Cystatin F (Cst7) drives sex-dependent changes in microglia in an amyloid-driven model of Alzheimer’s disease
Curation statements for this article:-
Curated by eLife

eLife assessment
This study presents a valuable finding on the function of the gene Cst7 in sex-divergent pathological changes in microglia in a mouse model of AB-driven Alzheimer's disease. The evidence supporting the claims of the authors is solid, although the study would be strengthened by validation of some of the key differentially expressed genes identified in RNA-sequencing experiments, and the inclusion of key controls and additional timepoints to address whether Cst7 drives disease progression or is simply upregulated as a result. The work will be of interest to neuroimmunologists and neuroscientists working on microglia and neurodegenerative disease.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Microglial endolysosomal (dys)function is strongly implicated in neurodegenerative disease. Transcriptomic studies show that a microglial state characterised by a set of genes involved in endolysosomal function is induced in both mouse Alzheimer’s disease (AD) models and human AD brain, and that the emergence of this state is emphasised in females. Cst7 (encoding cystatin F) is among the most highly upregulated genes in these microglia. However, despite such striking and robust upregulation, the function of Cst7 in neurodegenerative disease is not understood. Here, we crossed Cst7 -/- mice with the App NL-G-F mouse to test the role of Cst7 in a model of amyloid-driven AD. Surprisingly, we found that Cst7 plays a sexually dimorphic role regulating microglia in this model. In females, Cst7 -/- App NL-G-F microglia had greater endolysosomal gene expression, lysosomal burden, and amyloid beta (Aβ) burden in vivo and were more phagocytic in vitro . However, in males, Cst7 -/- App NL-G-F microglia were less inflammatory and had a reduction in lysosomal burden but had no change in Aβ burden. Overall, our study reveals functional roles for one of the most commonly upregulated genes in microglia across disease models, and the sex-specific profiles of Cst7 -/- -altered microglial disease phenotypes. More broadly, the findings raise important implications for AD including crucial questions on sexual dimorphism in neurodegenerative disease and the interplay between endolysosomal and inflammatory pathways in AD pathology.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
This paper evaluates the effect of knocking out CST7(Cystatin 5) on the APPNL-G-F Alzheimer's disease mouse model. They found sexually dimorphic outcomes, with differential transcriptional responses, increased phagocytosis (but interestingly a higher plaque burden) in females and suppressed inflammatory microglial activation in males (but interestingly no change in plaque burden). This study offers new insight into the functional role of CST7 that is upregulated in a subset of disease- associated microglia in AD models and human brain. Despite the discovery of disease-associated microglia several years ago, there has been little effort in understanding the function of the different genes that make up this profile, making this paper especially timely. Overall, the experiments are …
Author Response
Reviewer #1 (Public Review):
This paper evaluates the effect of knocking out CST7(Cystatin 5) on the APPNL-G-F Alzheimer's disease mouse model. They found sexually dimorphic outcomes, with differential transcriptional responses, increased phagocytosis (but interestingly a higher plaque burden) in females and suppressed inflammatory microglial activation in males (but interestingly no change in plaque burden). This study offers new insight into the functional role of CST7 that is upregulated in a subset of disease- associated microglia in AD models and human brain. Despite the discovery of disease-associated microglia several years ago, there has been little effort in understanding the function of the different genes that make up this profile, making this paper especially timely. Overall, the experiments are well-controlled and the data support the main conclusions and the manuscript could be strengthened by addressing the below comments and clarifying questions that could impact the interpretation of their data/ findings.
- In the first section discussing CST7 expression levels in AD models, it would be good to involve a discussion of levels of CST7 change in human AD samples. There are sufficient available datasets to look at this, and it would help us understand how comparable the animal models are to human patients. For example, while in mice CST7 is highly enriched in microglia/macrophages, in human datasets it seems like it is not quite so specific to microglia - it is equally expressed in endothelial cells. This might have a significant impact on the interpretation of the data, and it would be good to introduce and assess the findings in mice through the human subjects lens. There is a discussion of the human data in the discussion section, but it would be more appropriately assessed in the same way as the mouse data and comparatively presented in the results section. The authors could also include the data from Gerrits et al. 2021 in their first figure.
