems involving cell culture, or with in-vitro systems with purified recombinant proteins, or indeed using computational and modelling approaches. None of these methods fall within the scope of our current investigation which is to understand how by Orai mediated Ca2+ entry regulates developmental maturation of Drosophila flight promoting dopaminergic neurons.
-
eLife assessment
In Drosophila melanogaster, the SOCE channel Orai is required for the development of flight promoting dopaminergic neurons. The Hasan laboratory has previously shown that disabling Orai function impairs Drosophila flight due to aberrant neuronal development at the pupal stage. In this fundamental study, Mitra et al show that SOCE drives a transcriptional feedback loop via the homeobox transcription factor, 'Trithorax-like' (Trl), and histone modifiers, Set2 and E(z), to regulate the expression of key genes required for the function of dopaminergic flight neurons, including the muscarinic acetylcholine receptor and the inositol 1,4,5-trisphosphate receptor. This solid study is carefully performed with validated methodology and most of the analyses are rigorous.
-
Joint Public Review
In this study, Mitra and coworkers extend their previous analyses of the functional role of Orai in the excitability of central dopaminergic neurons in Drosophila. The authors show that a dominant-negative mutant of Orai (OraiE180A) significantly alters the gene expression profile of flight-promoting dopaminergic neurons (fpDANs), including that of Set2, E(z), and Trl, thereby shifting the level of epigenetic signatures that modulate gene expression. The Orai-Trl-Set2 pathway modulates the expression of voltage gated calcium channels, which, in turn, are involved in dopamine release. The study is generally well-done, is in-depth, and comprehensive. The finding that SOCE regulates a wide range of neuronal genes necessary for neuronal excitability and effector signaling by controlling chromatin remodeling genes is a …
Joint Public Review
In this study, Mitra and coworkers extend their previous analyses of the functional role of Orai in the excitability of central dopaminergic neurons in Drosophila. The authors show that a dominant-negative mutant of Orai (OraiE180A) significantly alters the gene expression profile of flight-promoting dopaminergic neurons (fpDANs), including that of Set2, E(z), and Trl, thereby shifting the level of epigenetic signatures that modulate gene expression. The Orai-Trl-Set2 pathway modulates the expression of voltage gated calcium channels, which, in turn, are involved in dopamine release. The study is generally well-done, is in-depth, and comprehensive. The finding that SOCE regulates a wide range of neuronal genes necessary for neuronal excitability and effector signaling by controlling chromatin remodeling genes is a noteworthy discovery.
The authors have adequately answered the previous concerns.
-
-
Author Response
The following is the authors’ response to the current reviews.
- The main issue relates to Set2, and how STIM1 expression rescues Set2-dependent functions in Set2 KO flies. If Set2 is downstream of STIM1, how would STIM1 over-expression rescue a Set2-dependent effect?
STIM rescue is of Set2 knockdown (RNAi) and NOT Set2 Knockout flies. Over expression of STIM raises SOCE in primary cultures of Drosophila neurons (as demonstrated in previous publications from our group: Agrawal et al., 2010; Chakraborty et al, 2016; Deb et al., 2016). The higher SOCE drives greater expression of Set2 from the endogenous locus thus reducing the efficacy of Set2 RNAi. Hence the rescue by STIM of Set2 KD flies in Figure S2E. We have explained this in lines 227-234.
- There is still no characterization of SOCE in fpDANs from flies expressing …
Author Response
The following is the authors’ response to the current reviews.
- The main issue relates to Set2, and how STIM1 expression rescues Set2-dependent functions in Set2 KO flies. If Set2 is downstream of STIM1, how would STIM1 over-expression rescue a Set2-dependent effect?
STIM rescue is of Set2 knockdown (RNAi) and NOT Set2 Knockout flies. Over expression of STIM raises SOCE in primary cultures of Drosophila neurons (as demonstrated in previous publications from our group: Agrawal et al., 2010; Chakraborty et al, 2016; Deb et al., 2016). The higher SOCE drives greater expression of Set2 from the endogenous locus thus reducing the efficacy of Set2 RNAi. Hence the rescue by STIM of Set2 KD flies in Figure S2E. We have explained this in lines 227-234.
- There is still no characterization of SOCE in fpDANs from flies expressing native Orai or the dominant negative OraiE180A mutant.
Measurement of SOCE is not technically feasible in ex-vivo preps due to the presence of extracellular calcium in the brain milieu. In the past we have measured SOCE from primary cultures of central dopaminergic neurons expressing either native Orai OR OraiE180A mutant (Pathak et al., 2015) where we found that all dopaminergic neurons expressing OraiE180A exhibit very low SOCE. This is the reason we have not measured SOCE in the fewer cells of the fpDAN subset marked by THD' GAL4. This point has been specifically mentioned and explained in the section on “limitations of the study” at the end of the manuscript.
- The revised version does not include an analysis of the STIM:Orai stoichiometry, which has been demonstrated to be essential for SOCE.
To measure such stoichiometry we would need to perform direct measurements of STIM and Orai levels by protein extraction from the fpDANs of all appropriate genotypes. This is not feasible due to the small number of cells available from each brain.
I confirm that there are no changes to the text OR figures from the previous version of the manuscript.
The following is the authors’ response to the original reviews.
[…]
The manuscript by Mitra and coworkers analyses the functional role of Orai in the excitability of central dopaminergic neurons in Drosophila. The authors show that a dominant-negative mutant of Orai (OraiE180A) significantly alters the gene expression profile of flight-promoting dopaminergic neurons (fpDANs). Among them, OraiE180A attenuates the expression of Set2 and enhances that of E(z) shifting the level of epigenetic signatures that modulate gene expression. The present results also demonstrate that Set2 expression via Orai involves the transcription factor Trl. The Orai-Trl-Set1 pathway modulates the expression of VGCC, which, in turn, are involved in dopamine release. The topic investigated is interesting and timely and the study is carefully performed and technically sound; however, there are several major concerns that need to be addressed:
- In Figure S2E, STIM is overexpressed in the absence of Set2 and this leads to rescue. It is presumed that STIM overexpression causes excess SOCE, yet this is rarely the case. Perhaps the bigger concern, however, is how excess SOCE might overcome the loss of SET2 if SET2 mediates SOCE-induced development of flight. These data are more consistent with something other than SET2 mediating this function.
Our statement that STIM overexpression overcomes deficits in SOCE is based on the following published work, which has been highlighted in the revised version of the manuscript (see Lines 226-233):
Studies of SOCE in wildtype cultured larval Drosophila neurons demonstrated that overexpression of STIM raised SOCE to the same extent as co-expression of STIM and Orai in the WT background (Chakraborty et al, 2016; Figure 1D).
Both Carbachol-induced IP3-mediated Ca2+ release and SOCE (measured by Ca2+ add back after Thapsigargin-induced store depletion) were rescued in primary cultures of IP3R hypomorphic mutant (itprku) Drosophila neurons by overexpression of STIM (Agrawal et al., 2010; Figure 8A-G).
Deb et al., 2016 (Supplementary Figure 2h,i) reaffirmed that overexpression of STIM significantly improves SOCE after Thapsigargin-induced passive store-depletion in Drosophila neurons expressing IP3RRNAi.
Consistent with the cellular rescue of SOCE, defects in flight initiation and physiology observed in the heteroallelic IP3R hypomorphic background (itprku) could be rescued by overexpression of STIM (Agrawal et al., 2010; Figure 3A-E) as well as Orai (Venkiteswaran and Hasan, 2009; Figure 3).
In Figure S2E, we show that flight deficits arising from THD’> Set2RNAi are rescued upon overexpression of STIM (i.e. THD’>Set2RNAi; STIMOE). Here and in another recent publication (Mitra et al., 2021) we show that neurons expressing Set2RNAi exhibit reduced expression of the IP3R and reduced ER-Ca2+ release presumably leading to reduced SOCE. As mentioned above we have consistently found that STIM overexpression raises both IP3-mediated Ca2+ release and SOCE in Drosophila neurons.
In this study, we propose that Ca2+ release through the IP3R followed by SOCE are part of a positive feedback loop (described in the revised manuscript- see Lines 302-307) driving expression of Set2 which in turn upregulates expression of mAChR and IP3R (Figure 3F) to regulate dopaminergic neuron function. Our observation that loss of Set2 (THD’>Set2RNAi) can be rescued by STIM overexpression is consistent with this model because:
Loss of Set2 (THD’>Set2RNAi) results in downregulation of several genes including mAChR and IP3R leading to decreased SOCE.
As evident from our previous studies increased STIM expression in the Set2RNAi background (THD’>Set2RNAi; STIMOE) is expected to enhance SOCE which we predict would rescue Set2 expression leading to rescue of other Set2 dependent downstream functions like flight (Figure 2D).
- In Figure 3, data is provided linking SET2 expression and Cch-induced Ca2+ responses. The presentation of these data is confusing. In addition, the results may be a simple side effect of SET2-dependent expression of IP3R. Given that this article is about SOCE, why isn't SOCE shown here? More generally, there are no measurements of SOCE in this entire article. Measuring SOCE (not what is measured in response to Cch) could help eliminate some of this confusion.
