Phosphate starvation signaling increases mitochondrial membrane potential through respiration-independent mechanisms
Curation statements for this article:-
Curated by eLife

eLife assessment
This study presents important findings on the regulation of the phosphate export cycle and identify the phosphatase Sit4 as a crucial player in regulation of the inner membrane potential of mitochondria. Whereas some of the data are convincing, the analyses will profit from deeper insights concerning metabolism alterations (carbon sources, amino acids). The major strength however is a new insight on how the cells use alternative ways for maintaining a critical mitochondrial inner membrane potential, and therefore this study is interesting to the broad audience with interests spanning from bioenergetics, metabolism and organellar and cell biology.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Mitochondrial membrane potential directly powers many critical functions of mitochondria, including ATP production, mitochondrial protein import, and metabolite transport. Its loss is a cardinal feature of aging and mitochondrial diseases, and cells closely monitor membrane potential as an indicator of mitochondrial health. Given its central importance, it is logical that cells would modulate mitochondrial membrane potential in response to demand and environmental cues, but there has been little exploration of this question. We report that loss of the Sit4 protein phosphatase in yeast increases mitochondrial membrane potential, both by inducing the electron transport chain and the phosphate starvation response. Indeed, a similarly elevated mitochondrial membrane potential is also elicited simply by phosphate starvation or by abrogation of the Pho85-dependent phosphate sensing pathway. This enhanced membrane potential is primarily driven by an unexpected activity of the ADP/ATP carrier. We also demonstrate that this connection between phosphate limitation and enhancement of mitochondrial membrane potential is observed in primary and immortalized mammalian cells as well as in Drosophila . These data suggest that mitochondrial membrane potential is subject to environmental stimuli and intracellular signaling regulation and raise the possibility for therapeutic enhancement of mitochondrial function even in defective mitochondria.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
In this study, the authors describe an elegant genetic screen for mutants that suppress defects of MCT1 deletions which are deficient in mitochondrial fatty acid synthesis. This screen identified many genes, including that for Sit4. In addition, genes for retrograde signaling factors (Rtg1, Rtg2 and Rtg3), proteins influencing proteasomal degradation (Rpn4, Ubc4) or ribosomal proteins (Rps17A, Rps29A) were found. From this mix of components, the authors selected Sit4 for further analysis. In the first part of the study, they analyzed the effect of Sit4 in context of MCT1 mutant suppression. This more specific part is very detailed and thorough, the experiments are well controlled and convincing. The second, more general part of the study focused on the effect of Sit4 on the level of the …
Author Response
Reviewer #1 (Public Review):
In this study, the authors describe an elegant genetic screen for mutants that suppress defects of MCT1 deletions which are deficient in mitochondrial fatty acid synthesis. This screen identified many genes, including that for Sit4. In addition, genes for retrograde signaling factors (Rtg1, Rtg2 and Rtg3), proteins influencing proteasomal degradation (Rpn4, Ubc4) or ribosomal proteins (Rps17A, Rps29A) were found. From this mix of components, the authors selected Sit4 for further analysis. In the first part of the study, they analyzed the effect of Sit4 in context of MCT1 mutant suppression. This more specific part is very detailed and thorough, the experiments are well controlled and convincing. The second, more general part of the study focused on the effect of Sit4 on the level of the mitochondrial membrane potential. This part is of high general interest, but less well developed. Nevertheless, this study is very interesting as it shows for the first time that phosphate export from mitochondrial is of general relevance for the membrane potential even in wild type cells (as long as they live from fermentation), that the Sit4 phosphatase is critical for this process and that the modulation of Sit4 activity influences processes relying on the membrane potential, such as the import of proteins into mitochondria. However, some aspects should be further clarified.
- It is not clear whether Sit4 is only relevant under fermentative conditions. Does Sit4 also influence the membrane potential in respiring cells? Fig. S2D shows the membrane potential in glucose and raffinose. Both carbon sources lead to fermentative growths. The authors should also test whether Sit4 levels influence the membrane potential when cells are grown under respirative conditions, such in ethanol, lactate or glycerol. Even if deletions of Sit4 affect respiration, mutants with altered activity can be easily analyzed.
sit4Δ cells fail to grow on nonfermentable media as shown by us (Figure 2—figure supplement 1C) and others (Arndt et al., 1989; Dimmer et al., 2002; Jablonka et al., 2006). In our opinion, the exact reason is unclear, but there is an interesting observation that addition of aspartate can partially restore growth on ethanol (Jablonka et al., 2006). Despite the lack of thorough investigation on this sit4Δ defect, an early study speculated that this defect could be related to the cAMP-PKA pathway (Sutton et al., 1991). This study pointed out genetic interactions of SIT4 with multiple genes in cAMP-PKA (Sutton et al., 1991). In addition, sit4Δ cells have similar phenotypes as those cAMP-PKA null mutants, such as glycogen accumulation, caffeine resistant, and failure to grow on nonfermentable media (Sutton et al., 1991). We have not found sit4Δ mutants that could grow on nonfermentable media based on literature search.
- The authors should give a name to the pathway shown in Fig. 4D. This would make it easier to follow the text in the results and the discussion. This pathway was proposed and characterized in the 90s by George Clark-Walker and others, but never carefully studied on a mechanistic level. Even if the flux through this pathway cannot be measured in this study, the regulatory role of Sit4 for this process is the most important aspect of this manuscript.
We now refer this mechanism as the mitochondrial ATP hydrolysis pathway.
- To further support their hypothesis, the authors should show that deletion of Pic1 or Atp1 wipes out the effect of a Sit4 deletion. In these petite-negative mutants, the phosphate export cycle cannot be carried out and thus, Sit4, should have no effect.