We agree with the reviewer on the importance of considering the work in the context of human disease. While CST7 is not as strongly upregulated in human AD brain as it is in mouse expression is observed predominantly in myeloid cells in the brain with very minimal expression detected in endothelial cells (see screenshots in Author response image 1 from Brain Myeloid Landscape platform (http://research-pub.gene.com/BrainMyeloidLandscape/BrainMyeloidLandscape2/) and is enriched in AD clusters vs homeostatic in scRNASeq studies (Gerrits et al., 2021). We attempted immunostaining for human CF (CST7) in AD brains to assess expression and co-localisation with microglial markers but failed to validate any of the antibodies tested. Additionally, King et al., 2023 (PMID: 36547260) recently showed increase in CST7 expression in bulk hippocampal RNASeq in AD vs mid-life controls suggesting an ageing/AD mechanism. CST7 has also been shown to be expressed following overexpression of TREM2 in human microglia in vitro and that siRNA-mediated knockdown of expression leads to an increase in phagocytosis (Popescu et al., 2023 - PMID: 36480007), mirroring our data and suggesting a conserved role in human cells. Overall, we believe that, even in the context of mouse models, the understanding of the function of genes upregulated in disease is of importance to the field and that this study paves the way for further work investigating human CST7 in disease. We have added this (with citations to the datasets mentioned) to the discussion (highlighted).
Author response image 1

- The differential RNAseq data is perhaps one of the most striking results of this paper; however it is difficult to see exactly how similar the male v female APPNL-G-F profiles are, in addition to the genes shared or not between the KO condition. Venn diagrams, in addition to statistical tests, would enhance this part of the paper and add more clarity.
We have added Venn diagrams to show DEGs between male and female AppNL-G-F microglia vs WT control to show how similar the male v female APPNL-G-F profiles are. Additionally, to exemplify the Cst7KO-Sex interaction, a Venn showing DEGs between male and female AppNL-G-F microglia vs. AppNL-G-FCst7-/- microglia (Fig. 2 – Fig. supplement 3). We confirm we have derived all differential gene expression changes reported (including those represented in the Venn diagrams) using appropriate Padj statistical approaches (see Methods).
- A major argument in the paper is a continuation of Sala-Frigerio 2019 which says that the female phenotype is an acceleration of the male phenotype. Does this mean that if males were assessed at later timepoints, they would be more similar to the females? Or are there intrinsic differences that never resolve? It would be helpful to see a later timepoint for males to get at the difference between these two options
This is an interesting question and while we acknowledge that empirically addressing with a later timepoint could add insight, we believe it would actually need multiple closely-spaced timepoints as choosing what single later timepoint would be optimal is difficult to judge (and likely not possible at all) for reasons below. We also believe data already published combined with our observations show it is most-likely a cell-intrinsic effect that explains our sex-specific differences.
First, we emphasize the acceleration of the microglial phenotype in female AppNL-G-F mice previously published is fairly subtle and relative rather than absolute e.g. the DAM/ARM microglia state represents ~50% of all microglia in male and ~55% of all microglia in females at 12 months old therefore both sexes have similarly abundant microglia in the state that most highly express Cst7. Indeed, after the age at which DAM/ARM state microglia appear in appreciable numbers (~ 6 months), both females and males both have an abundance of them. It is important to note that a 12-month male is far more “progressed” than a 6-month female hence the stepped age effect is temporally short.
Second, Cst7 deletion in the AppNL-G-F mice condition caused qualitative differences affecting distinct genes and/or overlapping genes moving in different directions between female and male mice - if a stepped age effect explained sex differences from Cst7 deletion, given that it could only be stepped by a very short timeframe (several weeks maximum) from reasoning above, we would expect to see similar qualitative changes but of different magnitude in female and male mice arising from Cst7 deletion; this is not the pattern we see.
Third, beyond 12 months old, regression from ARM/DAM actually occurs, again making it unlikely males would “catch up” with females to show the same profile from Cst7 deletion but just at an older age – practically, this also complicates choosing a single later timepoint (and age-related systemic morbidity emerges as a potential confounder as well).
In summary, while the acceleration of the DAM signature in female microglia offers an intriguing possible explanation to our observation of sexual dimorphism in response to deletion of one of the key genes in this signature, we believe it more likely that intrinsic effects are responsible for the Cst7 deletion sex-related impact. Taking the alternative perspective, even if a stepped age effect in the underlying progression of the model could explain our findings, this would need multiple timepoints with short gaps between (e.g. monthly at 12, 13, 14, 15 months old) to provide the temporal resolution to expose this pattern; we would not have the resources to conduct such a resource-intensive and lengthy study. We hope this reasoning appears logical and conscious of the importance to convey this in our manuscript we have revised the Discussion to as concisely as possible capture some key points outlined above.