This section has been re-written in the revised version for better clarity and we have explained how Set2-dependent IP3R expression is an important component of Orai-mediated Ca2+ entry in fpDANs (see Lines 302-307). Here, we propose that IP3-mediated Ca2+ release and SOCE, through Orai, are together part of a positive feedback loop (see Lines 286-307) driving transcription of Set2 which in turn upregulates mAChR and IP3R expression (Figure 3F). We hypothesized that the observed loss of CCh-induced Ca2+ response in the Set2RNAi background (Figure 3B-D; THD’>Set2RNAi) results from decreased itpr and mAChR expression and verified this in Figure 3E. This is further validated by the rescue of CCh-induced Ca2+ response and itpr/mAChR expression in the OraiE180A background upon Set2 overexpression (Figure 3B-E; THD’>OraiE180A; Set2OE). We were constrained to measure CCh-induced Ca2+ responses in OraiE180A expressing neurons for the following reasons (highlighted in the revised version of the manuscript- (See Lines 307-313; ‘Limitations of the study’-Lines 719-735):
SOCE measurements through Tg mediated store Ca2+ release followed by Ca2+ add back require a 0 Ca2+ environment that can only be achieved in culture. The Drosophila brain is bathed in hemolymph which contains Ca2+ and there do not exist any methods to readily deplete Ca2+ from the tissue to create a 0 Ca2+ environment without also effecting the health of the neurons.
Cultures of the subset of dopaminergic neurons (THD’) we have focused on in this study were not feasible due to the small number of neurons being studied from the total number of dopaminergic neurons in the brain (~35/400). In previous studies we have shown that SOCE post-Tg induced store depletion is abrogated in cultured dopaminergic neurons from Drosophila upon expression of OraiE180A (Pathak et al., 2015). Furthermore, Carbachol-induced IP3-mediated Ca2+ release is tightly coupled to SOCE in Drosophila neurons (Venkiteswaran and Hasan, 2009) and Ca2+ release from the IP3R is physiologically relevant for flight behavior in THD’ neurons (Sharma and Hasan, 2020).
- A significant gap in the study relates to the conclusion that trl is a SOCE-regulated transcription factor. This conclusion is entirely based on genetic analysis of STIMKO heterozygous flies in which a copy of the trl13C hypomorph allele is introduced. While these results suggest a genetic interaction between the expression of the two genes, the evidence that expression translates into a functional interaction that places trl immediately downstream of SOCE is not rigorous or convincing. All that can be said is that the double mutant shows a defect in flight which could arise from an interruption of the circuit. Further, it is not clear whether the trl13C hypomorph is only introduced during the critical 72-96 hour time window when the Orai1E180E phenotype shows up. The same applies to the over-expression of Set2 and the other genes. If the expression is not temporally controlled, then the phenotype could be due to the blockade of an entirely different aspect of flight neuron function.
The idea that Trl functions downstream of Orai-mediated Ca2+ entry in THD’ neurons is based on the following genetic evidence (highlighted in the revised version; see Lines 339-341; 351-367; 647-65; ‘Limitations of the study’: 736-739)
In Figure 4D, we show evidence of genetic interaction between trl-STIM and trl-Set2. The rescue of trl13c/STIMKO with STIM overexpression in THD’ neurons indicates that excess SOCE (driven by STIMOE) may activate the residual Trl (there exists a WT Trl copy in this genetic background) to rescue THD’ flight function. This is further supported by the rescue of trl/STIMKO with Set2 overexpression in THD’ neurons, which is consistent with the feedback loop model proposed in Figure 5C (see Lines 390-396) where we propose that reduced SOCE leads to reduced ‘activated’ Trl and thus reduced Set2 expression, and the latter is rescued by SET2OE . The manner in which SOCE ‘activates’ Trl is the subject of ongoing investigations.
The trl hypomorphic alleles (including trl13C) exist as genetic mutants and they affect Trl function in all tissues throughout development. While we concede that these mutant alleles would affect multiple functions at other stages of development, which may impinge on the phenotypes noted in Figure S4B, we have used a targeted RNAi approach to validate Trl function specifically in the THD’ neurons (see Figure 4C; Lines 339-341).
Overexpression mediated rescues (including Set2) were not induced only during the critical 72-96 hrs APF developmental window. Having established that Orai function drives critical gene expression during this window (Figure 1), it is reasonable to assume that Set2 rescue of loss of flight in OraiE180A occurs in the same time window where flight is disrupted (see Lines 221-224).
- In Figure 4, data is shown that SOCE compensates for the loss of Trl, the presumed mediator of SOCE-dependent flight. The fact that flight deficits are rescued by raising SOCE in the absence of Trl is very inconsistent with this conclusion.
We apologise for this confusion and have clarified in the revision (see Lines 346-367). trl13c is a recessive allele of Trl and has been written as such throughout the text and in the figures (i.e trl13c and NOT Trl13c). In all cases of Trl mutant rescue by STIMOE and Set2OE there exists residual Trl that can be activated by excess SOCE thus leading to the rescue. This is true for trl13C/ STIMKO where each mutant is present as a heterozygote (the complete genotype of this strain is STIMKO/+; trl13c/+; this has been corrected in the revision). Similarly, for TrlRNAi we expect reduced levels (but not complete loss) of Trl. Thus the SOCE rescue of loss of Trl occurs in conditions where Trl levels are reduced but NOT absent. Homozygous trl null mutants are lethal.
- In Figure 5 (A-C), data is provided that Trl transcripts are unaffected by loss of SOCE and that overexpression cannot rescue flightlessness. From this, the authors conclude that this gene "must" be calcium responsive. While that is one possibility, it is also possible that these genes are not functionally linked.
The idea that Trl is functionally linked to SOCE is based on the following evidence (included in the revised version- see Lines 339-341; 346-367; 391-396)
In Figure 4C we show that flight defects caused by partial loss of Trl (THD’>TrlRNAi) were rescued by STIM overexpression (THD’>TrlRNAi; STIMOE). As mentioned above we have found that STIM overexpression raises SOCE.
Heteroalleles of the trl13C hypomorph exhibit a strong genetic interaction with a single copy of the null allele of STIMKO as shown by the flight deficit of trl13c/+; STIMKO/+ (trl13C/STIMKO ) flies (Figure 4D). The genotypes will be corrected in the revision.
Flight defects in trl13C/STIMKO flies could be rescued by STIM overexpression in the THD’ neurons (trl13C/STIMKO; THD’>STIMOE)
In Figure 4E, we show that partial loss of Trl in THD’ neurons (THD’>TrlRNAi) leads to decreased expression of the Ca2+ responsive genes mAChR, itpr, and Set2 genes indicating that Trl is a constituent of the SOCE-driven transcriptional feedback loop (see Figure 5C).
Since we could not detect a well-defined Ca2+ binding domain in Trl, we hypothesize that it could be activated by a Ca2+ dependent post-translational modification. Phosphoproteome analysis of Trl demonstrated that it does indeed undergo phosphorylation at a Threonine residue (T237; Zhai et al., 2008), which lies within a potential site for CaMKII. Independently, CaMKII has been identified as a binding partner of Trl from a Trl interactome study (Lomaev et al., 2018). Past work from our group (Ravi et al., 2018) identified a role for CaMKII in THD’ neurons in the context of flight. We are currently testing if CaMKII functions downstream of SOCE in THD’ neurons to mediate flight and will update this information in the next version of the manuscript.
Now included in the revised version of the manuscript as Figure S5; Lines 397-424)
- There is no characterization of SOCE in fpDANs from flies expressing native Orai or the dominant negative OraiE180A mutant. While the authors refer to previous studies, as the manuscript is essentially based on Orai function thapsigargin-induced SOCE should be tested using the Ca2+ add-back protocol in order to assess the release of Ca2+ from the ER in response to thapsigargin as well as the subsequent SOCE.
The fpDANs consist of 16-19 neurons in each hemisphere (PPL1 are 10-12 and PPM3 are 6-7 cells; Pathak et al., 2015). Measuring SOCE from these neurons in vivo is not possible due to the presence of abundant extracellular Ca2+ in the brain. Given their sparse number, it proved technically challenging to isolate the fpDANs in culture to perform SOCE measurements using the Ca2+ add back protocol. Due to these reasons, we have relied upon using Carbachol to elicit IP3-mediated Ca2+ release and SOCE as a proxy for in vivo SOCE. In previous studies we have shown that Carbachol treatment of cultured Drosophila neurons elicits IP3-mediated Ca2+ release and SOCE (Agrawal et al., 2010; Figure 8). Moreover, expression of OraiE180A completely blocks SOCE as measured in primary cultures of dopaminergic neurons (Pathak et al., 2015; Figure 1E). Hence we have not repeated SOCE measurements from all dopaminergic neurons in this work. In the revised version we have explicitly stated this weakness of our study and the reasons for it (See Lines 307-313; ‘Limitations of the study’-Lines 719-735).
- In the experiments performed to rescue flight duration in Set2RNAi individuals the authors overexpress STIM and attribute the effect to "Excess STIM presumably drives higher SOCE sufficient to rescue flight bout durations caused by deficient Set2 levels.". This should be experimentally tested as the STIM:Orai stoichiometry has been demonstrated as essential for SOCE.
The assumption that STIM overexpression drives higher SOCE is based upon previously published work from Drosophila neurons (Agrawal et al., 2010; Chakraborty et al, 2016; Deb et al., 2016) which demonstrates that excess WT STIM overcomes IP3R deficiencies (RNAi or hypomorphic mutants) to rescue SOCE. We agree that STIM-Orai stoichiometry is essential for SOCE, and propose that the rescue backgrounds possess sufficient WT Orai, which is recruited by the excess STIM to mediate the rescue. We have referenced the earlier work to validate our use of STIMOE for rescue of SOCE (See Lines 226-233).
