Dysregulation of mTOR signaling mediates common neurite and migration defects in both idiopathic and 16p11.2 deletion autism neural precursor cells
Curation statements for this article:-
Curated by eLife

eLife assessment:
This important study describes converging cellular phenotypes in human neural progenitor cells derived from individuals with differing genetic forms of autism spectrum disorders. These convincing data demonstrate that altered mTOR signaling occurs in all cases of autism examined in the study, providing a common starting point for understanding the etiology of neuronal deficits in autism. The work will be of broad interest to neurobiologists especially those studying molecular mechanisms of brain development and disease.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Autism spectrum disorder (ASD) is defined by common behavioral characteristics, raising the possibility of shared pathogenic mechanisms. Yet, vast clinical and etiological heterogeneity suggests personalized phenotypes. Surprisingly, our iPSC studies find that six individuals from two distinct ASD subtypes, idiopathic and 16p11.2 deletion, have common reductions in neural precursor cell (NPC) neurite outgrowth and migration even though whole genome sequencing demonstrates no genetic overlap between the datasets. To identify signaling differences that may contribute to these developmental defects, an unbiased phospho-(p)-proteome screen was performed. Surprisingly despite the genetic heterogeneity, hundreds of shared p-peptides were identified between autism subtypes including the mTOR pathway. mTOR signaling alterations were confirmed in all NPCs across both ASD subtypes, and mTOR modulation rescued ASD phenotypes and reproduced autism NPC-associated phenotypes in control NPCs. Thus, our studies demonstrate that genetically distinct ASD subtypes have common defects in neurite outgrowth and migration which are driven by the shared pathogenic mechanism of mTOR signaling dysregulation.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
“A sample size of 3 idiopathic seems underpowered relative to the many types of genetic changes that can occur in ASD. Since the authors carried out WGS, it would be useful to know what potential causative variants were found in these 3 individuals and even if not overlapping if they might expect to be in a similar biological pathway.
If the authors randomly selected 3 more idiopathic cell lines from individuals with autism, would these cell lines also have altered mTOR signaling? And could a line have the same cell biology defects without a change in mTOR signaling? The authors argue that the sample size could be the reason for lack of overlap of the proteomic changes (unlike the phosphor-proteomic overlaps), which makes the overlapping cell biology findings even more remarkable. Or is …
Author Response
Reviewer #1 (Public Review):
“A sample size of 3 idiopathic seems underpowered relative to the many types of genetic changes that can occur in ASD. Since the authors carried out WGS, it would be useful to know what potential causative variants were found in these 3 individuals and even if not overlapping if they might expect to be in a similar biological pathway.
If the authors randomly selected 3 more idiopathic cell lines from individuals with autism, would these cell lines also have altered mTOR signaling? And could a line have the same cell biology defects without a change in mTOR signaling? The authors argue that the sample size could be the reason for lack of overlap of the proteomic changes (unlike the phosphor-proteomic overlaps), which makes the overlapping cell biology findings even more remarkable. Or is the phenotyping simply too crude to know if the phenotypes truly are the same?”
We appreciate these thoughtful comments and also agree that of several models, our studies indicate the possibility of mTOR alteration in multiple forms of ASD. As above, we are currently pursuing this hypothesis with newly acquired DOD support. With regard to the I-ASD population, we agree that there are a large variety of genetic changes that can occur in genetically undefined ASDs. Indeed, this is precisely why we expected to see “personalized” phenotypes in each I-ASD individual when we embarked on this study. At that time, several years ago, we had planned to expand the analyses to more I-ASD individuals to assess for additional personalized phenotypes. However, as our studies progressed, we were surprised to find convergence in our I-ASD population in terms of neurite outgrowth and migration and later proteomic results showing convergence in mTOR. We found it particularly remarkable that despite a sample size of 3 that this convergence was noted. When we had the opportunity to extend our studies to the 16p11.2 deletion population, we were thrilled to conduct the first comparison between I-ASD and a genetically defined ASD and, as such, the scope of the paper turned towards this comparison. We do agree that analyses of the other I-ASD individuals would be a beneficial endeavor, both to understand how pervasive NPC migration and neurite deficits are in autism and to assess the presence of mTOR dysregulation. Furthermore, it would be important to see whether alterations in other pathways could also lead to similar cell biological deficits, though we know that other studies of neurodevelopmental disorders have found such cellular dysregulations without reporting concurrent mTOR dysregulation. Given our current grant funding to extend these analyses, such experiments within this manuscript would not be feasible.