- If the central argument is that CST7 in females decreases phagocytosis and in males increases microglia activation, are there changes in amyloid plaque burden or structure in the APPNL-G-F /CST 7 KO mice compared to APPNL-G-F/CST7 WT that reflect these changes? Please address. If not, how does this affect the functional interpretation of differential expression observed in phagocytic/reactive microglia genes? Pieces of this are discussed but it could be clearer.
We emphasise the data already presented in Fig 6 and Fig. 6 – Fig. Supplement 2 showing altered Aβ burden (6E10 staining) and plaque count (MeX04) but no change in plaque area. Regarding the functional interpretation of Cst7-dependent gene changes in microglia beyond the endolysosomal function we present in figures 3-5, we have included additional data using simple immunohistochemistry, as suggested by the reviewer, to assess synapse abundance. We show loss of Sy38 coverage around plaques (Fig. 6I) and a moderate but significant decrease in coverage between AppNL-G-F/Cst7-/- vs AppNL-G-F brains only in females (Fig. 6J). This reflects the effect observed with plaque coverage whereby we observe increased burden in AppNL-G-F/Cst7-/- vs AppNL-G-F females but not males (Fig. 6B-F) suggesting the increased plaque burden in Cst7-/- female mice may lead to increased synapse loss. We would also emphasise that altered expression of phagolysosomal genes could affect disease in ways beyond interactions with amyloid and synapses.
- It is confusing that increased phagocytosis in the APPNL-G-F/CST7 KO females leads to greater plaque burden, considering proteolysis is not affected. What might explain this observation? Additionally, it is interesting that suppression of microglial activation doesn't lead to an increase in plaques in the male APPNL-G-F/CST7 KO mice. How does the profile of phagocytic microglia in the male APPNL-G-F/CST7 KO mice differ from the APPNL-G-F males?
We emphasize our comments on this topic in the discussion where we speculate that the greater plaque burden in females is linked to increased uptake of Aβ (which we observe in Fig. 4B&C) and deposition into plaques as suggested by Huang et al., 2021 (PMID: 33859405), d’Errico et al., 2022 (PMID: 34811521) and Shabestari et al., 2022 (PMID: 35705056). Regarding the lack of effect in males despite the suppression of inflammatory genes, we agree this is a curious observation, although may point to as yet ill-defined mechanisms for how inflammatory pathways influence plaque pathology. Unfortunately, we were not able to specifically compare the profile of phagocytic microglia in AppNL-G-F vs AppNL-G-FCst7-/- as we did not perform single-cell RNASeq. However, our bulk RNASeq profiling suggests modest downregulation of phagocytic/endolysosomal genes (eg Lilrb4a, Fig. 2I) and reduced expression of LAMP2 in microglia by immunostaining. We have added further comment on this in the discussion.
- Seems that the authors have potentially discovered an unusual mechanism for how CST7 could regulate cell autonomous function without impacting its canonical protease target. The authors deal with this extensively in the discussion but an ELISA or ICC to localize CST7 to microglia in vitro or in vitro would help address this point.
We have added FISH data localising Cst7 expression to IBA1+ cells specifically around plaques in App brains (Fig. 1B-E). We agree that assessing the subcellular localisation and any non-microglial expression of Cystatin-F (the protein coded by Cst7) would offer valuable insight into the protease target and may reveal details on the precise mechanism by which CF deletion leads the phenotype we observe in this study. However, despite attempting numerous commercially available and gifted antibodies to detect CF we were unable to validate (using Cst7-/- as controls) any methods other than FISH.
- The authors focus on plaques in their final figure, however dysregulated microglial phagocytosis could impact many other aspects of brain health. Simple immunohistochemistry for synapses and myelin/oligodendrocytes (especially given the results of the in vitro phagocytosis assay) could provide more insight here.
We fully agree with the reviewer. As also outlined in our responses elsewhere, phagocytic changes could have multiple consequences, and we have included additional data using immunohistochemistry as advised for synapses in WT, AppNL-G-F, and AppNL-G-F/Cst7-/- brains. We show loss of Sy38 coverage around plaques (Fig. 6I) and a moderate but significant decrease in coverage between AppNL-G-F/Cst7-/- vs AppNL-G-F brains only in females (Fig. 6J). This reflects the effect observed with plaque coverage whereby we observe increased burden in AppNL-G-F/Cst7-/- vs AppNL-G-F females but not males (Fig. 6B-F) suggesting the increased plaque burden in Cst7-/- female mice may lead to increased synapse loss.
