Context-Aware Amino Acid Embedding Advances Analysis of TCR-Epitope Interactions
Curation statements for this article:-
Curated by eLife

eLife assessment
This study provides an important tool for predicting binding between immune cells receptors and antigens based on protein sequence data. The analysis convincingly showed the tool's effectiveness in both supervised TCR binding prediction and unsupervised clustering, surpassing existing methods in accuracy and reducing annotation costs. This study will be of interest to immunologists and computational biologists.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Abstract
Accurate prediction of binding interaction between T cell receptors (TCRs) and host cells is fundamental to understanding the regulation of the adaptive immune system as well as to developing data-driven approaches for personalized immunotherapy. While several machine learning models have been developed for this prediction task, the question of how to specifically embed TCR sequences into numeric representations remains largely unexplored compared to protein sequences in general. Here, we investigate whether the embedding models designed for protein sequences, and the most widely used BLOSUM-based embedding techniques are suitable for TCR analysis. Additionally, we present our context-aware amino acid embedding models (<monospace>catELMo</monospace>) designed explicitly for TCR analysis and trained on 4M unlabeled TCR sequences with no supervision. We validate the effectiveness of <monospace>catELMo</monospace> in both supervised and unsupervised scenarios by stacking the simplest models on top of our learned embeddings. For the supervised task, we choose the binding affinity prediction problem of TCR and epitope sequences and demonstrate notably significant performance gains (up by at least 14% AUC) compared to existing embedding models as well as the state-of-the-art methods. Additionally, we also show that our learned embeddings reduce more than 93% annotation cost while achieving comparable results to the state-of-the-art methods. In TCR clustering task (unsupervised), <monospace>catELMo</monospace> identifies TCR clusters that are more homogeneous and complete about their binding epitopes. Altogether, our <monospace>catELMo</monospace> trained without any explicit supervision interprets TCR sequences better and negates the need for complex deep neural network architectures in downstream tasks.
Article activity feed
-

-
-

Author response:
The following is the authors’ response to the original reviews.
Thank you very much for the careful and positive reviews of our manuscript. We have addressed each comment in the attached revised manuscript. We describe the modifications below. To avoid confusion, we've changed supplementary figure and table captions to start with "Supplement Figure" and "Supplementary Table," instead of "Figure" and "Table."
We have modified/added:
● Supplementary Table S1: AUC scores for the top 10 frequent epitope types (pathogens) in the testing set of epitope split.
● Supplementary Table S5: AUCs of TCR-epitope binding affinity prediction models with BLOSUM62 to embed epitope sequences.
● Supplementary Table S6: AUCs of TCR-epitope binding affinity prediction models trained on catELMo TCR embeddings and random-initialized …
Author response:
The following is the authors’ response to the original reviews.
Thank you very much for the careful and positive reviews of our manuscript. We have addressed each comment in the attached revised manuscript. We describe the modifications below. To avoid confusion, we've changed supplementary figure and table captions to start with "Supplement Figure" and "Supplementary Table," instead of "Figure" and "Table."
We have modified/added:
● Supplementary Table S1: AUC scores for the top 10 frequent epitope types (pathogens) in the testing set of epitope split.
● Supplementary Table S5: AUCs of TCR-epitope binding affinity prediction models with BLOSUM62 to embed epitope sequences.
● Supplementary Table S6: AUCs of TCR-epitope binding affinity prediction models trained on catELMo TCR embeddings and random-initialized epitope embeddings.
● Supplementary Table S7: AUCs of TCR-epitope binding affinity prediction models trained on catELMo and BLOSUM62 embeddings.
● Supplementary Figure 4: TCR clustering performance for the top 34 abundant epitopes representing 70.55% of TCRs in our collected databases.
● Section Discussion.
● Section 4.1 Data: TCR-epitope pairs for binding affinity prediction.
● Section 4.4.2 Epitope-specific TCR clustering.
Public Reviews:
Reviewer #1 (Public Review):
In this manuscript, the authors described a computational method catELMo for embedding TCR CDR3 sequences into numeric vectors using a deep-learning-based approach, ELMo. The authors applied catELMo to two applications: supervised TCR-epitope binding affinity prediction and unsupervised epitope-specific TCR clustering. In both applications, the authors showed that catELMo generated significantly better binding prediction and clustering performance than other established TCR embedding methods. However, there are a few major concerns that need to be addressed.
(1) There are other TCR CDR3 embedding methods in addition to TCRBert. The authors may consider incorporating a few more methods in the evaluation, such as TESSA (PMCID: PMC7799492), DeepTCR (PMCID: PMC7952906) and the embedding method in ATM-TCR (reference 10 in the manuscript). TESSA is also the embedding method in pMTnet, which is another TCR-epitope binding prediction method and is the reference 12 mentioned in this manuscript.