Regarding the phenotyping methods used, we decided to assess neurite outgrowth and migration as they are both cytoskeleton dependent processes that are critical for neurodevelopment and are often regulated by the same genes. Furthermore, similar analyses have been applied to Fragile-X Syndrome, 22q11.2 deletion syndrome, and schizophrenia NPCs (Shcheglovitov A. et al., 2013; Mor-Shaked H. et al., 2016; Urbach A. et al., 2010; Kelley D. J. et al., 2008; Doers M. E. et al., 2014; Brennand K. et al., 2015; Lee I. S. et al., 2015; Marchetto M. C. et al., 2011). As such, it seems that multiple underlying etiologies can lead to similar dysregulated cellular phenotypes that can contribute to a variety of neurodevelopmental disorders. On a more global level, there are only a few different cellular functions a developing neuron can undergo, and these include processes such as proliferation, survival, migration, and differentiation. Thus, to understand neurodevelopmental disorders, it is important to study the more “crude” or “global” cellular functions occurring during neurodevelopment to determine whether they are disrupted in disorders such as ASD. In our studies we find that there are indeed dysregulations in many of these basic developmental processes, indicating that the typical steps that occur for normal brain cytoarchitecture may be disrupted in ASD. To understand why, we then further utilized molecular studies to “zoom” in on potential mechanisms which implicated common dysregulation in mTOR signaling as one driver for these common cellular phenotypes. As suggested, we did complete WGS on all the I-ASD individuals and did not see any overlapping genetic variants between the three I-ASD individuals as mentioned in our manuscript. The genetic data was published in a larger manuscript incorporating the data (Zhou A. et al., 2023). However, there were variants that were unique to each I-ASD individual which were not seen in their unaffected family members, and it is possible these variants could be contributing to the I-ASD phenotypes. We also utilized IPA to conduct pathway analysis on the WGS data utilizing the same approach we did in analysis of p- proteome and proteome data. From WGS data, we selected high read-quality variants that were found only in I-ASD individuals and had a functional impact on protein (ie excluding synonymous variants). The enriched pathways obtained from this data were strikingly different from the pathways we found in the p-proteome analysis and are now included in supplemental Figure 6 in the manuscript. Briefly, the top 5 enriched pathways were: O-linked glycosylation, MHC class 1 signaling, Interleukin signaling, Antigen presentation, and regulation of transcription.
Reviewer #2 (Public Review):
- I found that interpreting how differential EF sensitivity is connected to the rest of the story difficult at times. First, it is unclear why these extracellular factors were picked. These are seemingly different in nature (a neuropeptide, a growth factor and a neuromodulator) targeting largely different pathways. This limits the interpretation of the ASD subtype-specific rescue results. One way of reframing that could help is that these are pro-migratory factors instead of EFs broadly defined that fail to promote migration in I-ASD lines due to a shared malfunctioning of the intracellular migration machinery or cell-cell interactions (possibly through tight junction signaling, Fig S2A). Yet, this doesn't explain the migration/neurite phenotypes in 16p11 lines where EF sensitivity is not altered, overall implying that divergent EF sensitivity independent of underlying mTOR state. What is the proposed model that connects all three findings (divergent EF sensitivity based on ASD subtypes, 2 mTOR classes, convergent cellular phenotypes)?
We thank you for the kind assessment of our manuscript and for the thought-provoking questions posed. In terms of extracellular factors, for our study, we defined extracellular factor as any growth factor, amino acid, neurotransmitter, or neuropeptide found in the extracellular environment of the developing cells. The EFs utilized were selected due to their well-established role in regulation of early neurodevelopmental phenotypes, their expression during the “critical window” of mid-fetal development (as determined by Allan Brain Atlas), and in the case of 5-HT, its association with ASD (Abdulamir H. A. et al., 2018; Adamsen D. et al., 2014; Bonnin A. et al., 2011; Bonnin A. et al., 2007; Chen X. et al., 2015; El Marroun H. et al., 2014; Hammock E. et al., 2012; Yang C. J. et al., 2014; Dicicco-Bloom E. et al., 1998; Lu N. et al., 1998; Suh J. et al., 2001; Watanabe J. et al., 2016; Gilmore J. H. et al., 2003; Maisonpierre P. C. et al., 1990; Dincel N. et al., 2013; Levi- Montalcini R., 1987). Lastly, prior experiments in our lab with a mouse model of neurodevelopmental disorders, had