The mitochondrial phosphate transport activity is electroneutral as it also pumps a proton together with inorganic phosphate. The F1 subunit of the ATP synthase (Atp1 and Atp2) is suggested among many literatures to be responsible for the ATP hydrolysis. We performed tetrad dissection to generate atp1Δ or atp2Δ in pho85Δ background. After streaking the single colony to a fresh plate, we noticed that atp1Δ mct1Δ and atp2Δ mct1Δ cells are lethal, and knocking out PHO85 rescued this synthetic lethality. It is not surprising that atp1Δ mct1Δ or atp2Δ mct1 Δ cells are lethal since the F1 subunit is important to generate a minimum of MMP in mct1 Δ cells when the ETC is absent (i.e., rho0 cells). However, knocking out PHO85 can generate MMP independent of F1 subunit of ATP synthase, which is suggested by the viable atp1Δ mct1Δ pho85Δ and atp2Δ mct1Δ pho85Δ cells. There are many ATPases in the mitochondrial matrix that could hydrolyze ATP for ADP/ATP carrier to generate MMP theoretically. However, we do not currently know exactly which ATPase(s) is activated by phosphate starvation. This data is now included as Figure 5—figure supplement 1F-G.
- What is the relevance of Sit4 for the Hap complex which regulates OXPHOS gene expression in yeast? The supplemental table suggests that Hap4 is strongly influenced by Sit4. Is this downstream of the proposed role in phosphate metabolism or a parallel Sit4 activity? This is a crucial point that should be addressed experimentally.
To investigate the role of the Hap complex in MMP generation in sit4Δ cells, we overexpressed and knocked out HAP4, the catalytic subunit of the Hap complex, separately in wild-type and sit4Δ cells. We confirmed the HAP4 overexpression by the enriched abundance of ETC complexes as shown in the BN-PAGE (Figure 2—figure supplement 1E). However, we did not observe any rescue of ETC or ATP synthase in mct1Δ cells when HAP4 was overexpressed. The enriched level of ETC complexes by HAP4 overexpress is not sufficient to rescue the MMP (Figure 2—figure supplement 1F).
Next, we knocked out HAP4 in sit4Δ cells. Knocking out SIT4 could still increase MMP in hap4Δ cells with a much-reduced magnitude, which phenocopied ETC subunit and RPO41 deletion in sit4Δ cells (Figure 2—figure supplement 1G).
In conclusion, the Hap complex is involved in the MMP increase when SIT4 is absent. However, it is not sufficient to increase MMP by overexpressing HAP4. The Hap complex discussion is now included in the manuscript, and the data is presented as Figure 2—figure supplement 1E-G.
- The authors use the accumulation of Ilv2 precursors as proxy for mitochondrial protein import efficiency. Ilv2 was reported before as a protein which, if import into mitochondria is slow, is deviated into the nucleus in order to be degraded (Shakya,..., Hughes. 2021, Elife). Is it possible that the accumulation of the precursor is the result of a reduced degradation of pre-Ilv2 in the nucleus rather than an impaired mitochondrial import? Since a number of components of the ubiquitin-proteasome system were identified with Sit4 in the same screen, a role of Sit4 in proteasomal degradation seems possible. This should be tested.
We thank the reviewer for pointing out this potential caveat with our Ilv2-FLAG reporter. With limited search and tests, we could not find another reporter that behaves like Ilv2FLAG. The reason Ilv2-FLAG is a perfect reporter for this study is because in wild-type cells, Ilv2-FLAG is not 100% imported. Therefore, we could demonstrate that mitochondria with higher MMP import more efficiently. Unfortunately, all of the mitochondrial proteins that we tested could efficiently import in wild-type cells. To identify other suitable mitochondrial proteins that behave like Ilv2-FLAG, we would need to conduct a more comprehensive screen.
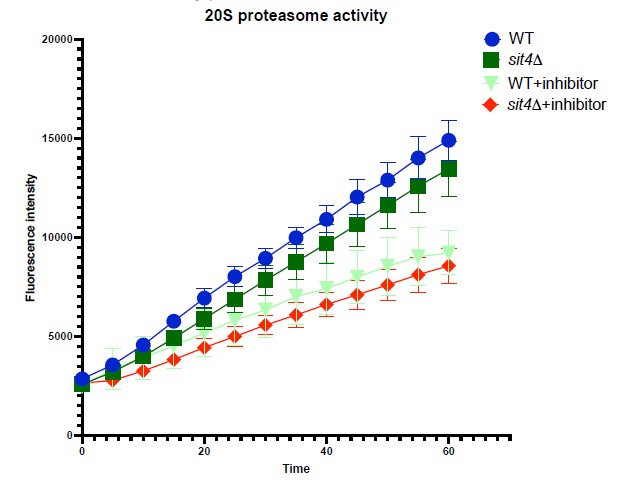
To address the concern of the involvement of protein degradation in obscuring the interpretation of Ilv2-FLAG import, we performed two experiments. First, we measured the proteasomal activity in wild-type and our mutants using a commercial kit (Cayman). We did not observe a statistically significant difference in 20S proteasomal activity between wild-type and sit4Δ cells.
In the second experiment, we reduced the MMP of sit4 cells using CCCP treatment and measured the Ilv2-FLAG import. We first treated sit4Δ cells with different dosage of CCCP for six hours and measured their MMP. sit4Δ cells treated with 75 µM CCCP had comparable MMP to wild-type cells. When we treated sit4Δ cells with higher concentrations of CCCP, most of the cells did not survive after six hours. Next, we performed the Ilv2-FLAG import assay. We observed similar level of unimported Ilv2FLAG (marked with *) in sit4Δ cells treated with 75 µM CCCP. This result confirms that sit4Δ cells have similar Ilv2-FLAG turnover mechanism and activity as the wild-type cells, because when we lower the MMP in sit4Δ background we observe a similar level of unimported Ilv2-FLAG. We thus feel confident in concluding that the Ilv2-FLAG import results are indeed an accurate proxy for MMP level. These data are now included as Figure 1—figure supplement 1H-J in the manuscript.
Author response image 1.
Reviewer #2 (Public Review):
This study reports interesting findings on the influence of a conserved phosphatase on mitochondrial biogenesis and function. In the absence of it, many nucleus-encoded mitochondrial proteins among which those involved in ATP generation are expressed much better than in normal cells. In addition to a better understanding of th mechanisms that regulate mitochondrial function, this work may help developing therapeutic strategies to diseases caused by mitochondrial dysfunction. However there are a number of issues that need clarification.