TESSA is designed for characterizing TCR repertoires, so we initially excluded it from the comparison. Our focus was on models developed specifically for amino acid embedding rather than TCR repertoire characterization. However, to address the reviewer's inquiry, we conducted further evaluations. Since both TESSA and DeepTCR used autoencoder-based models to embed TCR sequences, we selected one used in TESSA for evaluation in our downstream prediction task, conducting ten trials in total. It achieved an average AUC of 75.69 in TCR split and 73.3 in epitope split. Notably, catELMo significantly outperformed such performance with an AUC of 96.04 in TCR split and 94.10 in epitope split.
Regarding the embedding method in ATM-TCR, it simply uses BLOSUM as an embedding matrix which we have already compared in Section 2.1. Furthermore, we have provided the comparison results between our prediction model trained on catELMo embeddings with the state-of-the-art prediction models such as netTCR and ATM-TCR in Table 6 of the Discussion section.
(2) The TCR training data for catELMo is obtained from ImmunoSEQ platform, including SARS-CoV2, EBV, CMV, and other disease samples. Meanwhile, antigens related to these diseases and their associated TCRs are extensively annotated in databases VDJdb, IEDB and McPAS-TCR. The authors then utilized the curated TCR-epitope pairs from these databases to conduct the evaluations for eptitope binding prediction and TCR clustering. Therefore, the training data for TCR embedding may already be implicitly tuned for better representations of the TCRs used in the evaluations. This seems to be true based on Table 4, as BERT-Base-TCR outperformed TCRBert. Could catELMo be trained on PIRD as TCRBert to demonstrate catELMo's embedding for TCRs targeting unseen diseases/epitopes?
We would like to note that catELMo was trained exclusively on TCR sequences in an unsupervised manner, which means it has never been exposed to antigen information. We also ensured that the TCRs used in catELMo's training did not overlap with our downstream prediction data. Please refer to the section 4.1 Data where we explicitly stated, “We note that it includes no identical TCR sequences with the TCRs used for training the embedding models.”. Moreover, the performance gap (~1%) between BERT-Base-TCR and TCRBert, as observed in Table 4, is relatively small, especially when compared to the performance difference (>16%) between catELMo and TCRBert.
To further address this concern, we conducted experiments using the same number of TCRs, 4,173,895 in total, sourced exclusively from healthy ImmunoSeq repertoires. This alternative catELMo model demonstrated a similar prediction performance (based on 10 trials) to the one reported in our paper, with an average AUC of 96.35% in TCR split and an average AUC of 94.03% in epitope split.
We opted not to train catELMo on the PIRD dataset for several reasons. First, approximately 7.8% of the sequences in PIRD also appear in our downstream prediction data, which could be a potential source of bias. Furthermore, PIRD encompasses sequences related to diseases such as Tuberculosis, HIV, CMV, among others, which the reviewer is concerned about.
(3) In the application of TCR-epitope binding prediction, the authors mentioned that the model for embedding epitope sequences was catElMo, but how about for other methods, such as TCRBert? Do the other methods also use catELMo-embedded epitope sequences as part of the binding prediction model, or use their own model to embed the epitope sequences? Since the manuscript focuses on TCR embedding, it would be nice for other methods to be evaluated on the same epitope embedding (maybe adjusted to the same embedded vector length).
Furthermore, the authors found that catELMo requires less training data to achieve better performance. So one would think the other methods could not learn a reasonable epitope embedding with limited epitope data, and catELMo's better performance in binding prediction is mainly due to better epitope representation.
Review 1 and 3 have raised similar concerns regarding the epitope embedding approach employed in our binding affinity prediction models. We address both comments together on page 6 where we discuss the epitope embedding strategies in detail.
(4) In the epitope binding prediction evaluation, the authors generated the test data using TCR-epitope pairs from VDJdb, IEDB, McPAS, which may be dominated by epitopes from CMV. Could the authors show accuracy categorized by epitope types, i.e. the accuracy for TCR-CMV pair and accuracy for TCR-SARs-CoV2 separately?
The categorized AUC scores have been added in Supplementary Table 7. We observed significant performance boosts from catELMo compared with other embedding models.
(5) In the unsupervised TCR clustering evaluation, since GIANA and TCRdist direct outputs the clustering result, so they should not be affected by hierarchical clusters. Why did the curves of GIANA and TCRdist change in Figure 4 when relaxing the hierarchical clustering threshold?
For fair comparisons, we performed GIANA and TCRdist with hierarchical clustering instead of the nearest neighbor search. We have clarified it in the revised manuscript as follows.
“Both methods are developed on the BLOSUM62 matrix and apply nearest neighbor search to cluster TCR sequences. GIANA used the CDR3 of TCRβ chain and V gene, while TCRdist predominantly experimented with CDR1, CDR2, and CDR3 from both TCRα and TCRβ chains. For fair comparisons, we perform GIANA and TCRdist only on CDR3 β chains and with hierarchical clustering instead of the nearest neighbor search.”