Here, we propose that Set2 is part of a positive feedback loop (see Lines 286-307) driving transcription of mAChR and IP3R (Figure 3F). In keeping with this hypothesis, we posit that the phenotypes observed in the Set2RNAi background (Figure 2D) result from decreased itpr and mAChR expression (validated in Figure 3E). This is further validated by the Set2 overexpression mediated rescue of OraiE180A (Figure 2D) and rescue of itpr/mAChR expression in the OraiE180A background (Figure 3B-E; THD’>OraiE180A; Set2OE).
- The authors show that overexpression of OraiE108A results in Stim downregulation at a mRNA level. What about the protein level? And more important, how does OraiE108A downregulate Stim expression? Does it promote Stim degradation? Does it inhibit Stim expression?
We hypothesize that changes in STIM mRNA observed in the THD’ > OraiE180A neurons stems from an overall reduction in IP3-mediated Ca2+ release and SOCE due to loss of Trl-Set2 driven gene expression detailed in our transcriptional feedback loop model (Figure 5C; see Lines 286-307; 581-591). We have attempted to explain this aspect more clearly in the revised version of the manuscript. While we agree that measuring levels of STIM protein would be helpful, estimation of protein levels from a limited number of neurons (~35 cells per brain) is technically challenging. The STIM antibody does not work well in immunohistochemistry. In the absence of any experimental evidence we cannot comment on how expression of OraiE180A might affect STIM protein turnover (see Lines 307-313).
- Lines 271-273, the authors state "whereas overexpression of a transgene encoding Set2 in THD' neurons either with loss of SOCE (OraiE180A) or with knockdown of the IP3R (itprRNAi), lead to significant rescue of the Ca2+ response". This is attributed to a positive effect of Set2 expression on IP3R expression and the authors show a positive correlation between these two parameters; however, there is no demonstration that Set2 expression can rescue IP3R expression in cells where the IP3R is knocked down (itprRNAi). This should be further demonstrated.
The rescue of IP3R expression by Set2 overexpression in itprRNAi was demonstrated in a different set of Drosophila neurons in an earlier study (Mitra et al., 2021) and has not been repeated specifically in THD’ neurons (see Lines 286-307). Similar to the previous study, here we tested CCh stimulated Ca2+ responses of THD’ neurons with itprRNAi and itprRNAi; SetOE (Fig S3), which are indeed rescued by SET2OE see Lines 280-285)
- The data presented in Figure 3E should be functionally demonstrated by analyzing the ability of CCh to release Ca2+ from the intracellular stores in the absence of extracellular Ca2+.
CCh-mediated Ca2+ release from the intracellular stores in the absence of extracellular Ca2+ has been described in primary cultures of Drosophila neurons in previously published work (Venkiteswaran and Hasan, 2009; Agrawal et al., 2010) This work focuses on a set of 16-19 dopaminergic neurons in a hemisphere of the Drosophila central brain. It is technically challenging to generate a 0 Ca2+ environment in vivo, which is essential for measuring store Ca2+ release. Given their meagre numbers, primary cultures of these neurons is not readily feasible. (see Lines 307-313; ‘Limitations of the study’-Lines 719-735)
- The conclusion that SOCE regulates the neuronal excitability threshold is based entirely on either partial behavioral rescue of flight, or measurements of KCl-induced Ca2+ rises monitored by GCaMP6m in DAN neurons. The threshold for neuronal excitability is a precise parameter based on rheobase measurements of action potentials in current-clamp. Measurements of slow calcium signals using a slow dye such as GCaMp6m should not be equated with neuronal excitability. What is measured is a loss of the calcium response in high K depolarization experiments, which occurs due to the loss of expression of Cav channels. Hence, the use of this term is not accurate and will confuse readers. The use of terms referring to neuronal excitability needs to be changed throughout the manuscript. As such, the conclusions regarding neuronal excitability should be strongly tempered and the data reinterpreted as there are no true measurements of neuronal excitability in the manuscript. All that can be said is that expression of certain ion channel genes is suppressed. Since both Na+ channels and K+ channel expression is down-regulated, it is hard to say precisely how membrane excitability is altered without action potential analysis.
The claim that SOCE influences neuronal excitability is based on the following observations:
Interruption of the transcriptional feedback loop involving SOCE, Trl, and Set2 through loss of any of its constituents, results in the downregulation of VGCCs (Figure 5G, 6H), which are essential components of action potentials.
OraiE180A mediated loss of SOCE in THD’ neurons abrogates the KCl-evoked depolarization response (Figure 6B, C) measured using GCaMP6m. We verified that this response requires VGCC function using pharmacological inhibition of L-type VGCCs (Figure 6E, F).
SOCE deficient THD’ neurons, which were presumably compromised in their ability to evoke action potentials could be rescued to undergo KCl-evoked depolarisation by expression of NachBac, which lowers the depolarization threshold (Figure 7C, D) or through optogenetic stimulation using CsChrimson (Figure 7F).
We agree that ‘neuronal excitability threshold’ is a precise electrophysiological parameter that has not been directly investigated here by measurement of action potentials. Therefore, references to neuronal excitability have been tempered throughout the revised manuscript and be replaced with a more generic reference to ‘neuronal activity’. In this context we have included further evidence supporting reduced activity of THD’ neurons upon loss of SOCE in the revision.
Since one of the key functional outcomes of activity during critical developmental periods such as the 72-96 hrs APF developmental window identified in this study, is remodelling of neuronal morphology, we decided to investigate the same in our context. Neuronal activity can drive changes in neurite complexity and axonal arborization (Depetris-Chauvin et al., 2011) especially during critical developmental periods (Sachse et al., 2007). To understand if Orai mediated Ca2+ entry and downstream gene expression through Set2 affects this activity-driven parameter, we investigated the morphology of fpDANs, and specifically measured the complexity of presynaptic terminals within the 2’1 lobe MB using super-resolution microscopy. We found striking changes in the neurite volume upon expression of OraiE180A which could be rescued by restoring either Set2 (OraiE180A; Set2OE) or by inducing hyperactivity through NachBac expression (OraiE180A ; NachBacOE). These data have been included in the revised manuscript (Figure 8 B, C, D; see Lines 481-482; 519-534; 584-591; 701-704).
- Related, since trl does not contain any molecular domains that could be regulated by Ca2+ signaling, it is unclear whether trl is directly regulated by SOCE or the regulation is highly indirect. Reporter assays evaluating trl activation upon Ca2+ rises would provide much stronger and more direct evidence for the conclusion that trl is a SOCE-regulated TF. As such the evidence is entirely based on RNAi downregulation of trl which indicates that trl is essential but has no bearing on exactly what point of the signaling cascade it is involved.
We agree that luciferase Trl reporters would provide a direct method to test SOCE-mediated activation. Future investigations will be targeted in this direction. Regarding possible mechanisms of Trl activation - since we could not detect a well-defined Ca2+ binding domain in Trl, we hypothesize that it may be phosphorylation by a Ca2+ sensitive kinase. Phosphoproteome analysis of Trl indicates that it does indeed undergo phosphorylation at a Threonine reside (T237; Zhai et al., 2008), which may be mediated by the Ca2+ sensitive kinase-CaMKII based on binding partners identified in the Trl interactome (Lomaev et al., 2018; Past work (Ravi et al., 2018) has indeed demonstrated a requirement for CaMKII in THD’ neurons for flight. We are currently testing whether CaMKII functions downstream of SOCE in these neurons to mediate flight, and will be updating this information in the next version of the manuscript.
New data and analysis has been included - see Figure S5; ‘Limitations of the study’- Lines 397-424; 736-739).
- Are NFAT levels altered in the Orai1 loss of function mutant? If not, this should be explicitly stated. It would seem based on previous literature that some gene regulation may be related to the downregulation of this established Ca2+-dependent transcription factor. Same for NFkb.
As mentioned in the revised version of the manuscript (see Lines 315-326), Drosophila NFAT lacks a calcineurin binding site and is therefore not sensitive to Ca2+ (Keyser et al., 2007). In the past we tested if knockdown of NF-kB in dopaminergic neurons gave a flight phenotype and did not observe any measurable deficit. From the RNAseq data we find a slight downregulation of NFAT (0.49 fold, p value=0.048) and NF-kb (0.26 fold, p value =0.258) the significance of which is unclear at this point. We did not find any consensus binding sites for these two factors in the regulatory regions of downregulated genes from THD’ neurons.
- Does over-expression of Set2 restore ion channel expression especially those of the VGCCs? This would provide rigorous, direct evidence that SOCE-mediated regulation of VGCCs through Set2 controls voltage-gated calcium channel signaling.
Set2 overexpression in the OraiE180A background indeed restores the expression of VGCC genes (see Figure 6H; Lines 461-468).
- All 6 representative panels from Figure 3B are duplicated in Figure 4G. Likewise, 2 representative panels from Figure 5H are duplicated in Figure 6D. Although these panels all represent the results from control experiments, the relevant experiments were likely not conducted at the same time and under the same conditions. Thus, control images from other experiments should not be used simply because they correspond to controls. This situation should be clarified.
We regret the confusion caused by the same representative images for the control experiments. These have been replaced by new representative images for Figure 4G and 6D in the updated version of the manuscript.