- The rationale of the screening assay to identify genes required for the gene expression modifications observed in mct1 mutant is not clear. Indeed, after crossing with the gene deletion libray, the cells become heterozygote for the mct1 deletion and should no longer be deficient in mtFAS. Thank you for clarifying this and if needed adjust the figure S1D to indicate that the mated cells are heterozygous for the mct1 and xxx mutations.
We updated the methods section and the graphic for the genetic screen to clarify these points within the SGA workflow overview. After we created the heterozygote by mating mct1Δ cells with the individual KO cells in the collection, these diploids underwent sporulation and selection for the desired double KO haploid. As a result, the luciferase assay was performed in haploid cells with MCT1 and one additional non-essential gene deleted.
- The tests shown in Fig. S1E should be repeated on individual subclones (at least 100) obtained after plating for single colonies a glucose culture of mct1 mutant, to determine the proportion of cells with functional (rho+) mtDNA in the mct1 glucose and raffinose cultures. With for instance a 50% proportion of rho- cells, this could substantially influence the results of the analyses made with these cells (including those aiming to evaluate the MMP).
We agree that this would provide a more confident estimate for population-level characterization of these colonies. It is important to note that we randomly chose 10 individual subclones, and 100% of these colonies were verified to be rho+. This suggests the population has functional mtDNA, and thus felt confident in the identity of our populations.
- The mitochondria area in mct1 cells (Fig.S1G) does not seem to be consistent with the tests in Fig. 1C. that indicate a diminished mitochondrial content in mct1 cells vs wild-type yeast. A better estimate (by WB for instance) of the mitochondrial content in the analyzed strains would enable to better evaluate MMP changes monitored with Mitotracker since the amount of mitochondria in cells correlate with the intensity of the fluorescence signal.
As this reviewer pointed out, we quantified mitochondrial area based on Tom70-GFP signal. This measurement is quantified by mitochondrial area over cell size. Cell size is an important parameter when measuring organelle size as most of the organelles scale up and down with the cell size. mct1Δ cells generally have smaller cell size than WT cells. Therefore, the mitochondrial area of mct1Δ cells was not significantly different from WT cells when scaled to cell size. We believe this is the best method to compare mitochondrial area. As for quantifying MMP from these microscopy images, we measured the average MitoTracker Red fluorescence intensity of each mitochondria defined by Tom70-GFP. This method inherently normalizes to subtract the influence of mitochondria area when quantifying MMP.
- Page 12: "These data demonstrate that loss of SIT4 results in a mitochondrial phenotype suggestive of an enhanced energetic state: higher membrane potential, hyper-tubulated morphology and more effective protein import." Furthermore, the sit4 mutant shows higher levels of OXPHOS complexes compared to WT yeast.
Despite these beneficial effects on mitochondria, the sit4 deletion strain fails to grow on respiratory substrates. It would be good to know whether the authors have some explanation for this apparent contradiction.
We agree that this was initially puzzling. We provide a more complete explanation above (see comments to reviewer #1 - major concern #1). Briefly, the growth deficiency in non-fermentable media with sit4Δ cells was reported and studied by multiple groups (Arndt et al., 1989; Dimmer et al., 2002; Jablonka et al., 2006). These seems to indicate that sit4Δ cells contain more ETC complexes and more OCR but cannot respire on nonfermentable carbon source. However, we do not think there is yet a clear explanation for this phenotype. One interesting observation reported is the addition of aspartate partly restoring cells’ growth on ethanol (Jablonka et al., 2006). One early study speculates that this defect could be related to the cAMP-PKA pathway. Sutton et al. pointed out genetic interactions with sit4 and multiple genes in cAMP-PKA (Sutton et al., 1991). In addition, sit4Δ cells have similar phenotypes as those cAMP-PKA null mutants, such as glycogen accumulation, caffeine resistance, and failure to grow on non-fermentable media. However, to keep this manuscript succinct, we opted to stay focused on MMP.
Reviewer #3 (Public Review):
In this study, the authors investigate the genetic and environmental causes of elevated Mitochondrial Membrane Potential (MMP) in yeast, and also some physiological effects correlated with increased MMP.
The study begins with a reanalysis of transcriptional data from a yeast mutant lacking the gene MCT1 whose deletion has been shown to cause defects in mitochondrial fatty acid synthesis. The authors note that in raffinose mct1del cells, unlike WT cells, fail to induce expression of many genes that code for subunits of the Electron Transport Chain (ETC) and ATP synthase. The deletion of MCT1 also causes induction of genes involved in acetyl-CoA production after exposure to raffinose. The authors therefore conduct a screen to identify mutants that suppress the induction of one of these acetylCoA genes, Cit2. They then validate the hits from this screen to see which of their suppressor mutants also reduce expression in four other genes induced in a mct1del strain. This yielded 17 genes that abolished induction of all 5 genes tested in an mct1del background during growth on raffinose.
The authors chose to focus on one of these hits, the gene coding for the phosphatase SIT4 (related to human PP6) which also caused an increase in expression of two respiratory chain genes. The authors then investigated MMP and mitochondrial morphology in strains containing SIT4 and MCT1 deletions and surprisingly saw that sit4del cells had highly elevated MMP, more reticular mitochondria, and were able to fully import the acetolactate synthase protein Ilv2p and form ETC and ATP synthase complexes, even in cells with an mct1del background, rescuing the low MMP, fragmented mitochondria, low import of Ilv2 and an inability to form ETC and ATP synthase complexes phenotypes of the mct1del strain. Surprisingly, the authors find that even though MMP is high and ETC subunits are present in the sit4del mct1del double deletion strain, that strain has low oxygen consumption and cannot grow under respiratory conditions, indicating that the elevated MMP cannot come from fully functional ETC subunits. The authors also observe that deleting key subunits of ETC complex III (QCR2) and IV (COX5) strongly reduced the MMP of the sit4del mutant, which would suggest that the majority of the increase in MMP of the sit4del mutant was dependant on a partially functional ETC. The authors note that there was still an increase in MMP in the qcr2del sit4del and cox4del sit4del strains relative to qcr2del and cox4del strains indicating that some part of the increase in MMP was not dependent on the ETC.