(6 & 7) In the unsupervised TCR clustering evaluation, the authors examined the TCR related to the top eight epitopes. However, there are much more epitopes curated in VDJdb, IEDB and McPAS-TCR. In real application, the potential epitopes is also more complex than just eight epitopes. Could the authors evaluate the clustering result using all the TCR data from the databases? In addition to NMI, it is important to know how specific each TCR cluster is. Could the authors add the fraction of pure clusters in the results? Pure cluster means all the TCRs in the cluster are binding to the same epitope, and is a metric used in the method GIANA.
We would like to note that there is a significant disparity in TCR binding frequencies across different epitopes in current databases. For instance, the most abundant epitope (KLGGALQAK) has approximately 13k TCRs binding to it, while 836 out of 982 epitopes are associated with fewer than 100 TCRs in our dataset. Furthermore, there are 9347 TCRs having the ability to bind multiple epitopes. In order to robustly evaluate the clustering performance, we originally selected the top eight frequent epitopes from McPAS and removed TCRs binding multiple epitopes to create a more balanced dataset.
We acknowledge that the real-world scenario is more complex than just eight epitopes. Therefore, we conducted clustering experiments using the top most abundant epitopes whose combined cognate TCRs make up at least 70% of TCRs across three databases (34 epitopes). This is illustrated in Supplementary Figure 5. Furthermore, we extended our analysis by clustering all TCRs after filtering out those that bind to multiple epitopes, resulting in 782 unique epitopes. We found that catELMo achieved the 3rd and 2nd best performance in NMI and Purity, respectively (see Table below). These are aligned with our previous observations of the eight epitopes.
Author response table 1.
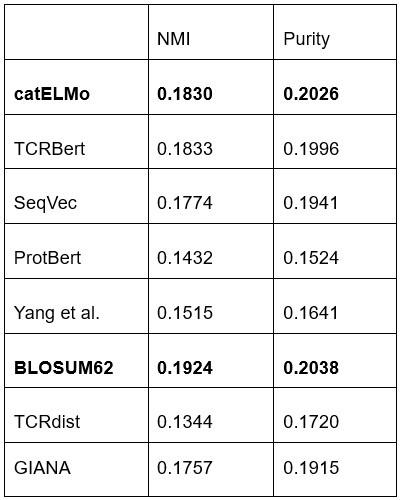
Reviewer #2 (Public Review):
In the manuscript, the authors highlighted the importance of T-cell receptor (TCR) analysis and the lack of amino acid embedding methods specific to this domain. The authors proposed a novel bi-directional context-aware amino acid embedding method, catELMo, adapted from ELMo (Embeddings from Language Models), specifically designed for TCR analysis. The model is trained on TCR sequences from seven projects in the ImmunoSEQ database, instead of the generic protein sequences. They assessed the effectiveness of the proposed method in both TCR-epitope binding affinity prediction, a supervised task, and the unsupervised TCR clustering task. The results demonstrate significant performance improvements compared to existing embedding models. The authors also aimed to provide and discuss their observations on embedding model design for TCR analysis: 1) Models specifically trained on TCR sequences have better performance than models trained on general protein sequences for the TCR-related tasks; and 2) The proposed ELMo-based method outperforms TCR embedding models with BERT-based architecture. The authors also provided a comprehensive introduction and investigation of existing amino acid embedding methods. Overall, the paper is well-written and well-organized.
The work has originality and has potential prospects for immune response analysis and immunotherapy exploration. TCR-epitope pair binding plays a significant role in T cell regulation. Accurate prediction and analysis of TCR sequences are crucial for comprehending the biological foundations of binding mechanisms and advancing immunotherapy approaches. The proposed embedding method presents an efficient context-aware mathematical representation for TCR sequences, enabling the capture and analysis of their structural and functional characteristics. This method serves as a valuable tool for various downstream analyses and is essential for a wide range of applications. Thank you.
Reviewer #3 (Public Review):
Here, the authors trained catElMo, a new context-aware embedding model for TCRβ CDR3 amino acid sequences for TCR-epitope specificity and clustering tasks. This method benchmarked existing work in protein and TCR language models and investigated the role that model architecture plays in the prediction performance. The major strength of this paper is comprehensively evaluating common model architectures used, which is useful for practitioners in the field. However, some key details were missing to assess whether the benchmarking study is a fair comparison between different architectures. Major comments are as follows:
- It is not clear why epitope sequences were also embedded using catELMo for the binding prediction task. Because catELMO is trained on TCRβ CDR3 sequences, it's not clear what benefit would come from this embedding. Were the other embedding models under comparison also applied to both the TCR and epitope sequences? It may be a fairer comparison if a single method is used to encode epitope sequence for all models under comparison, so that the performance reflects the quality of the TCR embedding only.
In our study, we indeed used the same embedding model for both TCRs and epitopes in each prediction model, ensuring a consistent approach throughout.
Recognizing the importance of evaluating the impact of epitope embeddings, we conducted experiments in which we used BLOSUM62 matrix to embed epitope sequences for all models. The results (Supplementary Table 5) are well aligned with the performance reported in our paper. This suggests that epitope embedding may not play as critical a role as TCR embedding in the prediction tasks. To further validate this point, we conducted two additional experiments.