- The figures are unusually busy and difficult to follow. In part this is because they usually have many panels (Fig. 1: A-I; Fig. 2, A-J, etc) but also because the arrangement of the panels is not consistent: sometimes the following panel is found to the right, other times it is below. It would help the reader to make the order of the panels consistent, and, if possible, reduce the number of panels and/or move some of the panels to new figures (eLife does not limit the number of display items).
The image panels have been rearranged for ease of reading in the updated version of the manuscript.
- As a final recommendation, the reviewers suggest that the authors a- Reword the text that refers to membrane excitability since membrane excitability was not directly measured here. b-Explain why STIM1 rescues the partial loss of flight in Set2 RNAi flies (Fig. S2E); and c- Explain how/why trl is calcium regulated and test using luciferase (or other) reporter assays whether Orai activation leads to trl activation.
a. Textual references to membrane excitability have been appropriately modified and some new data has been included in this regard (see Figure 8 B, C, D; Lines 481-483; 519-534; 584-591; 701-704).
b. We have provided a detailed explanation for how STIM overexpression might rescue the phenotypes caused by Set2RNAi in Point 1 (see Lines 226-233). In short, these phenotypes depend upon IP3R mediated Ca2+ entry driving a transcriptional feedback loop. We relied upon past reports that STIM overexpression upregulates IP3R-mediated Ca2+ release and SOCE in Drosophila itpr mutant neurons (Agrawal et al., 2010; Chakraborty et al, 2016; Deb et al, 2016). We therefore propose that STIM overexpression in the Set2RNAi background rescues IP3R mediated Ca2+ release followed by SOCE, which drives enhanced Set2 transcription, counteracting the effects of the RNAi. We will explain this more clearly with past references in the next revision.
c. We have provided a detailed response to this comment in Point 12. Briefly, we agree that building luciferase reporters for Trl could be an ideal strategy to test for its responsiveness to SOCE and needs to be done in future. As an alternate strategy, we have looked at data from existing studies of interacting partners of Trl (Lomaev et al., 2017) and identified CamKII, which is both Ca2+ responsive (Braun and Schulman, 1995; Yasuda et al., 2022), and thus might activate Trl through a phosphorylation-switch like mechanism (see Figure S5; ‘Limitations of the study’-736-739; Lines 397-424). Moreover, a previous publication identified a requirement for CamKII in THD’ neurons for Drosophila flight (Ravi et al., 2018). We have tested the ability of a dominant active version of CamKII to rescue THD’>E180A flight deficits and have included this information in the next version of the manuscript.
References
Agrawal N, Venkiteswaran G, Sadaf S, Padmanabhan N, Banerjee S, Hasan G. Inositol 1,4,5-Trisphosphate Receptor and dSTIM Function in Drosophila Insulin-Producing Neurons Regulates Systemic Intracellular Calcium Homeostasis and Flight. J Neurosci. 2010;30:1301-1313. doi:10.1523/jneurosci.3668-09.2010
Braun AP, Schulman H. A non-selective cation current activated via the multifunctional Ca(2+)-calmodulin-dependent protein kinase in human epithelial cells. J Physiol. 1995. 488:37-55. doi:10.1113/jphysiol.1995.sp020944
Chakraborty S, Deb BK, Chorna T, Konieczny V, Taylor CW, Hasan G. Mutant IP3 receptors attenuate store-operated Ca2+ entry by destabilizing STIM-Orai interactions in Drosophila neurons. J Cell Sci. 2016. 129:3903-3910. doi:10.1242/jcs.191585
Deb BK, Pathak T, Hasan G. Store-independent modulation of Ca2+ entry through Orai by Septin 7. Nat Commun. 2016. 7:11751. doi:10.1038/ncomms11751
Depetris-Chauvin A, Berni J, Aranovich EJ, Muraro NI, Beckwith EJ, Ceriani MF. Adult-specific electrical silencing of pacemaker neurons uncouples molecular clock from circadian outputs. Curr Biol. 2011. 21:1783-1793. doi: 10.1016/j.cub.2011.09.027.
Keyser P, Borge-Renberg K, Hultmark D. The Drosophila NFAT homolog is involved in salt stress tolerance. Insect Biochem Mol Biol. 2007. 37:356-362. doi:10.1016/j.ibmb.2006.12.009
Kilo L, Stürner T, Tavosanis G, Ziegler AB. Drosophila Dendritic Arborisation Neurons: Fantastic Actin Dynamics and Where to Find Them. Cells. 2021. 10:2777. doi:10.3390/cells10102777
Lomaev D, Mikhailova A, Erokhin M, et al. The GAGA factor regulatory network: Identification of GAGA factor associated proteins. PLoS One. 2017. 12:e0173602. doi:10.1371/journal.pone.0173602
Mitra R, Richhariya S, Jayakumar S, Notani D, Hasan G. IP3/Ca2+ signals regulate larval to pupal transition under nutrient stress through the H3K36 methyltransferase dSET2. Development. 2021. 148:dev199018. doi:10.1101/2020.11.25.399329
Pathak T, Agrawal T, Richhariya S, Sadaf S, Hasan G. Store-Operated Calcium Entry through Orai Is Required for Transcriptional Maturation of the Flight Circuit in Drosophila. J Neurosci. 2015. 35:13784-13799. doi:10.1523/jneurosci.1680-15.2015
Ravi P, Trivedi D, Hasan G. FMRFa receptor stimulated Ca2+ signals alter the activity of flight modulating central dopaminergic neurons in Drosophila melanogaster. Barsh GS, ed. PLOS Genet. 2018. 14:e1007459. doi:10.1371/journal.pgen.1007459
Sachse S, Rueckert E, Keller A, Okada R, Tanaka NK, Ito K, Vosshall LB. Activity-dependent plasticity in an olfactory circuit. Neuron. 2007. 56:838-50. doi: 10.1016/j.neuron.2007.10.035.
Sharma A, Hasan G. Modulation of flight and feeding behaviours requires presynaptic IP3Rs in dopaminergic neurons. Elife. 2020;9. e62297.doi:10.7554/elife.62297
Venkiteswaran G, Hasan G. Intracellular Ca2+ signalling and store operated Ca2+ entry are required in Drosophila neurons for flight. Proc Natl Acad Sci. 2009.106:10326-10331. doi: 10.1073/pnas.0902982106
Yasuda R, Hayashi Y, Hell JW. CaMKII: a central molecular organizer of synaptic plasticity, learning and memory. Nat Rev Neurosci. 2022. 23: 666-682 doi:10.1038/s41583-022-00624-2
Zhai B, Villén J, Beausoleil SA, Mintseris J, Gygi SP. Phosphoproteome Analysis of Drosophila melanogaster Embryos. J Proteome Res. 2008. 7:1675-1682. doi:10.1021/pr700696a
-
eLife assessment
In Drosophila melanogaster, the Store-operated Ca2+ entry (SOCE) channel, Orai, is required for the development of flight-promoting dopaminergic neurons. Here, Mitra et al. determine that expression of a loss-of-function Orai1 mutant during the 72-96 hour window of pupal development impairs gene expression in dopaminergic flight neurons in part through the expression of Set2, a histone methyltransferase. The authors identify a large number of genes that are controlled by Set2, and show that Set2 is controlled by the Trl/GAF transcription factor. Although the findings reported here are important, the evidence supporting some of the claims is incomplete.
-
Joint Public Review:
The manuscript by Mitra and coworkers analyses the functional role of Orai in the excitability of central dopaminergic neurons in Drosophila. The authors show that a dominant-negative mutant of Orai (OraiE180A) significantly alters the gene expression profile of flight-promoting dopaminergic neurons (fpDANs). Among them, OraiE180A attenuates the expression of Set2 and enhances that of E(z) shifting the level of epigenetic signatures that modulate gene expression. The present results also demonstrate that Set2 expression via Orai involves the transcription factor Trl. The Orai-Trl-Set2 pathway modulates the expression of VGCC, which, in turn, are involved in dopamine release. The topic investigated is interesting and timely and the study is carefully performed and technically sound.
The reviewers appreciate the …
Joint Public Review:
The manuscript by Mitra and coworkers analyses the functional role of Orai in the excitability of central dopaminergic neurons in Drosophila. The authors show that a dominant-negative mutant of Orai (OraiE180A) significantly alters the gene expression profile of flight-promoting dopaminergic neurons (fpDANs). Among them, OraiE180A attenuates the expression of Set2 and enhances that of E(z) shifting the level of epigenetic signatures that modulate gene expression. The present results also demonstrate that Set2 expression via Orai involves the transcription factor Trl. The Orai-Trl-Set2 pathway modulates the expression of VGCC, which, in turn, are involved in dopamine release. The topic investigated is interesting and timely and the study is carefully performed and technically sound.
The reviewers appreciate the authors' efforts to revise the manuscript in order to address many of their concerns. Nevertheless, there remain a few important issues:
The main issue relates to Set2, and how STIM1 expression rescues Set2-dependent functions in Set2 KO flies. If Set2 is downstream of STIM1, how would STIM1 over-expression rescue a Set2-dependent effect?
There is still no characterization of SOCE in fpDANs from flies expressing native Orai or the dominant negative OraiE180A mutant.
The revised version does not include an analysis of the STIM:Orai stoichiometry, which has been demonstrated to be essential for SOCE.