The authors dismiss the possibility that the increase in MMP could have been through the reversal of ATP synthase because they observe that inhibition of ATP synthase with oligomycin led to an increase of MMP in sit4del cells. Indicating that ATP synthase is operating in a forward direction in sit4del cells.
Noting that genes for phosphate starvation are induced in sit4del cells, the authors investigate the effects of phosphate starvation on MMP. They found that phosphate starvation caused an increase in MMP and increased Ilv2p import even in the absence of a mitochondrial genome. They find that inhibition of the ADP/ATP carrier (AAC) with bongkrekic acid (BKA) abolishes the increase of MMP in response to phosphate starvation. They speculate that phosphate starvation causes an increase in MMP through the import and conversion of ATP to ADP and subsequent pumping of ADP and inorganic phosphate out of the mitochondria.
They further show that MMP is also increased when the cyclin dependent kinase PHO85 which plays a role in phosphate signaling is deleted and argue that this indicates that it is not a decrease in phosphate which causes the increase in MMP under phosphate starvation, but rather the perception of a decrease in phosphate as signalled through PHO85. Unlike in the case of SIT4 deletion, the increase in MMP caused by the deletion of pho85 is abolished when MCT1 is deleted.
Finally they show an increase in MMP in immortalized human cell lines following phosphate starvation and treatment with the phosphate transporter inhibitor phosphonoformic acid (PFA). They also show an increase in MMP in primary hepatocytes and in midgut cells of flies treated with PFA.
The link between phosphate starvation and elevated MMP is an important and novel finding and the evidence is clear and compelling. Based on their experiments in various mammalian contexts, this link appears likely to be generalizable, and they propose and begin to test an interesting hypothesis for how MMP might occur in response to phosphate starvation in the absence of the Electron Transport Chain.
The link between phosphate starvation and deletion of the conserved phosphatase SIT4 is also interesting and important, and while the authors' experiments and analysis suggest some connection between the two observations, that connection is still unclear.
Major points
Mitotracker is great fluorescent dye, but it measures membrane potential only indirectly. There is a danger when cells change growth rates, ion concentrations, or when the pH changes, all MMP indicating dyes change in fluorescence: their signal is confounded Change in phosphate levels can possibly do both, alter pH and ion concentrations. Because all conclusions of the manuscript are based on a change in MMP, it would be a great precaution to use a dye-independent measure of membrane potential, and confirm at least some key results.
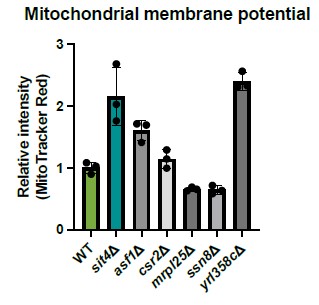
Mitochondrial MMP does strongly influence amino acid metabolism, and indeed the SIT4 knockout has a quite striking amino acid profile, with histidine, lysine, arginine, tyrosine being increased in concentration. http://ralser.charite.de/metabogenecards/Chr_04/YDL047W.html Could this amino acid profile support the conclusions of the authors? At least lysine and arginine are down in petites due to a lack of membrane potential and iron sulfur cluster export.- and here they are up. Along these lines, according to the same data resource, the knock-outs CSR2, ASF1, SSN8, YLR0358 and MRPL25 share the same metabolic profile. Due to limited time I did not re-analyse the data provided by the authors- but it would be worth checking if any of these genes did come up in the screens of the authors.
We tested the mutants within the same cluster as SIT4 shown in this paper from the deletion collection and measured their MMP. yrl358cΔ cells have similar high MMP as observed in sit4Δ cells. However, this gene has a yet undefined function. Beyond YRL358C, we did not observe similar MMP increases in other gene deletions from this panel, which does not support the notion that amino acids such as histidine, lysine, arginine, or tyrosine play a determining effect in driving MMP.
The media condition and strain used in the suggested paper is very different from what we used in our study. Instead of growing prototrophic cells in minimal media without any amino acids, we used auxotrophic yeast strains and grew them in media containing complete amino acids. So far, none of the other defects or signaling associated with SIT4 deletion could influence MMP as much as the phosphate signaling. We interpret these data to support the hypothesis that the MMP observation in sit4Δ cells is connected with the phosphate signaling as illustrated by the second half of the story in our manuscript.
Author reponse image 2.
One important claim in the manuscript attempts to explain a mechanism for the MMP increase in response to phosphate starvation which is independent of the ETC and ATP synthase.
It seems to me the only direct evidence to support this claim is that inhibition of the AAC with BKA stops the increase of mitotracker fluorescence in response to phosphate starvation in both WT and rho0 cells (Figs 4B and 4C). It would strengthen the paper if the authors could provide some orthogonal evidence.
This is a similar comment as raised by reviewer #1 - major concern #3. We refer the reviewer to our discussion and the new data above. Briefly, we do not think F1 subunit is responsible for the ATP hydrolysis activity to generate MMP in phosphate depleted situation. We believe there are additional ATPase(s) in the mitochondrial matrix that can be utilized to couple to ADP/ATP carrier for MMP generation during phosphate starvation. However, we have not identified the relevant ATPase(s) at this point, and it is likely that multiple ATPases could contribute to this activity.
Introduction/Discussion The author might want to make the reader of the article aware that the 'reversal' of the ATP synthase directionality -i.e. ATP hydrolysis by the ATP synthase as a mechanism to create a membrane potential (in petites), has always been a provocative idea - but one that thus far could never be fully substantiated. Indeed some people that are very familiar with the topic, are skeptical this indeed happens. For instance, Vowinckel et al 2021 (PMID: 34799698) measured precise carbon balances for peptide cells, and found no evidence for a futile cycle - peptides grow slower, but accumulate the same biomass from glucose as peptides that re-evolve at a fast growth rate . Perhaps the manuscript could be updated accordingly.
We thank the reviewer for pointing out this additional relevant study. We have rephased the referenced sentence in the introduction. The MMP generation in phosphate starvation is independent of the F1 portion of ATP synthase. Therefore, our data neither supports or refutes either of these arguments.
In the introduction and conclusion there is discussion of MMP set points. In particular the authors state:
"Critically, we find that cells often prioritize this MMP setpoint over other bioenergetic priorities, even in challenging environments, suggesting an important evolutionary benefit."