Firstly, we used catELMo to embed TCRs while employing randomly initialized embedding matrices with trainable parameters for epitope sequences. It yielded similar prediction performance as when catELMo was used for both TCR and epitope embedding (Supplementary Table 6). Secondly, we utilized BLOSUM62 to embed TCRs but employed catELMo for epitope sequence embedding, resulting in performance comparable to using BLOSUM62 for both TCRs and epitopes (Supplementary Table 4). These experiment results confirmed the limited impact of epitope embedding on downstream performance.
We conjecture that these results may be attributed to the significant disparity in data scale between TCRs (~290k) and epitopes (less than 1k). Moreover, TCRs tend to exhibit high similarity, whereas epitopes display greater distinctiveness from one another. These features of TCRs require robust embeddings to facilitate effective separation and improve downstream performance, while epitope embedding primarily serves as a categorical encoding.
We have included a detailed discussion of these findings in the revised manuscript to provide a comprehensive understanding of the role of epitope embeddings in TCR binding prediction.
- The tSNE visualization in Figure 3 is helpful. It makes sense that the last hidden layer features separate well by binding labels for the better performing models. However, it would be useful to know if positive and negative TCRs for each epitope group also separate well in the original TCR embedding space. In other words, how much separation between these groups is due to the neural network vs just the embedding?
It is important to note that we used the same downstream prediction model, a simple three-linear-layer network, for all the discussed embedding methods. We believe that the separation observed in the t-SNE visualization effectively reflects the ability of our embedding model. Also, we would like to mention that it can be hard to see a clear distinction between positive and negative TCRs in the original embedding space because embedding models were not trained on positive/negative labels. Please refer to the t-SNE of the original TCR embeddings below.
Author response image 1.
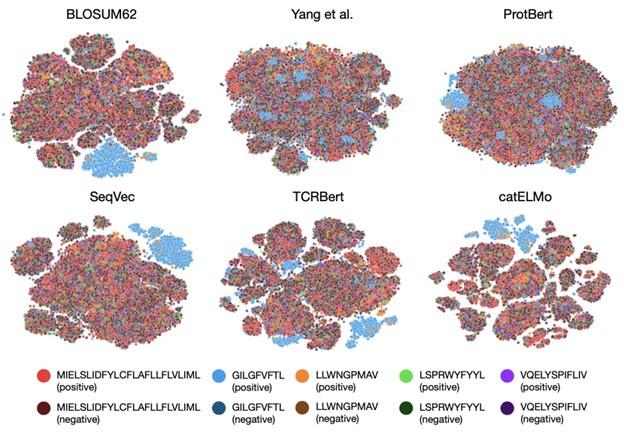
- To generate negative samples, the author randomly paired TCRs from healthy subjects to different epitopes. This could produce issues with false negatives if the epitopes used are common. Is there an estimate for how frequently there might be false negatives for those commonly occurring epitopes that most populations might also have been exposed to? Could there be a potential batch effect for the negative sampled TCR that confounds with the performance evaluation?
Thank you for bringing this valid and interesting point up. Generating negative samples is non-trivial since only a limited number of non-binding TCR-pairs are publicly available and experimentally validating non-binding pairs is costly [1]. Standard practices for generating negative pairs are (1) paring epitopes with healthy TCRs [2, 3], and (2) randomly shuffling existing TCR-epitope pairs [4,5]. We used both approaches (the former included in the main results, and the latter in the discussion). In both scenarios, catELMo embeddings consistently demonstrated superior performance.
We acknowledge the possibility of false negatives due to the finite-sized TCR database from which we randomly selected TCRs, however, we believe that the likelihood of such occurrences is low. Given the vast diversity of human TCR clonotypes, which can exceed 10^15[6], the chance of randomly selecting a TCR that specifically recognizes a target epitope is relatively small.
In order to investigate the batch effect, we generated new negative pairs using different seeds and observed consistent prediction performance across these variations. However, we agree that there could still be a potential batch effect for the negative samples due to potential data bias.
We have discussed the limitation of generative negative samples in the revised manuscript.
- Most of the models being compared were trained on general proteins rather than TCR sequences. This makes their comparison to catELMO questionable since it's not clear if the improvement is due to the training data or architecture. The authors partially addressed this with BERT-based models in section 2.4. This concern would be more fully addressed if the authors also trained the Doc2vec model (Yang et al, Figure 2) on TCR sequences as baseline models instead of using the original models trained on general protein sequences. This would make clear the strength of context-aware embeddings if the performance is worse than catElmo and BERT.
We agree it is important to distinguish between the effects of training data and architecture on model performance.
In Section 2.4, as the reviewer mentioned, we compared catELMo with BERT-based models trained on the same TCR repertoire data, demonstrating that architecture plays a significant role in improving performance. Furthermore, in Section 2.5, we compared catELMo-shallow with SeqVec, which share the same architecture but were trained on different data, highlighting the importance of data on the model performance.