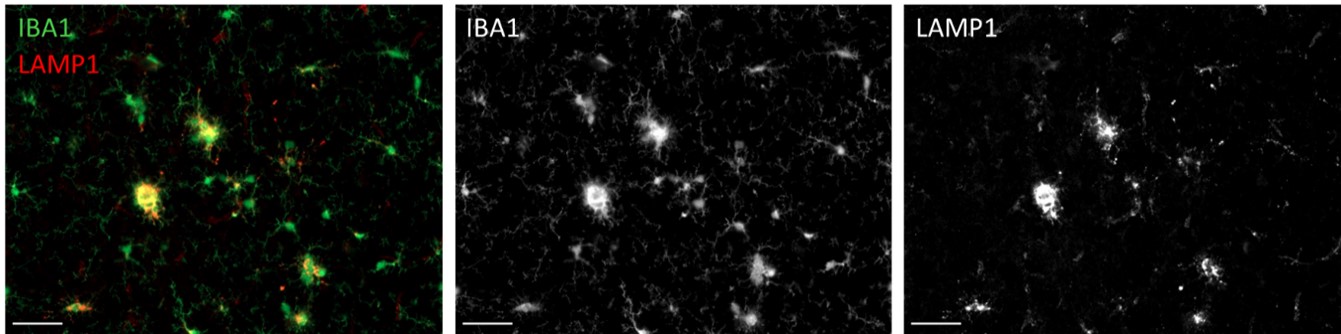
We also performed immunohistochemistry for myelin makers MAG and MBP but found no plaque-associated pathology. Finally, we searched for dystrophic neurites using LAMP1 but found that the antibody stained microglial lysosomes rather than dystrophic neurites in this model (see Author response image 2), an observation that has been made by others (Sharoar et al., 2021 - PMID: 34215298).
Overall, our data suggest Cst7 may play a protective role in females, limiting phagocytosis, reducing plaque burden and blunting synapse loss.
Author response image 2.
Reviewer #3 (Public Review):
In this manuscript, Daniels et al explored the role of Cystatin F in an A-driven mouse model of Alzheimer's disease. By crossing a constitutive knockout mouse lacking the gene that encodes Cystatin F, Cst7, to the AppNL-G-F mouse line, the authors describe impairments in microglial gene expression and phagocytic function that emerge more prominently in females versus males lacking Cst7. A strength of the study is its focus: given mounting evidence that microglia are a hub of neurological dysfunction with particular potential to trigger or exacerbate neurodegenerative disorders, it is essential to determine the changes in microglia that occur pathologically to promote disease progression. Similarly, the wide-spread identification of the gene in question, Cst7, as upregulated in AD models makes this gene a good target for mechanistic studies.
The paper in its current form also has several weaknesses which limit the insights derived, weaknesses that are largely related to the experimental tools and approaches chosen by the authors to test their hypotheses. For example, the paper begins with a figure replotting data from previous studies showing that Cst7 is upregulated in mouse models of Alzheimer's disease. Though relevant to the current study, there are no new insights provided here. Next, the authors perform bulk RNA-sequencing on microglia isolated from male and female mice in the Cst7-/-; AppNL-G-F mouse line. In the methods, it is unclear whether the authors took precautions to preserve the endogenous transcriptional state of these cells given evidence that microglia can acquire a DAM-like signature simply due to the process of dissociation (Marsh et al, Nature Neuroscience, 2022). If the authors did not control for this, their results may not support the conclusions they draw from the data. Relatedly, it appears the authors pooled all microglia together here, instead of just isolating DAMs specifically or analyzing microglia at single-cell resolution, which could reveal the heterogeneous nature of the role of Cst7 in microglia. In addition to losing information about heterogeneity, another concern is that they could be diluting out the major effects of the model on microglial function by including all microglia. Overall, the biggest issue I have with the RNA-sequencing data is the lack of validation of the gene expression changes identified using a different method that does not require dissociation, like immunohistochemistry or fluorescence in situ hybridization. Especially given the limited number of genes they found to be mis-regulated (see Fig. 2 E and G), I worry that these changes might simply be noise, especially since the authors provide no further evidence of their mis-regulation. Without further validation, the data presented are not sufficient to support the authors' claims.