This does not seem to be consistent with the central finding of the manuscript that MMP changes under phosphate starvation. MMP doesn't seem so much to have a 'set point' but rather be an important physiological variable that reacts to stimuli such as phosphate starvation.
The reviewer raises a rational alternative hypothesis to the one that we have proposed. In reality, both of these are complete speculations to explain the data and we can’t think of any way to test the evolutionary basis for the mechanisms that we describe. We recognize that untested/untestable speculative arguments have limitations and there are viable alternative hypotheses. We have softened our language to ensure that it is clear that this is only a speculation.
The authors suggest that deletion of Pho85 causes an increase in MMP because of cellular signaling. However, they also state in the conclusion:
"Unlike phosphate starvation, the pho85D mutant has elevated intracellular phosphate concentrations. This suggests that the phosphate effect on MMP is likely to be elicited by cellular signaling downstream of phosphate sensing rather than some direct effect of environmental depletion of phosphate on mitochondrial energetics."
The authors should cite the study that shows deletion of PHO85 causes increased intracellular phosphate concentrations. It also seems possible that the 'cellular signaling' that causes the increase in MMP could be a result of this increase in intracellular phosphate concentrations, which could constitute a direct effect of an environmental overload of phosphate on mitochondrial energetics.
We now cited the literature that shows higher intracellular phosphate in pho85Δ cells (Gupta et al., 2019; Liu et al., 2017). Depleting phosphate in the media drastically reduced intracellular phosphate concentration, which is the opposing situation as pho85Δ cells. Nevertheless, we observed higher MMP in either situation. We concluded from these two observations that the increase in MMP is a response to the signaling activated by phosphate depletion rather than the intracellular phosphate abundance.
Related to this point, in the conclusion, the authors state:
"We now show that intracellular signaling can lead to an increased MMP even beyond the wild-type level in the absence of mitochondrial genome."
In sum, the data shows that signaling is important here- but signaling alone is only the message - not the biophysical process that creates a membrane potential. The authors then could revise this slightly.
We have rephrased this sentence as suggested, which now reads “We now show that intracellular signaling triggers a process that can lead to an increased MMP even beyond the wild-type level in the absence of mitochondrial genome”.
The authors state in the conclusion that
"We first made the observation that deletion of the SIT4 gene, which encodes the yeast homologue of the mammalian PP6 protein phosphatase, normalized many of the defects caused by loss of mtFAS, including gene expression programs, ETC complex assembly, mitochondrial morphology, and especially MMP (Fig. 1)"
The data shown though indicates that a defect in mtFAS in terms of MMP, deletion of SIT4 causes a huge increase (and departure away from normality) whether or not mct1 is present (Fig 1D)
We changed the word “normalized” to “reversed”. In the discussion section, we also emphasized that many of these increases are independent of mitochondrial dysfunction induced by loss of mtFAS.
The language "SIT4 is required for both the positive and negative transcriptional regulation elicited by mitochondrial dysfunction" feels strong. SIT4 seems to influence positive transcriptional regulation in response to mitochondrial dysfunction caused by MCT1 deletion (but may not be the only thing as there appears to be an increase in CIT2 expression in a sit4del background following a further deletion of MCT1). In terms of negative regulation, SIT4 deletion clearly affects the baseline, but MCT1 deletion still causes down regulation of both examples shown in Fig 1B, showing that negative transcriptional regulation can still occur in the absence of SIT4. The authors might consider showing fold change of expression as they do in later figures (Figs 4B and C) to help the reader evaluate the quantitative changes they demonstrate.
We now displayed the fold change as suggested. This sentence now reads “These data suggest that SIT4 positively and negatively influences transcriptional regulation elicited by mitochondrial dysfunction”.
The authors induce phosphate starvation by adding increasing amounts of potassium phosphate monobasic at a pH of 4.1 to phosphate dropout media supplemented with potassium. The authors did well to avoid confounding effects of removing potassium. The final pH of YNB is typically around 5.2. Is it possible that the authors are confounding a change in pH with phosphate starvation? One would expect the media in the phosphate starvation condition to have a higher pH than the phosphate replacement or control media. Is a change in pH possibly a confounding factor when interpreting phosphate starvation? Perhaps the authors could quantify the pH of the media they use for the experiment to understand how much of a factor that could be. One needs to be careful with Miotracker and any other fluorescent dye when pH changes. Albeit having constraints on its own, MitoLoc as a protein rather than small molecule marker of MMP might be a good complement.
We followed the protocol used by many other studies that depleted phosphate in the media. The reason we and others adjusted the media without inorganic phosphate to a pH of 4.1 is because that is the pH of phosphate monobasic. From there, we could add phosphate monobasic to create +Pi media without changing the media pH. Therefore, media containing different concentrations of phosphate all have the exact same pH. We now emphasize that all media containing different levels of inorganic phosphate have the same pH to the manuscript to eliminate such concern (see page 18).
Even though all media have the similar pH, we also provided complementary data using a parallel approach to measure the MMP by assessing mitochondrial protein import as demonstrated previously with Ilv2-FLAG, which shares the same principle as mitoLoc.
Reference
Arndt, K. T., Styles, C. A., & Fink, G. R. (1989). A suppressor of a HIS4 transcriptional defect encodes a protein with homology to the catalytic subunit of protein phosphatases. Cell, 56(4), 527–537. https://doi.org/10.1016/00928674(89)90576-X
Dimmer, K. S., Fritz, S., Fuchs, F., Messerschmitt, M., Weinbach, N., Neupert, W., & Westermann, B. (2002). Genetic basis of mitochondrial function and morphology in Saccharomyces cerevisiae. Molecular Biology of the Cell, 13(3), 847–853. https://doi.org/10.1091/mbc.01-12-0588
Gupta, R., Walvekar, A. S., Liang, S., Rashida, Z., Shah, P., & Laxman, S. (2019). A tRNA modification balances carbon and nitrogen metabolism by regulating phosphate homeostasis. ELife, 8, e44795. https://doi.org/10.7554/eLife.44795
Jablonka, W., Guzmán, S., Ramírez, J., & Montero-Lomelí, M. (2006). Deviation of carbohydrate metabolism by the SIT4 phosphatase in Saccharomyces cerevisiae. Biochimica et Biophysica Acta (BBA) - General Subjects, 1760(8), 1281–1291. https://doi.org/10.1016/j.bbagen.2006.02.014
Liu, N.-N., Flanagan, P. R., Zeng, J., Jani, N. M., Cardenas, M. E., Moran, G. P., & Köhler, J. R. (2017). Phosphate is the third nutrient monitored by TOR in Candida albicans and provides a target for fungal-specific indirect TOR inhibition. Proceedings of the National Academy of Sciences, 114(24), 6346–6351. https://doi.org/10.1073/pnas.1617799114
Sutton, A., Immanuel, D., & Arndt, K. T. (1991). The SIT4 protein phosphatase functions in late G1 for progression into S phase. Molecular and Cellular Biology, 11(4), 2133–2148.