To further address the reviewer's concern, we trained a Doc2Vec model on the TCR sequences that have been used for catELMo training. We observed significantly lower prediction performance compared to catELMo, with an average AUC of 50.24% in TCR split and an average AUC of 51.02% in epitope split, making the strength of context-aware embeddings clear.
Recommendations for the authors:
Reviewer #1 (Recommendations For The Authors):
(1) It is known that TRB CDR3, the CDR1, CDR2 on TRBV gene and the TCR alpha chain also contribute to epitope recognition, but were not modeled in catELMo. It would be nice for the authors to add this as a current limitation for catELMo in the Discussion section.
We have discussed the limitation in the revised manuscript.
“Our study focuses on modeling the TCRβ chain CDR3 region, which is known as the primary determinant of epitope binding. Other regions, such as CDR1 and CDR2 on the TRB V gene, along with the TCRα chain, may also contribute to specificity in antigen recognition. However, a limited number of available samples for those additional features can be a challenge for training embedding models. Future work may explore strategies to incorporate these regions while mitigating the challenges of working with limited samples.”
(2) I tried to follow the instructions to train a binding affinity prediction model for TCR-epitope pairs, however, the cachetools=5.3.0 seems could not be found when running "pip install -r requirements.txt" in the conda environment bap. Is this cachetools version supported after Python 3.7 so the Python 3.6.13 suggested on the GitHub repo might not work?
This has been fixed. We have updated the README.md on our github page.
Reviewer #2 (Recommendations For The Authors):
The article is well-constructed and well-written, and the analysis is comprehensive.
The comments for minor issues that I have are as follows:
(1) In the Methods section, it will be clearer if the authors interpret more on how the standard deviation is calculated in all tables. How to define the '10 trials'? Are they based on different random training and test set splits?
‘10 trials' refers to the process of splitting the dataset into training, validation, and testing sets using different seeds for each trial. Different trials have different training, validation, and testing sets. For each trial, we trained a prediction model on its training set and measured performance on its testing set. The standard deviation was calculated from the 10 measurements, estimating model performance variation across different random splits of the data.
(2) The format of AUCs and the improvement of AUCs need to be consistent, i.e., with the percent sign.
We have updated the format of AUCs.
Reviewer #3 (Recommendations For The Authors):
In addition to the recommendations in the public review, we had the following more minor questions and recommendations:
- Could you provide some more background on the data, such as overlaps between the databases, and how the training and validation split was performed between the three databases? Also summary statistics on the length of TCR and epitope sequence data would be helpful.
We have provided more details about data in our revision.
- Could you comment on the runtime to train and embed using the catELMo and BERT models?
Our training data is TCR sequences with relatively short lengths (averaging less than 20 amino acid residues). Such characteristic significantly reduces the computational resources required compared to training large-scale language models on extensive text corpora. Leveraging standard machines equipped with two GeForce RTX 2080 GPUs, we were able to complete the training tasks within a matter of days. After training, embedding one sequence can be accomplished in a matter of seconds.
- Typos and wording:
- Table 1 first row of "source": "immunoSEQ" instead of "immuneSEQ"
This has been corrected.
- L23 of abstract "negates the need of complex deep neural network architecture" is a little confusing because ELMo itself is a deep neural network architecture. Perhaps be more specific and add that the need is for downstream tasks.
We have made it more specific in our abstract.
“...negates the need for complex deep neural network architecture in downstream tasks.”
References
(1) Montemurro, Alessandro, et al. "NetTCR-2.0 enables accurate prediction of TCR-peptide binding by using paired TCRα and β sequence data." Communications biology 4.1 (2021): 1060.
(2) Jurtz, Vanessa Isabell, et al. "NetTCR: sequence-based prediction of TCR binding to peptide-MHC complexes using convolutional neural networks." BioRxiv (2018): 433706.
(3) Gielis, Sofie, et al. "Detection of enriched T cell epitope specificity in full T cell receptor sequence repertoires." Frontiers in immunology 10 (2019): 2820.
(4) Cai, Michael, et al. "ATM-TCR: TCR-epitope binding affinity prediction using a multi-head self-attention model." Frontiers in Immunology 13 (2022): 893247.
(5) Weber, Anna, et al. "TITAN: T-cell receptor specificity prediction with bimodal attention networks." Bioinformatics 37 (2021): i237-i244.
(6) Lythe, Grant, et al. "How many TCR clonotypes does a body maintain?." Journal of theoretical biology 389 (2016): 214-224.
-
eLife assessment
This study provides an important tool for predicting binding between immune cells receptors and antigens based on protein sequence data. The analysis convincingly showed the tool's effectiveness in both supervised TCR binding prediction and unsupervised clustering, surpassing existing methods in accuracy and reducing annotation costs. This study will be of interest to immunologists and computational biologists.