We believe we have addressed this comment in the “Essential Revisions (for the authors)” section above. Please see again below:
We took standard precautions to minimise the risk of aberrant ex vivo cell activation, including maintaining cells on ice during non-enzyme steps of the procedure and carrying out preps in small batches to minimise time taken from removal of brain to purification of microglial RNA. Importantly, we also validated key expression data by in situ methods such as RNA FISH for Cst7 and Lilrb4a (Fig. 1B-E, Fig 2. - Fig. supplement 3) thus eliminating dissection-induced effects. Additionally, when performing qPCR on microglia from non-disease mice to test the disease-specific role of Cst7-dependent gene regulation we did not observe the same gene changes (Fig 2. - Fig. supplement 4) which, if such changes were dependent on tissue dissociation, we would expect to observe in WT or disease animals. We utilised the resources provided by Marsh et al. 2022 to search for overlap between enzyme-induced genes and our DEG lists from our key comparisons. We found the enzyme-induced gene set had very minimal overlap with any of our comparisons with overlap of only 4 genes between enzyme-induced genes and Cst7-dependent genes in males and no overlap between enzyme-induced genes and Cst7-dependent genes in females. We would further point out that the disease-induced microglial RNAseq profile in the AppNL-G-F Cst7+/+ (i.e. disease WT) condition mirrors those observed previously by multiple methods including in situ profiling (Zeng et al 2023 - PMID: 36732642) and RiboTag approaches (Kang et al 2018 - PMID: 30082275). We believe these combined approaches provide convincing validation of the RNAseq data.
In assessing the changes in microglial function and A pathology that occur in males and females of the Cst7-/-; AppNL-G-F line, the authors identify some differences between how females and males are affected by the loss of Cst7. While the statistical analyses the authors perform as given in the figure legends appear to be correct, the plots do not show significant changes between males and females for a given parameter. Take for example Figure 3H. Loss of Cst7 decreases IBA+Lamp+ microglia in males but increases this parameter in females. However, it does not appear that there is a significant difference in IBA+Lamp+ microglia in male versus female mice lacking Cst7. If there is no absolute difference between males and females, can the differential effects of Cst7 knockout on the sexes really be so relevant to the sexual dimorphism observed in the disease? I question this connection, but perhaps a greater discussion of what the result might mean by the authors would be helpful for placing this into context.
We understand the reviewer’s perspective and we agree that the interpretations could be presented and explained better in the text - we have updated the discussion as suggested to address this.
We designed our study initially to search for sex-specific effects of Cst7. Therefore, whilst our ANOVA does include main effects analysis for disease or sex, we carried out post-hoc analysis primarily to investigate effects of Cst7 deletion within sex. In the case of Fig. 3H pointed out by the reviewer, we observe a main effect for disease in the ANOVA and for disease-sex interaction but not for sex. Post-hoc analysis revealed the sex-specific effects of Cst7 we describe in the manuscript. This approach on analysis was also taken by Hoghooghi et al. (2020 - PMID: 33027652) who show related pathway gene Cstc is detrimental in EAE in females but not males (included in the discussion in this manuscript). The observation in Fig. 3H that there appears to be a Cst7 effect in males and females but not a sex effect in Cst7-/- is accurate but a relative anomaly in this study. Generally, we find that, alongside Cst7 deletion affecting females differently to males, we also see a sex effect in Cst7-/- animals but not in Cst7+/+ animals i.e. absolute levels in disease condition as well as relative changes from control to disease condition are different between males and females. This is exemplified in Fig. 4B&C where we observe increased microglial Aβ in female Cst7-/- animals vs male Cst7-/- animals and in Fig. 6D where we observe increased Aβ plaque burden in female Cst7-/- animals vs male Cst7-/- animals. This is most strikingly demonstrated in the case of our RNASeq data where we observe a difference in sex-dependent genes in AppNL-G-F vs AppNL-G-F/Cst7-/- (Fig. 2 – Fig. supplement 3B) implying removal of the Cst7 gene led to an ‘unlocking’ of sexual dimorphism in our cohort which we comment on in the discussion.
Finally, the use of in vitro assays of microglial function can be helpful as secondary analyses when coupled with in vivo or ex vivo approaches, but are not on their own sufficient to support the authors' conclusions. Quantitative engulfment assays (see Schafer et al, Neuron, 2012) on brain tissue showing that male and female microglia lacking Cst7 engulf different amounts of material (e.g. plaques, synapses, myelin) in the intact brain would be more convincing.
We agree that in vitro assays for microglial function are not always sufficient as standalone methods to support conclusions on functions in disease. The reviewer may have missed our in vivo MeX04 uptake assays (Fig 4A-D) which use measurements by flow cytometry on isolated microglia, this is a reflection of the microglial uptake in vivo following MeX04 injection pre-mortem – this experiment showed increased microglial Aβ in female Cst7-/- animals vs male Cst7-/- animals (Fig. 4B&C). Our in vitro assays complement and extend insight in ways not possible in vivo, for example they offer key insight into uptake/degradation kinetics that would be extremely challenging to carry out in vivo.