-
-
Author Response
Joint Public review
The manuscript by Mitra and coworkers analyses the functional role of Orai in the excitability of central dopaminergic neurons in Drosophila. The authors show that a dominant-negative mutant of Orai (OraiE180A) significantly alters the gene expression profile of flight-promoting dopaminergic neurons (fpDANs). Among them, OraiE180A attenuates the expression of Set2 and enhances that of E(z) shifting the level of epigenetic signatures that modulate gene expression. The present results also demonstrate that Set2 expression via Orai involves the transcription factor Trl. The Orai-Trl-Set1 pathway modulates the expression of VGCC, which, in turn, are involved in dopamine release. The topic investigated is interesting and timely and the study is carefully performed and technically sound; however, there …
Author Response
Joint Public review
The manuscript by Mitra and coworkers analyses the functional role of Orai in the excitability of central dopaminergic neurons in Drosophila. The authors show that a dominant-negative mutant of Orai (OraiE180A) significantly alters the gene expression profile of flight-promoting dopaminergic neurons (fpDANs). Among them, OraiE180A attenuates the expression of Set2 and enhances that of E(z) shifting the level of epigenetic signatures that modulate gene expression. The present results also demonstrate that Set2 expression via Orai involves the transcription factor Trl. The Orai-Trl-Set1 pathway modulates the expression of VGCC, which, in turn, are involved in dopamine release. The topic investigated is interesting and timely and the study is carefully performed and technically sound; however, there are several major concerns that need to be addressed:
- In Figure S2E, STIM is overexpressed in the absence of Set2 and this leads to rescue. It is presumed that STIM overexpression causes excess SOCE, yet this is rarely the case. Perhaps the bigger concern, however, is how excess SOCE might overcome the loss of SET2 if SET2 mediates SOCE-induced development of flight. These data are more consistent with something other than SET2 mediating this function.
Our statement that STIM overexpression overcomes deficits in SOCE is based on the following published work:
Studies of SOCE in wildtype cultured larval Drosophila neurons demonstrated that overexpression of STIM raised SOCE to the same extent as co-expression of STIM and Orai in the WT background (Chakraborty et al, 2016; Figure 1D).
Both Carbachol-induced IP3-mediated Ca2+ release and SOCE (measured by Ca2+ add back after Thapsigargin-induced store depletion) were rescued in primary cultures of IP3R hypomorphic mutant (itprku) Drosophila neurons by overexpression of STIM (Agrawal et al., 2010; Figure 8A-G).
Deb et al., 2016 (Supplementary Figure 2h,i) reaffirmed that overexpression of STIM significantly improves SOCE after Thapsigargin-induced passive store-depletion in Drosophila neurons expressing IP3RRNAi.
Consistent with the cellular rescue of SOCE, defects in flight initiation and physiology observed in the heteroallelic IP3R hypomorphic background (itprku) could be rescued by overexpression of STIM (Agrawal et al., 2010; Figure 3A-E) as well as Orai (Venkiteswaran and Hasan, 2009; Figure 3).
In Figure S2E, we show that flight deficits arising from THD’> Set2RNAi are rescued upon overexpression of STIM (i.e. THD’>Set2RNAi; STIMOE). Here and in another recent publication (Mitra et al., 2021) we show that neurons expressing Set2RNAi exhibit reduced expression of the IP3R and reduced ER-Ca2+ release presumably leading to reduced SOCE. As mentioned above we have consistently found that STIM overexpression raises both IP3-mediated Ca2+ release and SOCE in Drosophila neurons.
In this study, we propose that Ca2+ release through the IP3R followed by SOCE are part of a positive feedback loop driving expression of Set2 which in turn upregulates expression of mAChR and IP3R (Figure 3F) to regulate dopaminergic neuron function. Our observation that loss of Set2 (THD’>Set2RNAi) can be rescued by STIM overexpression is consistent with this model because:
Loss of Set2 (THD’>Set2RNAi) results in downregulation of several genes including mAChR and IP3R leading to decreased SOCE.
As evident from our previous studies increased STIM expression in the Set2RNAi background (THD’>Set2RNAi; STIMOE) is expected to enhance SOCE which we predict would rescue Set2 expression leading to rescue of other Set2 dependent downstream functions like flight (Figure 2D).
- In Figure 3, data is provided linking SET2 expression and Cch-induced Ca2+ responses. The presentation of these data is confusing. In addition, the results may be a simple side effect of SET2-dependent expression of IP3R. Given that this article is about SOCE, why isn't SOCE shown here? More generally, there are no measurements of SOCE in this entire article. Measuring SOCE (not what is measured in response to Cch) could help eliminate some of this confusion.
We will re-write this section in the revised version for better clarity and explain how Set2-dependent IP3R expression is an important component of Orai-mediated Ca2+ entry in fpDANs. Here, we propose that IP3-mediated Ca2+ release and SOCE, through Orai, are together part of a positive feedback loop driving transcription of Set2 which in turn upregulates mAChR and IP3R expression (Figure 3F). We hypothesized that the observed loss of CCh-induced Ca2+ response in the Set2RNAi background (Figure 3B-D; THD’>Set2RNAi) results from decreased itpr and mAChR expression and verified this in Figure 3E. This is further validated by the rescue of CCh-induced Ca2+ response and itpr/mAChR expression in the OraiE180A background upon Set2 overexpression (Figure 3B-E; THD’>OraiE180A; Set2OE). We were constrained to measure CCh-induced Ca2+ responses in OraiE180A expressing neurons for the following reasons:
SOCE measurements through Tg mediated store Ca2+ release followed by Ca2+ add back require a 0 Ca2+ environment that can only be achieved in culture. The Drosophila brain is bathed in hemolymph which contains Ca2+ and there do not exist any methods to readily deplete Ca2+ from the tissue to create a 0 Ca2+ environment without also effecting the health of the neurons.
Cultures of the subset of dopaminergic neurons (THD’) we have focused on in this study were not feasible due to the small number of neurons being studied from the total number of dopaminergic neurons in the brain (~35/400). In previous studies we have shown that SOCE post-Tg induced store depletion is abrogated in cultured dopaminergic neurons from Drosophila upon expression of OraiE180A (Pathak et al., 2015).
Furthermore, Carbachol-induced IP3-mediated Ca2+ release is tightly coupled to SOCE in Drosophila neurons (Venkiteswaran and Hasan, 2009) and Ca2+ release from the IP3R is physiologically relevant for flight behavior in THD’ neurons (Sharma and Hasan, 2020).
- A significant gap in the study relates to the conclusion that trl is a SOCE-regulated transcription factor. This conclusion is entirely based on genetic analysis of STIMKO heterozygous flies in which a copy of the trl13C hypomorph allele is introduced. While these results suggest a genetic interaction between the expression of the two genes, the evidence that expression translates into a functional interaction that places trl immediately downstream of SOCE is not rigorous or convincing. All that can be said is that the double mutant shows a defect in flight which could arise from an interruption of the circuit. Further, it is not clear whether the trl13C hypomorph is only introduced during the critical 72-96 hour time window when the Orai1E180E phenotype shows up. The same applies to the over-expression of Set2 and the other genes. If the expression is not temporally controlled, then the phenotype could be due to the blockade of an entirely different aspect of flight neuron function.
The idea that Trl functions downstream of Orai-mediated Ca2+ entry in THD’ neurons is based on the following genetic evidence:
In Figure 4D, we show evidence of genetic interaction between trl-STIM and trl-Set2. The rescue of trl13c/STIMKO with STIM overexpression in THD’ neurons indicates that excess SOCE (driven by STIMOE) may activate the residual Trl (there exists a WT Trl copy in this genetic background) to rescue THD’ flight function. This is further supported by the rescue of trl/STIMKO with Set2 overexpression in THD’ neurons, which is consistent with the feedback loop model proposed in Figure 5C - where we propose that reduced SOCE leads to reduced ‘activated’ Trl and thus reduced Set2 expression, and the latter is rescued by SET2OEThe manner in which SOCE ‘activates’ Trl is the subject of ongoing investigations.
The trl hypomorphic alleles (including trl13C) exist as genetic mutants and they affect Trl function in all tissues throughout development. While we concede that these mutant alleles would affect multiple functions at other stages of development, which may impinge on the phenotypes noted in Figure S4B, we have used a targeted RNAi approach to validate Trl function specifically in the THD’ neurons (Figure 4C).
Overexpression mediated rescues (including Set2) were not induced only during the critical 72-96 hrs APF developmental window. Having established that Orai function drives critical gene expression during this window (Figure 1), it is reasonable to assume that Set2 rescue of loss of flight in OraiE180A occurs in the same time window where flight is disrupted.
4- In Figure 4, data is shown that SOCE compensates for the loss of Trl, the presumed mediator of SOCE-dependent flight. The fact that flight deficits are rescued by raising SOCE in the absence of Trl is very inconsistent with this conclusion.
We apologise for this confusion and will clarify in the revision. trl13c is a recessive allele of Trl and should be written as such throughout the text and in the figures (i.e trl13c and NOT Trl13c). In all cases of Trl mutant rescue by STIMOE and Set2OE there exists residual Trl that can be activated by excess SOCE thus leading to the rescue. This is true for trl13C/ STIMKO where each mutant is present as a heterozygote (the complete genotype of this strain is STIMKO/+; trl13c/+; this will be corrected in the revision). Similarly, for TrlRNAi we expect reduced levels (but not complete loss) of Trl. Thus the SOCE rescue of loss of Trl occurs in conditions where Trl levels are reduced but NOT absent. Homozygous trl null mutants are lethal.