-
Reviewer #1 (Public Review):
In this manuscript, the authors described a computational method catELMo for embedding TCR CDR3 sequences into numeric vectors using a deep-learning-based approach, ELMo. The authors applied catELMo to two applications: supervised TCR-epitope binding affinity prediction and unsupervised epitope-specific TCR clustering. In both applications, the authors showed that catELMo generated significantly better binding prediction and clustering performance than other established TCR embedding methods.
The authors have addressed all of my concerns except for one as following:
(5) GIANA's result is like
– ## TIME:2020-12-14 14:45:14|cmd: GIANA4.py|COVID_test/rawData/hc10s10.txt|IsometricDistance_Thr=7.0|thr_v=3.7|thr_s=3.3|exact=True|Vgene=True|ST=3
– ## Column Info: CDR3 aa sequence, cluster id, other information in …
Reviewer #1 (Public Review):
In this manuscript, the authors described a computational method catELMo for embedding TCR CDR3 sequences into numeric vectors using a deep-learning-based approach, ELMo. The authors applied catELMo to two applications: supervised TCR-epitope binding affinity prediction and unsupervised epitope-specific TCR clustering. In both applications, the authors showed that catELMo generated significantly better binding prediction and clustering performance than other established TCR embedding methods.
The authors have addressed all of my concerns except for one as following:
(5) GIANA's result is like
– ## TIME:2020-12-14 14:45:14|cmd: GIANA4.py|COVID_test/rawData/hc10s10.txt|IsometricDistance_Thr=7.0|thr_v=3.7|thr_s=3.3|exact=True|Vgene=True|ST=3
– ## Column Info: CDR3 aa sequence, cluster id, other information in the input file
CAISDGTAASSTDTQYF 1 TRBV10-3*01 6.00384245917387e-05 0.930103216755186 COVID19:BS-EQ-0002-T1-replacement_TCRB.tsv
CAISDGTAASSTDTQYF 1 TRBV10-3*01 4.34559031223066e-05 0.918135389545364 COVID19:BS-EQ-0002-T2-replacement_TCRB.tsv
CANATLLQVLSTDTQYF 2 TRBV21-1*01 3.00192122958694e-05 0.878695260046097 COVID19:BS-EQ-0002-T1-replacement_TCRB.tsv
CANATLLQVLSTDTQYF 2 TRBV21-1*01 1.44853010407689e-05 0.768125375525736 COVID19:BS-EQ-0002-T2-replacement_TCRB.ts
...as in its example file at: https://raw.githubusercontent.com/s175573/GIANA/master/data/hc10s10--RotationEncodingBL62.txt
The results directly give the clustering results in the second column, and there is no direct distance metric for hierarchical clustering. Therefore, it is still not clear how the authors conducted the hierarchical clustering on GIANA's results. Did the hierarchical clustering apply to each of the original clusters on the CDR3 distances within the same original cluster?
-
Reviewer #2 (Public Review):
In the manuscript, the authors highlighted the importance of T-cell receptor (TCR) analysis and the lack of amino acid embedding methods specific to this domain. The authors proposed a novel bi-directional context-aware amino acid embedding method, catELMo, adapted from ELMo (Embeddings from Language Models), specifically designed for TCR analysis. The model is trained on TCR sequences from seven projects in the ImmunoSEQ database, instead of the generic protein sequences. They assessed the effectiveness of the proposed method in both TCR-epitope binding affinity prediction, a supervised task, and the unsupervised TCR clustering task. The results demonstrate significant performance improvements compared to existing embedding models. The authors also aimed to provide and discuss their observations on …
Reviewer #2 (Public Review):
In the manuscript, the authors highlighted the importance of T-cell receptor (TCR) analysis and the lack of amino acid embedding methods specific to this domain. The authors proposed a novel bi-directional context-aware amino acid embedding method, catELMo, adapted from ELMo (Embeddings from Language Models), specifically designed for TCR analysis. The model is trained on TCR sequences from seven projects in the ImmunoSEQ database, instead of the generic protein sequences. They assessed the effectiveness of the proposed method in both TCR-epitope binding affinity prediction, a supervised task, and the unsupervised TCR clustering task. The results demonstrate significant performance improvements compared to existing embedding models. The authors also aimed to provide and discuss their observations on embedding model design for TCR analysis: 1) Models specifically trained on TCR sequences have better performance than models trained on general protein sequences for the TCR-related tasks; and 2) The proposed ELMo-based method outperforms TCR embedding models with BERT-based architecture. The authors also provided a comprehensive introduction and investigation of existing amino acid embedding methods. Overall, the paper is well-written and well-organized.
The work has originality and has potential prospects for immune response analysis and immunotherapy exploration. TCR-epitope pair binding plays a significant role in T cell regulation. Accurate prediction and analysis of TCR sequences are crucial for comprehending the biological foundations of binding mechanisms and advancing immunotherapy approaches. The proposed embedding method presents an efficient context-aware mathematical representation for TCR sequences, enabling the capture and analysis of their structural and functional characteristics. This method serves as a valuable tool for various downstream analyses and is essential for a wide range of applications.