-
eLife assessment
This study presents important findings on the regulation of the phosphate export cycle and identify the phosphatase Sit4 as a crucial player in regulation of the inner membrane potential of mitochondria. Whereas some of the data are convincing, the analyses will profit from deeper insights concerning metabolism alterations (carbon sources, amino acids). The major strength however is a new insight on how the cells use alternative ways for maintaining a critical mitochondrial inner membrane potential, and therefore this study is interesting to the broad audience with interests spanning from bioenergetics, metabolism and organellar and cell biology.
-
Reviewer #1 (Public Review):
In this study, the authors describe an elegant genetic screen for mutants that suppress defects of MCT1 deletions which are deficient in mitochondrial fatty acid synthesis. This screen identified many genes, including that for Sit4. In addition, genes for retrograde signaling factors (Rtg1, Rtg2 and Rtg3), proteins influencing proteasomal degradation (Rpn4, Ubc4) or ribosomal proteins (Rps17A, Rps29A) were found. From this mix of components, the authors selected Sit4 for further analysis. In the first part of the study, they analyzed the effect of Sit4 in context of MCT1 mutant suppression. This more specific part is very detailed and thorough, the experiments are well controlled and convincing. The second, more general part of the study focused on the effect of Sit4 on the level of the mitochondrial …
Reviewer #1 (Public Review):
In this study, the authors describe an elegant genetic screen for mutants that suppress defects of MCT1 deletions which are deficient in mitochondrial fatty acid synthesis. This screen identified many genes, including that for Sit4. In addition, genes for retrograde signaling factors (Rtg1, Rtg2 and Rtg3), proteins influencing proteasomal degradation (Rpn4, Ubc4) or ribosomal proteins (Rps17A, Rps29A) were found. From this mix of components, the authors selected Sit4 for further analysis. In the first part of the study, they analyzed the effect of Sit4 in context of MCT1 mutant suppression. This more specific part is very detailed and thorough, the experiments are well controlled and convincing. The second, more general part of the study focused on the effect of Sit4 on the level of the mitochondrial membrane potential. This part is of high general interest, but less well developed. Nevertheless, this study is very interesting as it shows for the first time that phosphate export from mitochondrial is of general relevance for the membrane potential even in wild type cells (as long as they live from fermentation), that the Sit4 phosphatase is critical for this process and that the modulation of Sit4 activity influences processes relying on the membrane potential, such as the import of proteins into mitochondria. However, some aspects should be further clarified.
1. It is not clear whether Sit4 is only relevant under fermentative conditions. Does Sit4 also influence the membrane potential in respiring cells? Fig. S2D shows the membrane potential in glucose and raffinose. Both carbon sources lead to fermentative growths. The authors should also test whether Sit4 levels influence the membrane potential when cells are grown under respirative conditions, such in ethanol, lactate or glycerol. Even if deletions of Sit4 affect respiration, mutants with altered activity can be easily analyzed.
2. The authors should give a name to the pathway shown in Fig. 4D. This would make it easier to follow the text in the results and the discussion. This pathway was proposed and characterized in the 90s by George Clark-Walker and others, but never carefully studied on a mechanistic level. Even if the flux through this pathway cannot be measured in this study, the regulatory role of Sit4 for this process is the most important aspect of this manuscript.
3. To further support their hypothesis, the authors should show that deletion of Pic1 or Atp1 wipes out the effect of a Sit4 deletion. In these petite-negative mutants, the phosphate export cycle cannot be carried out and thus, Sit4, should have no effect.
4. What is the relevance of Sit4 for the Hap complex which regulates OXPHOS gene expression in yeast? The supplemental table suggests that Hap4 is strongly influenced by Sit4. Is this downstream of the proposed role in phosphate metabolism or a parallel Sit4 activity? This is a crucial point that should be addressed experimentally.
5. The authors use the accumulation of Ilv2 precursors as proxy for mitochondrial protein import efficiency. Ilv2 was reported before as a protein which, if import into mitochondria is slow, is deviated into the nucleus in order to be degraded (Shakya,..., Hughes. 2021, Elife). Is it possible that the accumulation of the precursor is the result of a reduced degradation of pre-Ilv2 in the nucleus rather than an impaired mitochondrial import? Since a number of components of the ubiquitin-proteasome system were identified with Sit4 in the same screen, a role of Sit4 in proteasomal degradation seems possible. This should be tested. -
Reviewer #2 (Public Review):
This study reports interesting findings on the influence of a conserved phosphatase on mitochondrial biogenesis and function. In the absence of it, many nucleus-encoded mitochondrial proteins among which those involved in ATP generation are expressed much better than in normal cells. In addition to a better understanding of th mechanisms that regulate mitochondrial function, this work may help developing therapeutic strategies to diseases caused by mitochondrial dysfunction. However there are a number of issues that need clarification.
The rationale of the screening assay to identify genes required for the gene expression modifications observed in mct1 mutant is not clear. Indeed, after crossing with the gene deletion libray, the cells become heterozygote for the mct1 deletion and should no longer be …
Reviewer #2 (Public Review):
This study reports interesting findings on the influence of a conserved phosphatase on mitochondrial biogenesis and function. In the absence of it, many nucleus-encoded mitochondrial proteins among which those involved in ATP generation are expressed much better than in normal cells. In addition to a better understanding of th mechanisms that regulate mitochondrial function, this work may help developing therapeutic strategies to diseases caused by mitochondrial dysfunction. However there are a number of issues that need clarification.