5- In Figure 5 (A-C), data is provided that Trl transcripts are unaffected by loss of SOCE and that overexpression cannot rescue flightlessness. From this, the authors conclude that this gene "must" be calcium responsive. While that is one possibility, it is also possible that these genes are not functionally linked.
The idea that Trl is functionally linked to SOCE is based on the following evidence:
In Figure 4C we show that flight defects caused by partial loss of Trl (THD’>TrlRNAi) were rescued by STIM overexpression (THD’>TrlRNAi; STIMOE). As mentioned above we have found that STIM overexpression raises SOCE.
Heteroalleles of the trl13C hypomorph exhibit a strong genetic interaction with a single copy of the null allele of STIMKO as shown by the flight deficit of trl13c/+; STIMKO/+ (trl13C/STIMKO ) flies (Figure 4D). The genotypes will be corrected in the revision.
Flight defects in trl13C/STIMKO flies could be rescued by STIM overexpression in the THD’ neurons (trl13C/STIMKO; THD’>STIMOE)
In Figure 4E, we show that partial loss of Trl in THD’ neurons (THD’>TrlRNAi) leads to decreased expression of the Ca2+ responsive genes mAChR, itpr, and Set2 genes indicating that Trl is a constituent of the SOCE-driven transcriptional feedback loop (Figure 5C).
Since we could not detect a well-defined Ca2+ binding domain in Trl, we hypothesize that it could be activated by a Ca2+ dependent post-translational modification. Phosphoproteome analysis of Trl demonstrated that it does indeed undergo phosphorylation at a Threonine residue (T237; Zhai et al., 2008), which lies within a potential site for CaMKII. Independently, CaMKII has been identified as a binding partner of Trl from a Trl interactome study (Lomaev et al., 2018). Past work from our group (Ravi et al., 2018) identified a role for CaMKII in THD’ neurons in the context of flight. We are currently testing if CaMKII functions downstream of SOCE in THD’ neurons to mediate flight and will update this information in the next version of the manuscript.
- There is no characterization of SOCE in fpDANs from flies expressing native Orai or the dominant negative OraiE180A mutant. While the authors refer to previous studies, as the manuscript is essentially based on Orai function thapsigargin-induced SOCE should be tested using the Ca2+ add-back protocol in order to assess the release of Ca2+ from the ER in response to thapsigargin as well as the subsequent SOCE.
The fpDANs consist of 16-19 neurons in each hemisphere (PPL1 are 10-12 and PPM3 are 6-7 cells; Pathak et al., 2015). Measuring SOCE from these neurons in vivo is not possible due to the presence of abundant extracellular Ca2+ in the brain. Given their sparse number, it proved technically challenging to isolate the fpDANs in culture to perform SOCE measurements using the Ca2+ add back protocol. Due to these reasons, we have relied upon using Carbachol to elicit IP3-mediated Ca2+ release and SOCE as a proxy for in vivo SOCE. In previous studies we have shown that Carbachol treatment of cultured Drosophila neurons elicits IP3-mediated Ca2+ release and SOCE (Agrawal et al., 2010; Figure 8). Moreover, expression of OraiE180A completely blocks SOCE as measured in primary cultures of dopaminergic neurons (Pathak et al., 2015; Figure 1E). Hence we have not repeated SOCE measurements from all dopaminergic neurons in this work. In the revised version we will explicitly state this weakness of our study and the reasons for it.
- In the experiments performed to rescue flight duration in Set2RNAi individuals the authors overexpress STIM and attribute the effect to "Excess STIM presumably drives higher SOCE sufficient to rescue flight bout durations caused by deficient Set2 levels.". This should be experimentally tested as the STIM:Orai stoichiometry has been demonstrated as essential for SOCE.
The assumption that STIM overexpression drives higher SOCE is based upon previously published work from Drosophila neurons (Agrawal et al., 2010; Chakraborty et al, 2016; Deb et al., 2016) which demonstrates that excess WT STIM overcomes IP3R deficiencies (RNAi or hypomorphic mutants) to rescue SOCE. We agree that STIM-Orai stoichiometry is essential for SOCE, and propose that the rescue backgrounds possess sufficient WT Orai, which is recruited by the excess STIM to mediate the rescue. We will reference the earlier work to validate our use of STIMOE for rescue of SOCE.
Here, we propose that Set2 is part of a positive feedback loop driving transcription of mAChR and IP3R (Figure 3F). In keeping with this hypothesis, we posit that the phenotypes observed in the Set2RNAi background (Figure 2D) result from decreased itpr and mAChR expression (validated in Figure 3E). This is further validated by the Set2 overexpression mediated rescue of OraiE180A (Figure 2D) and rescue of itpr/mAChR expression in the OraiE180A background (Figure 3B-E; THD’>OraiE180A; Set2OE).
- The authors show that overexpression of OraiE108A results in Stim downregulation at a mRNA level. What about the protein level? And more important, how does OraiE108A downregulate Stim expression? Does it promote Stim degradation? Does it inhibit Stim expression?
We hypothesize that changes in STIM mRNA observed in the THD’ > OraiE180A neurons stems from an overall reduction in IP3-mediated Ca2+ release and SOCE due to loss of Trl-Set2 driven gene expression detailed in our transcriptional feedback loop model (Figure 5C). We will attempt to explain this aspect more clearly in the next version of the manuscript. While we agree that measuring levels of STIM protein would be helpful, estimation of protein levels from a limited number of neurons (~35 cells per brain) is technically challenging. The STIM antibody does not work well in immunohistochemistry. In the absence of any experimental evidence we cannot comment on how expression of OraiE180A might affect STIM protein turnover.
- Lines 271-273, the authors state "whereas overexpression of a transgene encoding Set2 in THD' neurons either with loss of SOCE (OraiE180A) or with knockdown of the IP3R (itprRNAi), lead to significant rescue of the Ca2+ response". This is attributed to a positive effect of Set2 expression on IP3R expression and the authors show a positive correlation between these two parameters; however, there is no demonstration that Set2 expression can rescue IP3R expression in cells where the IP3R is knocked down (itprRNAi). This should be further demonstrated.
The rescue of IP3R expression by Set2 overexpression in itprRNAi was demonstrated in a different set of Drosophila neurons in an earlier study (Mitra et al., 2021) and has not been repeated specifically in THD’ neurons. Similar to the previous study, here we tested CCh stimulated Ca2+ responses of THD’ neurons with itprRNAi and itprRNAi; SetOE (Fig S3), which are indeed rescued by SET2OE.
- The data presented in Figure 3E should be functionally demonstrated by analyzing the ability of CCh to release Ca2+ from the intracellular stores in the absence of extracellular Ca2+.
CCh-mediated Ca2+ release from the intracellular stores in the absence of extracellular Ca2+ has been described in primary cultures of Drosophila neurons in previously published work (Venkiteswaran and Hasan, 2009; Agrawal et al., 2010) This work focuses on a set of 16-19 dopaminergic neurons in a hemisphere of the Drosophila central brain. It is technically challenging to generate a 0 Ca2+ environment in vivo, which is essential for measuring store Ca2+ release. Given their meagre numbers, primary cultures of these neurons is not readily feasible.
- The conclusion that SOCE regulates the neuronal excitability threshold is based entirely on either partial behavioral rescue of flight, or measurements of KCl-induced Ca2+ rises monitored by GCaMP6m in DAN neurons. The threshold for neuronal excitability is a precise parameter based on rheobase measurements of action potentials in current-clamp. Measurements of slow calcium signals using a slow dye such as GCaMp6m should not be equated with neuronal excitability. What is measured is a loss of the calcium response in high K depolarization experiments, which occurs due to the loss of expression of Cav channels. Hence, the use of this term is not accurate and will confuse readers. The use of terms referring to neuronal excitability needs to be changed throughout the manuscript. As such, the conclusions regarding neuronal excitability should be strongly tempered and the data reinterpreted as there are no true measurements of neuronal excitability in the manuscript. All that can be said is that expression of certain ion channel genes is suppressed. Since both Na+ channels and K+ channel expression is down-regulated, it is hard to say precisely how membrane excitability is altered without action potential analysis.
The claim that SOCE influences neuronal excitability is based on the following observations:
Interruption of the transcriptional feedback loop involving SOCE, Trl, and Set2 through loss of any of its constituents, results in the downregulation of VGCCs (Figure 5G, 6H), which are essential components of action potentials.
OraiE180A mediated loss of SOCE in THD’ neurons abrogates the KCl-evoked depolarization response (Figure 6B, C) measured using GCaMP6m. We verified that this response requires VGCC function using pharmacological inhibition of L-type VGCCs (Figure 6E, F).
SOCE deficient THD’ neurons, which were presumably compromised in their ability to evoke action potentials could be rescued to undergo KCl-evoked depolarisation by expression of NachBac, which lowers the depolarization threshold (Figure 7C, D) or through optogenetic stimulation using CsChrimson (Figure 7F).
We agree that ‘neuronal excitability threshold’ is a precise electrophysiological parameter that has not been directly investigated here by measurement of action potentials. Therefore, references to neuronal excitability will be tempered throughout the revised manuscript and be replaced with a more generic reference to ‘neuronal activity’. In this context we propose to include further evidence supporting reduced excitability of THD’ neurons upon loss of SOCE in the revision.