-
Reviewer #3 (Public Review):
In this study, Zhang and colleagues proposed an ELMo-based embedding model (catELMo) for TCRβ CDR3 amino acid sequences. They showed the effectiveness of catELMo in both supervised TCR binding prediction and unsupervised clustering, surpassing existing methods in accuracy and reducing annotation costs. The study provides insights on the effect of model architectures to TCR specificity prediction and clustering tasks.
The authors have addressed our prior critiques of the manuscript.
-
-
eLife assessment
This study provides a valuable tool for predicting binding between immune cells receptors and antigens based on protein sequence data. Its improvement over existing methods is supported by solid analysis, though more details on data, architectures and benchmarking are needed to fully justify this claim. This study will be of interest to immunologists and computational biologists.
-
Reviewer #1 (Public Review):
In this manuscript, the authors described a computational method catELMo for embedding TCR CDR3 sequences into numeric vectors using a deep-learning-based approach, ELMo. The authors applied catELMo to two applications: supervised TCR-epitope binding affinity prediction and unsupervised epitope-specific TCR clustering. In both applications, the authors showed that catELMo generated significantly better binding prediction and clustering performance than other established TCR embedding methods. However, there are a few major concerns that need to be addressed.
1. There are other TCR CDR3 embedding methods in addition to TCRBert. The authors may consider incorporating a few more methods in the evaluation, such as TESSA (PMCID: PMC7799492), DeepTCR (PMCID: PMC7952906) and the embedding method in ATM-TCR …
Reviewer #1 (Public Review):
In this manuscript, the authors described a computational method catELMo for embedding TCR CDR3 sequences into numeric vectors using a deep-learning-based approach, ELMo. The authors applied catELMo to two applications: supervised TCR-epitope binding affinity prediction and unsupervised epitope-specific TCR clustering. In both applications, the authors showed that catELMo generated significantly better binding prediction and clustering performance than other established TCR embedding methods. However, there are a few major concerns that need to be addressed.
1. There are other TCR CDR3 embedding methods in addition to TCRBert. The authors may consider incorporating a few more methods in the evaluation, such as TESSA (PMCID: PMC7799492), DeepTCR (PMCID: PMC7952906) and the embedding method in ATM-TCR (reference 10 in the manuscript). TESSA is also the embedding method in pMTnet, which is another TCR-epitope binding prediction method and is the reference 12 mentioned in this manuscript.
2. The TCR training data for catELMo is obtained from ImmunoSEQ platform, including SARS-CoV2, EBV, CMV, and other disease samples. Meanwhile, antigens related to these diseases and their associated TCRs are extensively annotated in databases VDJdb, IEDB and McPAS-TCR. The authors then utilized the curated TCR-epitope pairs from these databases to conduct the evaluations for eptitope binding prediction and TCR clustering. Therefore, the training data for TCR embedding may already be implicitly tuned for better representations of the TCRs used in the evaluations. This seems to be true based on Table 4, as BERT-Base-TCR outperformed TCRBert. Could catELMo be trained on PIRD as TCRBert to demonstrate catELMo's embedding for TCRs targeting unseen diseases/epitopes?
3. In the application of TCR-epitope binding prediction, the authors mentioned that the model for embedding epitope sequences was catElMo, but how about for other methods, such as TCRBert? Do the other methods also use catELMo-embedded epitope sequences as part of the binding prediction model, or use their own model to embed the epitope sequences? Since the manuscript focuses on TCR embedding, it would be nice for other methods to be evaluated on the same epitope embedding (maybe adjusted to the same embedded vector length). Furthermore, the authors found that catELMo requires less training data to achieve better performance. So one would think the other methods could not learn a reasonable epitope embedding with limited epitope data, and catELMo's better performance in binding prediction is mainly due to better epitope representation.
4. In the epitope binding prediction evaluation, the authors generated the test data using TCR-epitope pairs from VDJdb, IEDB, McPAS, which may be dominated by epitopes from CMV. Could the authors show accuracy categorized by epitope types, i.e. the accuracy for TCR-CMV pair and accuracy for TCR-SARs-CoV2 separately?
5. In the unsupervised TCR clustering evaluation, since GIANA and TCRdist direct outputs the clustering result, so they should not be affected by hierarchical clusters. Why did the curves of GIANA and TCRdist change in Figure 4 when relaxing the hierarchical clustering threshold?
6. In the unsupervised TCR clustering evaluation, the authors examined the TCR related to the top eight epitopes. However, there are much more epitopes curated in VDJdb, IEDB and McPAS-TCR. In real application, the potential epitopes is also more complex than just eight epitopes. Could the authors evaluate the clustering result using all the TCR data from the databases?
7. In addition to NMI, it is important to know how specific each TCR cluster is. Could the authors add the fraction of pure clusters in the results? Pure cluster means all the TCRs in the cluster are binding to the same epitope, and is a metric used in the method GIANA.