In general, a major limitation to the insights that can be derived in the study is the decision of the authors to perform all experiments at a single late-stage time point of 12 months of age. As this is quite far into disease progression for many AD models, phenotypic changes identified by the authors could arise due to the downstream effects of plaque deposition and therefore may not implicate Cst7 as a mechanism driving neurodegeneration rather than one of many inflammatory changes that accompany AD mouse models nearing the one-year time point. A related problem is that the study uses a constitutive KO mouse that has lacked Cst7 expression throughout life, not just during disease processes that increase with aging. In summary, the topic of the article is important and timely, but the connection between the data and the authors' conclusions is not as strong as it could be.
As described above, Cst7 expression is absent at steady-state and low until 6-12 months. Therefore, we predict that deletion would have little effect until 12+ months whereby cells expressing Cst7 have had the temporal window to affect disease pathology, as we find in the current study. This was a key part of the reasoning in our choice of the 12-month age for analyses. The negligible expression of Cst7 at baseline/early stages of disease suggests constitutive KO of the gene will not impact the phenotype until disease onset. This is substantiated by the lack of any genotype-related differences in the WT vs Cst7-/- comparisons in the non-disease condition.
-
eLife assessment
This study presents a valuable finding on the function of the gene Cst7 in sex-divergent pathological changes in microglia in a mouse model of AB-driven Alzheimer's disease. The evidence supporting the claims of the authors is solid, although the study would be strengthened by validation of some of the key differentially expressed genes identified in RNA-sequencing experiments, and the inclusion of key controls and additional timepoints to address whether Cst7 drives disease progression or is simply upregulated as a result. The work will be of interest to neuroimmunologists and neuroscientists working on microglia and neurodegenerative disease.
-
Reviewer #1 (Public Review):
This paper evaluates the effect of knocking out CST7(Cystatin 5) on the APPNL-G-F Alzheimer's disease mouse model. They found sexually dimorphic outcomes, with differential transcriptional responses, increased phagocytosis (but interestingly a higher plaque burden) in females and suppressed inflammatory microglial activation in males (but interestingly no change in plaque burden). This study offers new insight into the functional role of CST7 that is upregulated in a subset of disease- associated microglia in AD models and human brain. Despite the discovery of disease-associated microglia several years ago, there has been little effort in understanding the function of the different genes that make up this profile, making this paper especially timely. Overall, the experiments are well-controlled and the data …
Reviewer #1 (Public Review):
This paper evaluates the effect of knocking out CST7(Cystatin 5) on the APPNL-G-F Alzheimer's disease mouse model. They found sexually dimorphic outcomes, with differential transcriptional responses, increased phagocytosis (but interestingly a higher plaque burden) in females and suppressed inflammatory microglial activation in males (but interestingly no change in plaque burden). This study offers new insight into the functional role of CST7 that is upregulated in a subset of disease- associated microglia in AD models and human brain. Despite the discovery of disease-associated microglia several years ago, there has been little effort in understanding the function of the different genes that make up this profile, making this paper especially timely. Overall, the experiments are well-controlled and the data support the main conclusions and the manuscript could be strengthened by addressing the below comments and clarifying questions that could impact the interpretation of their data/ findings.
1. In the first section discussing CST7 expression levels in AD models, it would be good to involve a discussion of levels of CST7 change in human AD samples. There are sufficient available datasets to look at this, and it would help us understand how comparable the animal models are to human patients. For example, while in mice CST7 is highly enriched in microglia/macrophages, in human datasets it seems like it is not quite so specific to microglia - it is equally expressed in endothelial cells. This might have a significant impact on the interpretation of the data, and it would be good to introduce and assess the findings in mice through the human subjects lens. There is a discussion of the human data in the discussion section, but it would be more appropriately assessed in the same way as the mouse data and comparatively presented in the results section. The authors could also include the data from Gerrits et al. 2021 in their first figure.
2. The differential RNAseq data is perhaps one of the most striking results of this paper; however it is difficult to see exactly how similar the male v female APPNL-G-F profiles are, in addition to the genes shared or not between the KO condition. Venn diagrams, in addition to statistical tests, would enhance this part of the paper and add more clarity.