Since one of the key functional outcomes of activity during critical developmental periods such as the 72-96 hrs APF developmental window identified in this study, is remodelling of neuronal morphology, we decided to investigate the same in our context. Neuronal activity can drive changes in neurite complexity and axonal arborization (Depetris-Chauvin et al., 2011) especially during critical developmental periods (Sachse et al., 2007). To understand if Orai mediated Ca2+ entry and downstream gene expression through Set2 affects this activity-driven parameter, we investigated the morphology of fpDANs, and specifically measured the complexity of presynaptic terminals within the 2’1 lobe MB using super-resolution microscopy. We found striking changes in the neurite volume upon expression of OraiE180A which could be rescued by restoring either Set2 (OraiE180A; Set2OE) or by inducing hyperactivity through NachBac expression (OraiE180A ; NachBacOE). These data will be included in the revised manuscript.
- Related, since trl does not contain any molecular domains that could be regulated by Ca2+ signaling, it is unclear whether trl is directly regulated by SOCE or the regulation is highly indirect. Reporter assays evaluating trl activation upon Ca2+ rises would provide much stronger and more direct evidence for the conclusion that trl is a SOCE-regulated TF. As such the evidence is entirely based on RNAi downregulation of trl which indicates that trl is essential but has no bearing on exactly what point of the signaling cascade it is involved.
We agree that luciferase Trl reporters would provide a direct method to test SOCE-mediated activation. Future investigations will be targeted in this direction. Regarding possible mechanisms of Trl activation - since we could not detect a well-defined Ca2+ binding domain in Trl, we hypothesize that it may be phosphorylation by a Ca2+ sensitive kinase. Phosphoproteome analysis of Trl indicates that it does indeed undergo phosphorylation at a Threonine reside (T237; Zhai et al., 2008), which may be mediated by the Ca2+ sensitive kinase-CaMKII based on binding partners identified in the Trl interactome (Lomaev et al., 2018). Past work (Ravi et al., 2018) has indeed demonstrated a requirement for CaMKII in THD’ neurons for flight. We are currently testing whether CaMKII functions downstream of SOCE in these neurons to mediate flight, and will be updating this information in the next version of the manuscript.
- Are NFAT levels altered in the Orai1 loss of function mutant? If not, this should be explicitly stated. It would seem based on previous literature that some gene regulation may be related to the downregulation of this established Ca2+-dependent transcription factor. Same for NFkb.
As mentioned in the text in lines (307-309), Drosophila NFAT lacks a calcineurin binding site and is therefore not sensitive to Ca2+ (Keyser et al., 2007). In the past we tested if knockdown of NF-kB in dopaminergic neurons gave a flight phenotype and did not observe any measurable deficit. From the RNAseq data we find a slight downregulation of NFAT (0.49 fold, p value=0.048) and NF-kb (0.26 fold, p value =0.258) the significance of which is unclear at this point. We did not find any consensus binding sites for these two factors in the regulatory regions of downregulated genes from THD’ neurons.
- Does over-expression of Set2 restore ion channel expression especially those of the VGCCs? This would provide rigorous, direct evidence that SOCE-mediated regulation of VGCCs through Set2 controls voltage-gated calcium channel signaling.
Set2 overexpression in the OraiE180A background indeed restores the expression of VGCC genes (Figure 6H).
- All 6 representative panels from Figure 3B are duplicated in Figure 4G. Likewise, 2 representative panels from Figure 5H are duplicated in Figure 6D. Although these panels all represent the results from control experiments, the relevant experiments were likely not conducted at the same time and under the same conditions. Thus, control images from other experiments should not be used simply because they correspond to controls. This situation should be clarified.
We regret the confusion caused by the same representative images for the control experiments. These will be replaced by new representative images for Figure 5H in the next updated version of the manuscript.
- The figures are unusually busy and difficult to follow. In part this is because they usually have many panels (Fig. 1: A-I; Fig. 2, A-J, etc) but also because the arrangement of the panels is not consistent: sometimes the following panel is found to the right, other times it is below. It would help the reader to make the order of the panels consistent, and, if possible, reduce the number of panels and/or move some of the panels to new figures (eLife does not limit the number of display items).
The image panels will be rearranged for ease of reading in the next updated version of the manuscript.
- As a final recommendation, the reviewers suggest that the authors a- Reword the text that refers to membrane excitability since membrane excitability was not directly measured here. b-Explain why STIM1 rescues the partial loss of flight in Set2 RNAi flies (Fig. S2E); and c- Explain how/why trl is calcium regulated and test using luciferase (or other) reporter assays whether Orai activation leads to trl activation.
a. Textual references to membrane excitability will be appropriately modified.
b. We have provided a detailed explanation for how STIM overexpression might rescue the phenotypes caused by Set2RNAi in Point 1. In short, these phenotypes depend upon IP3R mediated Ca2+ entry driving a transcriptional feedback loop. We relied upon past reports that STIM overexpression upregulates IP3R-mediated Ca2+ release and SOCE in Drosophila itpr mutant neurons (Agrawal et al., 2010; Chakraborty et al, 2016; Deb et al, 2016). We therefore propose that STIM overexpression in the Set2RNAi background rescues IP3R mediated Ca2+ release followed by SOCE, which drives enhanced Set2 transcription, counteracting the effects of the RNAi. We will explain this more clearly with past references in the next revision.
c. We have provided a detailed response to this comment in Point 12. Briefly, we agree that building luciferase reporters for Trl could be an ideal strategy to test for its responsiveness to SOCE and needs to be done in future. As an alternate strategy, we have looked at data from existing studies of interacting partners of Trl (Lomaev et al., 2017) and identified CamKII, which is both Ca2+ responsive (Braun and Schulman, 1995; Yasuda et al., 2022), and thus might activate Trl through a phosphorylation-switch like mechanism. Moreover, a previous publication identified a requirement for CamKII in THD’ neurons for Drosophila flight (Ravi et al., 2018). We are testing the ability of a dominant active version of CamKII to rescue THD’>E180A flight deficits and will include this information in the next version of the manuscript.
References
- Agrawal N, Venkiteswaran G, Sadaf S, Padmanabhan N, Banerjee S, Hasan G. Inositol 1,4,5-Trisphosphate Receptor and dSTIM Function in Drosophila Insulin-Producing Neurons Regulates Systemic Intracellular Calcium Homeostasis and Flight. J Neurosci. 2010;30:1301-1313. doi:10.1523/jneurosci.3668-09.2010
- Braun AP, Schulman H. A non-selective cation current activated via the multifunctional Ca(2+)-calmodulin-dependent protein kinase in human epithelial cells. J Physiol. 1995. 488:37-55. doi:10.1113/jphysiol.1995.sp020944
- Chakraborty S, Deb BK, Chorna T, Konieczny V, Taylor CW, Hasan G. Mutant IP3 receptors attenuate store-operated Ca2+ entry by destabilizing STIM-Orai interactions in Drosophila neurons. J Cell Sci. 2016. 129:3903-3910. doi:10.1242/jcs.191585
- Deb BK, Pathak T, Hasan G. Store-independent modulation of Ca2+ entry through Orai by Septin 7. Nat Commun. 2016. 7:11751. doi:10.1038/ncomms11751
- Depetris-Chauvin A, Berni J, Aranovich EJ, Muraro NI, Beckwith EJ, Ceriani MF. Adult-specific electrical silencing of pacemaker neurons uncouples molecular clock from circadian outputs. Curr Biol. 2011. 21:1783-1793. doi: 10.1016/j.cub.2011.09.027.
- Keyser P, Borge-Renberg K, Hultmark D. The Drosophila NFAT homolog is involved in salt stress tolerance. Insect Biochem Mol Biol. 2007. 37:356-362. doi:10.1016/j.ibmb.2006.12.009
- Kilo L, Stürner T, Tavosanis G, Ziegler AB. Drosophila Dendritic Arborisation Neurons: Fantastic Actin Dynamics and Where to Find Them. Cells. 2021. 10:2777. doi:10.3390/cells10102777
- Lomaev D, Mikhailova A, Erokhin M, et al. The GAGA factor regulatory network: Identification of GAGA factor associated proteins. PLoS One. 2017. 12:e0173602. doi:10.1371/journal.pone.0173602
- Mitra R, Richhariya S, Jayakumar S, Notani D, Hasan G. IP3/Ca2+ signals regulate larval to pupal transition under nutrient stress through the H3K36 methyltransferase dSET2. Development. 2021. 148:dev199018. doi:10.1101/2020.11.25.399329
- Pathak T, Agrawal T, Richhariya S, Sadaf S, Hasan G. Store-Operated Calcium Entry through Orai Is Required for Transcriptional Maturation of the Flight Circuit in Drosophila. J Neurosci. 2015. 35:13784-13799. doi:10.1523/jneurosci.1680-15.2015
- Ravi P, Trivedi D, Hasan G. FMRFa receptor stimulated Ca2+ signals alter the activity of flight modulating central dopaminergic neurons in Drosophila melanogaster. Barsh GS, ed. PLOS Genet. 2018. 14:e1007459. doi:10.1371/journal.pgen.1007459
- Sachse S, Rueckert E, Keller A, Okada R, Tanaka NK, Ito K, Vosshall LB. Activity-dependent plasticity in an olfactory circuit. Neuron. 2007. 56:838-50. doi: 10.1016/j.neuron.2007.10.035.