The rationale of the screening assay to identify genes required for the gene expression modifications observed in mct1 mutant is not clear. Indeed, after crossing with the gene deletion libray, the cells become heterozygote for the mct1 deletion and should no longer be deficient in mtFAS. Thank you for clarifying this and if needed adjust the figure S1D to indicate that the mated cells are heterozygous for the mct1 and xxx mutations.
The tests shown in Fig. S1E should be repeated on individual subclones (at least 100) obtained after plating for single colonies a glucose culture of mct1 mutant, to determine the proportion of cells with functional (rho+) mtDNA in the mct1 glucose and raffinose cultures. With for instance a 50% proportion of rho- cells, this could substantially influence the results of the analyses made with these cells (including those aiming to evaluate the MMP).
The mitochondria area in mct1 cells (Fig.S1G) does not seem to be consistent with the tests in Fig. 1C. that indicate a diminished mitochondrial content in mct1 cells vs wild-type yeast. A better estimate (by WB for instance) of the mitochondrial content in the analyzed strains would enable to better evaluate MMP changes monitored with Mitotracker since the amount of mitochondria in cells correlate with the intensity of the fluorescence signal.
Page 12: "These data demonstrate that loss of SIT4 results in a mitochondrial phenotype suggestive of an enhanced energetic state: higher membrane potential, hyper-tubulated morphology and more effective protein import." Furthermore, the sit4 mutant shows higher levels of OXPHOS complexes compared to WT yeast.
Despite these beneficial effects on mitochondria, the sit4 deletion strain fails to grow on respiratory substrates. It would be good to know whether the authors have some explanation for this apparent contradiction.
-
Reviewer #3 (Public Review):
In this study, the authors investigate the genetic and environmental causes of elevated Mitochondrial Membrane Potential (MMP) in yeast, and also some physiological effects correlated with increased MMP.
The study begins with a reanalysis of transcriptional data from a yeast mutant lacking the gene MCT1 whose deletion has been shown to cause defects in mitochondrial fatty acid synthesis. The authors note that in raffinose mct1del cells, unlike WT cells, fail to induce expression of many genes that code for subunits of the Electron Transport Chain (ETC) and ATP synthase. The deletion of MCT1 also causes induction of genes involved in acetyl-CoA production after exposure to raffinose. The authors therefore conduct a screen to identify mutants that suppress the induction of one of these acetyl-CoA genes, Cit2. …
Reviewer #3 (Public Review):
In this study, the authors investigate the genetic and environmental causes of elevated Mitochondrial Membrane Potential (MMP) in yeast, and also some physiological effects correlated with increased MMP.
The study begins with a reanalysis of transcriptional data from a yeast mutant lacking the gene MCT1 whose deletion has been shown to cause defects in mitochondrial fatty acid synthesis. The authors note that in raffinose mct1del cells, unlike WT cells, fail to induce expression of many genes that code for subunits of the Electron Transport Chain (ETC) and ATP synthase. The deletion of MCT1 also causes induction of genes involved in acetyl-CoA production after exposure to raffinose. The authors therefore conduct a screen to identify mutants that suppress the induction of one of these acetyl-CoA genes, Cit2. They then validate the hits from this screen to see which of their suppressor mutants also reduce expression in four other genes induced in a mct1del strain. This yielded 17 genes that abolished induction of all 5 genes tested in an mct1del background during growth on raffinose.
The authors chose to focus on one of these hits, the gene coding for the phosphatase SIT4 (related to human PP6) which also caused an increase in expression of two respiratory chain genes. The authors then investigated MMP and mitochondrial morphology in strains containing SIT4 and MCT1 deletions and surprisingly saw that sit4del cells had highly elevated MMP, more reticular mitochondria, and were able to fully import the acetolactate synthase protein Ilv2p and form ETC and ATP synthase complexes, even in cells with an mct1del background, rescuing the low MMP, fragmented mitochondria, low import of Ilv2 and an inability to form ETC and ATP synthase complexes phenotypes of the mct1del strain. Surprisingly, the authors find that even though MMP is high and ETC subunits are present in the sit4del mct1del double deletion strain, that strain has low oxygen consumption and cannot grow under respiratory conditions, indicating that the elevated MMP cannot come from fully functional ETC subunits. The authors also observe that deleting key subunits of ETC complex III (QCR2) and IV (COX5) strongly reduced the MMP of the sit4del mutant, which would suggest that the majority of the increase in MMP of the sit4del mutant was dependant on a partially functional ETC. The authors note that there was still an increase in MMP in the qcr2del sit4del and cox4del sit4del strains relative to qcr2del and cox4del strains indicating that some part of the increase in MMP was not dependent on the ETC.
The authors dismiss the possibility that the increase in MMP could have been through the reversal of ATP synthase because they observe that inhibition of ATP synthase with oligomycin led to an increase of MMP in sit4del cells. Indicating that ATP synthase is operating in a forward direction in sit4del cells.
Noting that genes for phosphate starvation are induced in sit4del cells, the authors investigate the effects of phosphate starvation on MMP. They found that phosphate starvation caused an increase in MMP and increased Ilv2p import even in the absence of a mitochondrial genome. They find that inhibition of the ADP/ATP carrier (AAC) with bongkrekic acid (BKA) abolishes the increase of MMP in response to phosphate starvation. They speculate that phosphate starvation causes an increase in MMP through the import and conversion of ATP to ADP and subsequent pumping of ADP and inorganic phosphate out of the mitochondria.
They further show that MMP is also increased when the cyclin dependent kinase PHO85 which plays a role in phosphate signaling is deleted and argue that this indicates that it is not a decrease in phosphate which causes the increase in MMP under phosphate starvation, but rather the perception of a decrease in phosphate as signalled through PHO85. Unlike in the case of SIT4 deletion, the increase in MMP caused by the deletion of pho85 is abolished when MCT1 is deleted.
Finally they show an increase in MMP in immortalized human cell lines following phosphate starvation and treatment with the phosphate transporter inhibitor phosphonoformic acid (PFA). They also show an increase in MMP in primary hepatocytes and in midgut cells of flies treated with PFA.