-
Reviewer #2 (Public Review):
In the manuscript, the authors highlighted the importance of T-cell receptor (TCR) analysis and the lack of amino acid embedding methods specific to this domain. The authors proposed a novel bi-directional context-aware amino acid embedding method, catELMo, adapted from ELMo (Embeddings from Language Models), specifically designed for TCR analysis. The model is trained on TCR sequences from seven projects in the ImmunoSEQ database, instead of the generic protein sequences. They assessed the effectiveness of the proposed method in both TCR-epitope binding affinity prediction, a supervised task, and the unsupervised TCR clustering task. The results demonstrate significant performance improvements compared to existing embedding models. The authors also aimed to provide and discuss their observations on …
Reviewer #2 (Public Review):
In the manuscript, the authors highlighted the importance of T-cell receptor (TCR) analysis and the lack of amino acid embedding methods specific to this domain. The authors proposed a novel bi-directional context-aware amino acid embedding method, catELMo, adapted from ELMo (Embeddings from Language Models), specifically designed for TCR analysis. The model is trained on TCR sequences from seven projects in the ImmunoSEQ database, instead of the generic protein sequences. They assessed the effectiveness of the proposed method in both TCR-epitope binding affinity prediction, a supervised task, and the unsupervised TCR clustering task. The results demonstrate significant performance improvements compared to existing embedding models. The authors also aimed to provide and discuss their observations on embedding model design for TCR analysis: 1) Models specifically trained on TCR sequences have better performance than models trained on general protein sequences for the TCR-related tasks; and 2) The proposed ELMo-based method outperforms TCR embedding models with BERT-based architecture. The authors also provided a comprehensive introduction and investigation of existing amino acid embedding methods. Overall, the paper is well-written and well-organized.
The work has originality and has potential prospects for immune response analysis and immunotherapy exploration. TCR-epitope pair binding plays a significant role in T cell regulation. Accurate prediction and analysis of TCR sequences are crucial for comprehending the biological foundations of binding mechanisms and advancing immunotherapy approaches. The proposed embedding method presents an efficient context-aware mathematical representation for TCR sequences, enabling the capture and analysis of their structural and functional characteristics. This method serves as a valuable tool for various downstream analyses and is essential for a wide range of applications.
-
Reviewer #3 (Public Review):
Here, the authors trained catElMo, a new context-aware embedding model for TCRβ CDR3 amino acid sequences for TCR-epitope specificity and clustering tasks. This method benchmarked existing work in protein and TCR language models and investigated the role that model architecture plays in the prediction performance. The major strength of this paper is comprehensively evaluating common model architectures used, which is useful for practitioners in the field. However, some key details were missing to assess whether the benchmarking study is a fair comparison between different architectures. Major comments are as follows:
- It is not clear why epitope sequences were also embedded using catELMo for the binding prediction task. Because catELMO is trained on TCRβ CDR3 sequences, it's not clear what benefit would …
Reviewer #3 (Public Review):
Here, the authors trained catElMo, a new context-aware embedding model for TCRβ CDR3 amino acid sequences for TCR-epitope specificity and clustering tasks. This method benchmarked existing work in protein and TCR language models and investigated the role that model architecture plays in the prediction performance. The major strength of this paper is comprehensively evaluating common model architectures used, which is useful for practitioners in the field. However, some key details were missing to assess whether the benchmarking study is a fair comparison between different architectures. Major comments are as follows:
- It is not clear why epitope sequences were also embedded using catELMo for the binding prediction task. Because catELMO is trained on TCRβ CDR3 sequences, it's not clear what benefit would come from this embedding. Were the other embedding models under comparison also applied to both the TCR and epitope sequences? It may be a fairer comparison if a single method is used to encode epitope sequence for all models under comparison, so that the performance reflects the quality of the TCR embedding only.
- The tSNE visualization in Figure 3 is helpful. It makes sense that the last hidden layer features separate well by binding labels for the better performing models. However, it would be useful to know if positive and negative TCRs for each epitope group also separate well in the original TCR embedding space. In other words, how much separation between these groups is due to the neural network vs just the embedding?
- To generate negative samples, the author randomly paired TCRs from healthy subjects to different epitopes. This could produce issues with false negatives if the epitopes used are common. Is there an estimate for how frequently there might be false negatives for those commonly occurring epitopes that most populations might also have been exposed to? Could there be a potential batch effect for the negative sampled TCR that confounds with the performance evaluation?
- Most of the models being compared were trained on general proteins rather than TCR sequences. This makes their comparison to catELMO questionable since it's not clear if the improvement is due to the training data or architecture. The authors partially addressed this with BERT-based models in section 2.4. This concern would be more fully addressed if the authors also trained the Doc2vec model (Yang et al, Figure 2) on TCR sequences as baseline models instead of using the original models trained on general protein sequences. This would make clear the strength of context-aware embeddings if the performance is worse than catElmo and BERT. -