3. A major argument in the paper is a continuation of Sala-Frigiero 2019 which says that the female phenotype is an acceleration of the male phenotype. Does this mean that if males were assessed at later timepoints, they would be more similar to the females? Or are there intrinsic differences that never resolve? It would be helpful to see a later timepoint for males to get at the difference between these two options
4. If the central argument is that CST7 in females decreases phagocytosis and in males increases microglia activation, are there changes in amyloid plaque burden or structure in the APPNL-G-F /CST 7 KO mice compared to APPNL-G-F/CST7 WT that reflect these changes? Please address. If not, how does this affect the functional interpretation of differential expression observed in phagocytic/reactive microglia genes? Pieces of this are discussed but it could be clearer
5. It is confusing that increased phagocytosis in the APPNL-G-F/CST7 KO females leads to greater plaque burden, considering proteolysis is not affected. What might explain this observation? Additionally, it is interesting that suppression of microglial activation doesn't lead to an increase in plaques in the male APPNL-G-F/CST7 KO mice. How does the profile of phagocytic microglia in the male APPNL-G-F/CST7 KO mice differ from the APPNL-G-F males?
6. Seems that the authors have potentially discovered an unusual mechanism for how CST7 could regulate cell autonomous function without impacting its canonical protease target. The authors deal with this extensively in the discussion but an ELISA or ICC to localize CST7 to microglia in vitro or in vitro would help address this point.
7. The authors focus on plaques in their final figure, however dysregulated microglial phagocytosis could impact many other aspects of brain health. Simple immunohistochemistry for synapses and myelin/oligodendrocytes (especially given the results of the in vitro phagocytosis assay) could provide more insight here. -
Reviewer #2 (Public Review):
In this article, Daniels et al evaluate the function of Cst7, a gene previously shown to be strongly expressed when microglia respond to Alzheimer's-like pathology. The reported findings include evidence for a sexually dimorphic role of Cst7 in microglia, including differences in lysosomal activity and ability to phagocytose. Some questions remain as to how many of these effects are 1) disease-independent, 2) age-dependent, and 3) ultimately affecting cognition
Strengths:
-The approach taken here is sound, knocking out Cst7 in an animal model of Alzheimer's-like pathology, and analysing a range of variables associated with the pathology.
-The authors have made good use of existing datasets, evidencing the advantages of data sharing and open data mining.
-Data reporting is also excellent, as we can see the …Reviewer #2 (Public Review):
In this article, Daniels et al evaluate the function of Cst7, a gene previously shown to be strongly expressed when microglia respond to Alzheimer's-like pathology. The reported findings include evidence for a sexually dimorphic role of Cst7 in microglia, including differences in lysosomal activity and ability to phagocytose. Some questions remain as to how many of these effects are 1) disease-independent, 2) age-dependent, and 3) ultimately affecting cognition
Strengths:
-The approach taken here is sound, knocking out Cst7 in an animal model of Alzheimer's-like pathology, and analysing a range of variables associated with the pathology.
-The authors have made good use of existing datasets, evidencing the advantages of data sharing and open data mining.
-Data reporting is also excellent, as we can see the individual data points, and also observe how optimal group numbers were used. This adds solidity to the study.
-The results are very well connected, with experiments focusing on the in vivo and in vitro lysosomal/phagocytic function
-Exploring the effect of sex, as an independent variable, is a refreshing approach and clearly an important one by looking at the findings reported here.Weaknesses:
-The basis for the hypothesis of Cst7 displaying sexual dymorphism is not as strong as indicated by the text. Data presented in Figure 1 supports 1/2 models have statistically significant differences in expression of Cst7 between males and females.
-As presented, it is hard to disentangle the differential impact of sex, in isolation, compared to the accelerated pathology/ageing observed in females. In other words, Cst7 could be playing a differential role in females not because that particular gene has sexually dimorphic roles, but because female microglia are generally more advanced in their phenotype and prone to Cst7-dependent effects that their younger counterparts (or male microglia) would not suffer. We also lack context when it comes to baseline effects of Cst7-/- compared to disease-related effects, since a crucial control (non-AD Cst7-/-) is missing from analyses, key in Figure 2 for example.
-It is unclear how the knockout of Cst7 would selectively affect microglia. The expression of Cst7 is definitely very high in microglia in AD, but it's less clear whether other cells express this gene as well. If so, the effects of Cst7-/- could be microglia-independent in part.