- Sharma A, Hasan G. Modulation of flight and feeding behaviours requires presynaptic IP3Rs in dopaminergic neurons. Elife. 2020;9. e62297.doi:10.7554/elife.62297
- Venkiteswaran G, Hasan G. Intracellular Ca2+ signalling and store operated Ca2+ entry are required in Drosophila neurons for flight. Proc Natl Acad Sci. 2009.106:10326-10331. doi: 10.1073/pnas.0902982106
- Yasuda R, Hayashi Y, Hell JW. CaMKII: a central molecular organizer of synaptic plasticity, learning and memory. Nat Rev Neurosci. 2022. 23: 666-682 doi:10.1038/s41583-022-00624-2
- Zhai B, Villén J, Beausoleil SA, Mintseris J, Gygi SP. Phosphoproteome Analysis of Drosophila melanogaster Embryos. J Proteome Res. 2008. 7:1675-1682. doi:10.1021/pr700696a
-
eLife assessment
In Drosophila melanogater, the Store-operated Ca2+ entry (SOCE) channel, Orai, is required for the development of flight-promoting dopaminergic neurons. Here, Mitra et al. determine that expression of a loss-of-function Orai1 mutant during the 72-96 hour window of pupal development impairs gene expression in dopaminergic flight neurons in part through the expression of Set2, a histone methyltransferase. The authors identify a large number of genes that are controlled by Set2, and show that Set2 is controlled by the trl transcription factor. Although the findings reported here are important, the evidence supporting some of the claims is incomplete.
-
Joint Public Review
The manuscript by Mitra and coworkers analyses the functional role of Orai in the excitability of central dopaminergic neurons in Drosophila. The authors show that a dominant-negative mutant of Orai (OraiE180A) significantly alters the gene expression profile of flight-promoting dopaminergic neurons (fpDANs). Among them, OraiE180A attenuates the expression of Set2 and enhances that of E(z) shifting the level of epigenetic signatures that modulate gene expression. The present results also demonstrate that Set2 expression via Orai involves the transcription factor Trl. The Orai-Trl-Set1 pathway modulates the expression of VGCC, which, in turn, are involved in dopamine release. The topic investigated is interesting and timely and the study is carefully performed and technically sound; however, there are several major …
Joint Public Review
The manuscript by Mitra and coworkers analyses the functional role of Orai in the excitability of central dopaminergic neurons in Drosophila. The authors show that a dominant-negative mutant of Orai (OraiE180A) significantly alters the gene expression profile of flight-promoting dopaminergic neurons (fpDANs). Among them, OraiE180A attenuates the expression of Set2 and enhances that of E(z) shifting the level of epigenetic signatures that modulate gene expression. The present results also demonstrate that Set2 expression via Orai involves the transcription factor Trl. The Orai-Trl-Set1 pathway modulates the expression of VGCC, which, in turn, are involved in dopamine release. The topic investigated is interesting and timely and the study is carefully performed and technically sound; however, there are several major concerns that need to be addressed:
1- In Figure S2E, STIM is overexpressed in the absence of Set2 and this leads to rescue. It is presumed that STIM overexpression causes excess SOCE, yet this is rarely the case. Perhaps the bigger concern, however, is how excess SOCE might overcome the loss of SET2 if SET2 mediates SOCE-induced development of flight. These data are more consistent with something other than SET2 mediating this function.
2- In Figure 3, data is provided linking SET2 expression and Cch-induced Ca2+ responses. The presentation of these data is confusing. In addition, the results may be a simple side effect of SET2-dependent expression of IP3R. Given that this article is about SOCE, why isn't SOCE shown here? More generally, there are no measurements of SOCE in this entire article. Measuring SOCE (not what is measured in response to Cch) could help eliminate some of this confusion.
3- A significant gap in the study relates to the conclusion that trl is a SOCE-regulated transcription factor. This conclusion is entirely based on genetic analysis of STIMKO heterozygous flies in which a copy of the trl13C hypomorph allele is introduced. While these results suggest a genetic interaction between the expression of the two genes, the evidence that expression translates into a functional interaction that places trl immediately downstream of SOCE is not rigorous or convincing. All that can be said is that the double mutant shows a defect in flight which could arise from an interruption of the circuit. Further, it is not clear whether the trl13C hypomorph is only introduced during the critical 72-96 hour time window when the Orai1E180E phenotype shows up. The same applies to the over-expression of Set2 and the other genes. If the expression is not temporally controlled, then the phenotype could be due to the blockade of an entirely different aspect of flight neuron function.
4- In Figure 4, data is shown that SOCE compensates for the loss of Trl, the presumed mediator of SOCE-dependent flight. The fact that flight deficits are rescued by raising SOCE in the absence of Trl is very inconsistent with this conclusion.
5- In Figure 5 (A-C), data is provided that Trl transcripts are unaffected by loss of SOCE and that overexpression cannot rescue flightlessness. From this, the authors conclude that this gene "must" be calcium responsive. While that is one possibility, it is also possible that these genes are not functionally linked.
6- There is no characterization of SOCE in fpDANs from flies expressing native Orai or the dominant negative OraiE180A mutant. While the authors refer to previous studies, as the manuscript is essentially based on Orai function thapsigargin-induced SOCE should be tested using the Ca2+ add-back protocol in order to assess the release of Ca2+ from the ER in response to thapsigargin as well as the subsequent SOCE.
7- In the experiments performed to rescue flight duration in Set2RNAi individuals the authors overexpress STIM and attribute the effect to "Excess STIM presumably drives higher SOCE sufficient to rescue flight bout durations caused by deficient Set2 levels.". This should be experimentally tested as the STIM:Orai stoichiometry has been demonstrated as essential for SOCE.
8- The authors show that overexpression of OraiE108A results in Stim downregulation at a mRNA level. What about the protein level? And more important, how does OraiE108A downregulate Stim expression? Does it promote Stim degradation? Does it inhibit Stim expression?
9- Lines 271-273, the authors state "whereas overexpression of a transgene encoding Set2 in THD' neurons either with loss of SOCE (OraiE180A) or with knockdown of the IP3R (itprRNAi), lead to significant rescue of the Ca2+ response". This is attributed to a positive effect of Set2 expression on IP3R expression and the authors show a positive correlation between these two parameters; however, there is no demonstration that Set2 expression can rescue IP3R expression in cells where the IP3R is knocked down (itprRNAi). This should be further demonstrated.
10- The data presented in Figure 3E should be functionally demonstrated by analyzing the ability of CCh to release Ca2+ from the intracellular stores in the absence of extracellular Ca2+.
11- The conclusion that SOCE regulates the neuronal excitability threshold is based entirely on either partial behavioral rescue of flight, or measurements of KCl-induced Ca2+ rises monitored by GCaMP6m in DAN neurons. The threshold for neuronal excitability is a precise parameter based on rheobase measurements of action potentials in current-clamp. Measurements of slow calcium signals using a slow dye such as GCaMp6m should not be equated with neuronal excitability. What is measured is a loss of the calcium response in high K depolarization experiments, which occurs due to the loss of expression of Cav channels. Hence, the use of this term is not accurate and will confuse readers. The use of terms referring to neuronal excitability needs to be changed throughout the manuscript. As such, the conclusions regarding neuronal excitability should be strongly tempered and the data reinterpreted as there are no true measurements of neuronal excitability in the manuscript. All that can be said is that expression of certain ion channel genes is suppressed. Since both Na+ channels and K+ channel expression is down-regulated, it is hard to say precisely how membrane excitability is altered without action potential analysis.
12- Related, since trl does not contain any molecular domains that could be regulated by Ca2+ signaling, it is unclear whether trl is directly regulated by SOCE or the regulation is highly indirect. Reporter assays evaluating trl activation upon Ca2+ rises would provide much stronger and more direct evidence for the conclusion that trl is a SOCE-regulated TF. As such the evidence is entirely based on RNAi downregulation of trl which indicates that trl is essential but has no bearing on exactly what point of the signaling cascade it is involved.
13- Are NFAT levels altered in the Orai1 loss of function mutant? If not, this should be explicitly stated. It would seem based on previous literature that some gene regulation may be related to the downregulation of this established Ca2+-dependent transcription factor. Same for NFkb.
14- Does over-expression of Set2 restore ion channel expression especially those of the VGCCs? This would provide rigorous, direct evidence that SOCE-mediated regulation of VGCCs through Set2 controls voltage-gated calcium channel signaling.
15- All 6 representative panels from Figure 3B are duplicated in Figure 4G. Likewise, 2 representative panels from Figure 5H are duplicated in Figure 6D. Although these panels all represent the results from control experiments, the relevant experiments were likely not conducted at the same time and under the same conditions. Thus, control images from other experiments should not be used simply because they correspond to controls. This situation should be clarified.
16- The figures are unusually busy and difficult to follow. In part this is because they usually have many panels (Fig. 1: A-I; Fig. 2, A-J, etc) but also because the arrangement of the panels is not consistent: sometimes the following panel is found to the right, other times it is below. It would help the reader to make the order of the panels consistent, and, if possible, reduce the number of panels and/or move some of the panels to new figures.
17- As a final recommendation, the reviewers suggest that the authors a- Reword the text that refers to membrane excitability since membrane excitability was not directly measured here. b-Explain why STIM1 rescues the partial loss of flight in Set2 RNAi flies (Fig. S2E); and c- Explain how/why trl is calcium regulated and test using luciferase (or other) reporter assays whether Orai activation leads to trl activation.
-