The link between phosphate starvation and elevated MMP is an important and novel finding and the evidence is clear and compelling. Based on their experiments in various mammalian contexts, this link appears likely to be generalizable, and they propose and begin to test an interesting hypothesis for how MMP might occur in response to phosphate starvation in the absence of the Electron Transport Chain.
The link between phosphate starvation and deletion of the conserved phosphatase SIT4 is also interesting and important, and while the authors' experiments and analysis suggest some connection between the two observations, that connection is still unclear.
Major points
Mitotracker is great fluorescent dye, but it measures membrane potential only indirectly. There is a danger when cells change growth rates, ion concentrations, or when the pH changes, all MMP indicating dyes change in fluorescence: their signal is confounded Change in phosphate levels can possibly do both, alter pH and ion concentrations. Because all conclusions of the manuscript are based on a change in MMP, it would be a great precaution to use a dye-independent measure of membrane potential, and confirm at least some key results.
Mitochondrial MMP does strongly influence amino acid metabolism, and indeed the SIT4 knockout has a quite striking amino acid profile, with histidine, lysine, arginine, tyrosine being increased in concentration. http://ralser.charite.de/metabogenecards/Chr_04/YDL047W.html
Could this amino acid profile support the conclusions of the authors? At least lysine and arginine are down in petites due to a lack of membrane potential and iron sulfur cluster export.- and here they are up. Along these lines, according to the same data resource, the knock-outs CSR2, ASF1, SSN8, YLR0358 and MRPL25 share the same metabolic profile. Due to limited time I did not re-analyse the data provided by the authors- but it would be worth checking if any of these genes did come up in the screens of the authors.One important claim in the manuscript attempts to explain a mechanism for the MMP increase in response to phosphate starvation which is independent of the ETC and ATP synthase.
It seems to me the only direct evidence to support this claim is that inhibition of the AAC with BKA stops the increase of mitotracker fluorescence in response to phosphate starvation in both WT and rho0 cells (Figs 4B and 4C). It would strengthen the paper if the authors could provide some orthogonal evidence.
Introduction/Discussion The author might want to make the reader of the article aware that the 'reversal' of the ATP synthase directionality -i.e. ATP hydrolysis by the ATP synthase as a mechanism to create a membrane potential (in petites), has always been a provocative idea - but one that thus far could never be fully substantiated. Indeed some people that are very familiar with the topic, are skeptical this indeed happens. For instance, Vowinckel et al 2021 (PMID: 34799698) measured precise carbon balances for peptide cells, and found no evidence for a futile cycle - peptides grow slower, but accumulate the same biomass from glucose as peptides that re-evolve at a fast growth rate . Perhaps the manuscript could be updated accordingly.
In the introduction and conclusion there is discussion of MMP set points. In particular the authors state:
"Critically, we find that cells often prioritize this MMP setpoint over other bioenergetic priorities, even in challenging environments, suggesting an important evolutionary benefit."
This does not seem to be consistent with the central finding of the manuscript that MMP changes under phosphate starvation. MMP doesn't seem so much to have a 'set point' but rather be an important physiological variable that reacts to stimuli such as phosphate starvation.
The authors suggest that deletion of Pho85 causes an increase in MMP because of cellular signaling. However, they also state in the conclusion:
"Unlike phosphate starvation, the pho85D mutant has elevated intracellular phosphate concentrations. This suggests that the phosphate effect on MMP is likely to be elicited by cellular signaling downstream of phosphate sensing rather than some direct effect of environmental depletion of phosphate on mitochondrial energetics."
The authors should cite the study that shows deletion of PHO85 causes increased intracellular phosphate concentrations. It also seems possible that the 'cellular signaling' that causes the increase in MMP could be a result of this increase in intracellular phosphate concentrations, which could constitute a direct effect of an environmental overload of phosphate on mitochondrial energetics.
Related to this point, in the conclusion, the authors state:
"We now show that intracellular signaling can lead to an increased MMP even beyond the wild-type level in the absence of mitochondrial genome."
In sum, the data shows that signaling is important here- but signaling alone is only the message - not the biophysical process that creates a membrane potential. The authors then could revise this slightly.
The authors state in the conclusion that
"We first made the observation that deletion of the SIT4 gene, which encodes the yeast homologue of the mammalian PP6 protein phosphatase, normalized many of the defects caused by loss of mtFAS, including gene expression programs, ETC complex assembly, mitochondrial morphology, and especially MMP (Fig. 1)"
The data shown though indicates that a defect in mtFAS in terms of MMP, deletion of SIT4 causes a huge increase (and departure away from normality) whether or not mct1 is present (Fig 1D)
The language "SIT4 is required for both the positive and negative transcriptional regulation elicited by mitochondrial dysfunction" feels strong. SIT4 seems to influence positive transcriptional regulation in response to mitochondrial dysfunction caused by MCT1 deletion (but may not be the only thing as there appears to be an increase in CIT2 expression in a sit4del background following a further deletion of MCT1). In terms of negative regulation, SIT4 deletion clearly affects the baseline, but MCT1 deletion still causes down regulation of both examples shown in Fig 1B, showing that negative transcriptional regulation can still occur in the absence of SIT4. The authors might consider showing fold change of expression as they do in later figures (Figs 4B and C) to help the reader evaluate the quantitative changes they demonstrate.
The authors induce phosphate starvation by adding increasing amounts of potassium phosphate monobasic at a pH of 4.1 to phosphate dropout media supplemented with potassium. The authors did well to avoid confounding effects of removing potassium. The final pH of YNB is typically around 5.2. Is it possible that the authors are confounding a change in pH with phosphate starvation? One would expect the media in the phosphate starvation condition to have a higher pH than the phosphate replacement or control media. Is a change in pH possibly a confounding factor when interpreting phosphate starvation? Perhaps the authors could quantify the pH of the media they use for the experiment to understand how much of a factor that could be. One needs to be careful with Miotracker and any other fluorescent dye when pH changes. Albeit having constraints on its own, MitoLoc as a protein rather than small molecule marker of MMP might be a good complement.
-