-Considering the large number of mice used in these studies, and the effort that very likely went into these, it is disappointing that we do not have any measure of cognition or any other behavioural task associated with the molecular data. Ultimately, changes in amyloid, for example, could or could not correlate with real pathology in APP models. -
Reviewer #3 (Public Review):
In this manuscript, Daniels et al explored the role of Cystatin F in an A-driven mouse model of Alzheimer's disease. By crossing a constitutive knockout mouse lacking the gene that encodes Cystatin F, Cst7, to the AppNL-G-F mouse line, the authors describe impairments in microglial gene expression and phagocytic function that emerge more prominently in females versus males lacking Cst7. A strength of the study is its focus: given mounting evidence that microglia are a hub of neurological dysfunction with particular potential to trigger or exacerbate neurodegenerative disorders, it is essential to determine the changes in microglia that occur pathologically to promote disease progression. Similarly, the wide-spread identification of the gene in question, Cst7, as upregulated in AD models makes this gene a …
Reviewer #3 (Public Review):
In this manuscript, Daniels et al explored the role of Cystatin F in an A-driven mouse model of Alzheimer's disease. By crossing a constitutive knockout mouse lacking the gene that encodes Cystatin F, Cst7, to the AppNL-G-F mouse line, the authors describe impairments in microglial gene expression and phagocytic function that emerge more prominently in females versus males lacking Cst7. A strength of the study is its focus: given mounting evidence that microglia are a hub of neurological dysfunction with particular potential to trigger or exacerbate neurodegenerative disorders, it is essential to determine the changes in microglia that occur pathologically to promote disease progression. Similarly, the wide-spread identification of the gene in question, Cst7, as upregulated in AD models makes this gene a good target for mechanistic studies.
The paper in its current form also has several weaknesses which limit the insights derived, weaknesses that are largely related to the experimental tools and approaches chosen by the authors to test their hypotheses. For example, the paper begins with a figure replotting data from previous studies showing that Cst7 is upregulated in mouse models of Alzheimer's disease. Though relevant to the current study, there are no new insights provided here. Next, the authors perform bulk RNA-sequencing on microglia isolated from male and female mice in the Cst7-/-; AppNL-G-F mouse line. In the methods, it is unclear whether the authors took precautions to preserve the endogenous transcriptional state of these cells given evidence that microglia can acquire a DAM-like signature simply due to the process of dissociation (Marsh et al, Nature Neuroscience, 2022). If the authors did not control for this, their results may not support the conclusions they draw from the data. Relatedly, it appears the authors pooled all microglia together here, instead of just isolating DAMs specifically or analyzing microglia at single-cell resolution, which could reveal the heterogeneous nature of the role of Cst7 in microglia. In addition to losing information about heterogeneity, another concern is that they could be diluting out the major effects of the model on microglial function by including all microglia. Overall, the biggest issue I have with the RNA-sequencing data is the lack of validation of the gene expression changes identified using a different method that does not require dissociation, like immunohistochemistry or fluorescence in situ hybridization. Especially given the limited number of genes they found to be mis-regulated (see Fig. 2 E and G), I worry that these changes might simply be noise, especially since the authors provide no further evidence of their mis-regulation. Without further validation, the data presented are not sufficient to support the authors' claims.
In assessing the changes in microglial function and A pathology that occur in males and females of the Cst7-/-; AppNL-G-F line, the authors identify some differences between how females and males are affected by the loss of Cst7. While the statistical analyses the authors perform as given in the figure legends appear to be correct, the plots do not show significant changes between males and females for a given parameter. Take for example Figure 3H. Loss of Cst7 decreases IBA+Lamp+ microglia in males but increases this parameter in females. However, it does not appear that there is a significant difference in IBA+Lamp+ microglia in male versus female mice lacking Cst7. If there is no absolute difference between males and females, can the differential effects of Cst7 knockout on the sexes really be so relevant to the sexual dimorphism observed in the disease? I question this connection, but perhaps a greater discussion of what the result might mean by the authors would be helpful for placing this into context.
Finally, the use of in vitro assays of microglial function can be helpful as secondary analyses when coupled with in vivo or ex vivo approaches, but are not on their own sufficient to support the authors' conclusions. Quantitative engulfment assays (see Schafer et al, Neuron, 2012) on brain tissue showing that male and female microglia lacking Cst7 engulf different amounts of material (e.g. plaques, synapses, myelin) in the intact brain would be more convincing.
In general, a major limitation to the insights that can be derived in the study is the decision of the authors to perform all experiments at a single late-stage time point of 12 months of age. As this is quite far into disease progression for many AD models, phenotypic changes identified by the authors could arise due to the downstream effects of plaque deposition and therefore may not implicate Cst7 as a mechanism driving neurodegeneration rather than one of many inflammatory changes that accompany AD mouse models nearing the one-year time point. A related problem is that the study uses a constitutive KO mouse that has lacked Cst7 expression throughout life, not just during disease processes that increase with aging. In summary, the topic of the article is important and timely, but the connection between the data and the authors' conclusions is not as strong as it could be.
-