shown atypical responses to EFs (IGF-1, FGF, PACAP). As such, when we first chose to use EFs in human NPCs we wanted to know 1) whether human NPCs even responded to these EFs, 2) whether EFs regulated neurite outgrowth and migration and 3) would there be a differential response in NPCs derived from those with ASD. Our studies were initiated on the I-ASD cohort and given the heterogeneity of ASD we had hypothesized we would get “personalized” neurite and migration phenotypes. Due to this reason, we also wanted to select multiple types of EFs that worked on different signaling pathways. Ultimately, instead of personalized phenotypes we found that all the I-ASD NPCs did not respond to any of the EFs tested whereas the 16p11.2 deletion NPCS did – this was therefore the only difference we found between these two “forms” of ASD. As noted, in I-ASD the lack of response to EFs can be ameliorated by modulating mTOR. However, in the 16p11.2 deletion, despite similar mTOR dysregulation as seen in I-ASD, there is no EF impairment. We do not have a cohesive model to explain why the 16pDel individuals differ from the I-ASD model other than to point to the p- proteomes which do show that the 16pDel NPCs are distinct from the I-ASD NPCs. It seems that mTOR alteration can contribute to impaired EF responsiveness in some NPCs but perhaps there is an additional defect that needs to be present in order for this defect to manifest, or that 16p11.2 deletion NPCs have specific compensatory features. For example, as noted in the thoughtful comment, the p-proteome canonical pathway analysis shows tight junction malfunction in I-ASD which is not present in the 16pDel NPCs and it could be the combination of mTOR dysregulation + dysregulated tight junction signaling that has led to lack of response to EFs in I-ASD. Regardless, we do not think the differences between two genetically distinct ASDs diminish the convergent mTOR results we have uncovered. That is, regardless of whatever defects are present in the ASD NPCs, we are able to rescue it with mTOR modulation which has fascinating implications for treatment and conceptualization for ASD. Lastly, we see our EF studies as an important inclusion as it shows that in some subtypes of ASD, lack of response to appropriate EFs could be contributing to neurodevelopmental abnormalities. Moreover, lack of response to these EFs could have implications for treatment of individuals with ASD (for example, SSRI are commonly used to treat co-morbid conditions in ASD but if an individual is unresponsive to 5- HT, perhaps this treatment is less effective). We have edited the manuscript to include an additional discussion section to address the EFs more thoroughly and have included a few extra sentences in the introduction as well!
- A similar bidirectional migration phenotype has been described in hiSPC-derived human cortical interneurons generated from individuals with Timothy Syndrome (Birey et al 2022, Cell Stem Cell). Here, authors show that the intracellular calcium influx that is excessive in Timothy Syndrome or pharmacologically dampened in controls results in similar migration phenotypes. Authors can consider referring to this report in support of the idea that bimodal perturbations of cardinal signaling pathways can converge upon common cellular migration deficits.
We thank you for pointing out the similar migration phenotype in the Timothy Syndrome paper and have now cited it in our manuscript. We have also expanded on the concept of “too much or too little” of a particular signaling mechanism leading to common outcomes.
Given that authors have access to 8 I-ASD hiPSC lines, it'd very informative to assay the mTOR state (e.g. pS6 westerns) in NPCs derived from all 8 lines instead of the 3 presented, even without assessing any additional cellular phenotypes, which authors have shown to be robust and consistent. This can help the readers better get a sense of the proportion of high mTOR vs low- mTOR classes in a larger cohort.
We have already addressed this in response to reviewer 1 and the essential revisions section, providing our reasoning for not expanding the study to all 8 I-ASD individuals.
- Does the mTOR modulation rescue EF-specific responses to migration as well (Figure 7)
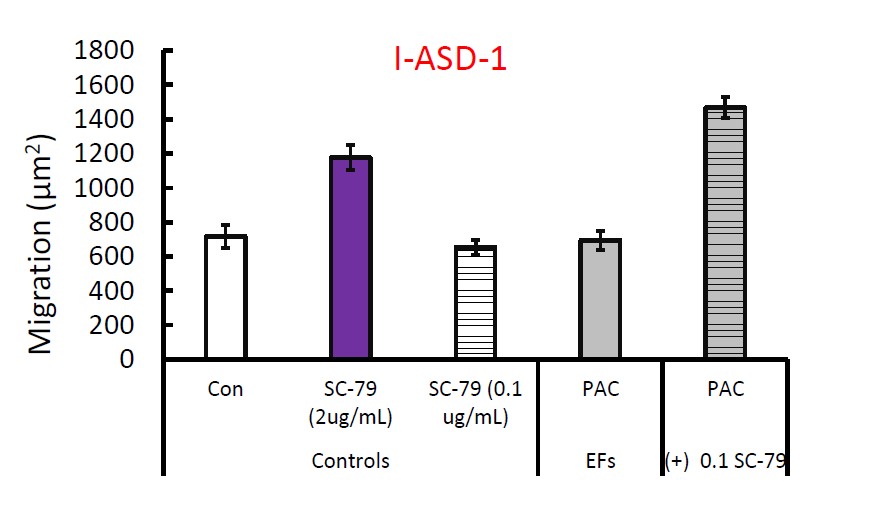
We did not conduct sufficient replicates of the rescue EF specific responses to migration due to the time consuming and resource intensive nature of the neurosphere experiments. Unlike the neurite experiments, the neurosphere experiments require significantly more cells, more time, selection of neurospheres based on a size criterion, and then manual trace measurements. We did one experiment in Family-1 where we utilized MK-2206 to abolish the response of Sib NPCs to PACAP. Likewise, adding SC-79 to I-ASD-1 neurospheres allowed for response to PACAP.
Author response image 1.
Author response image 2.
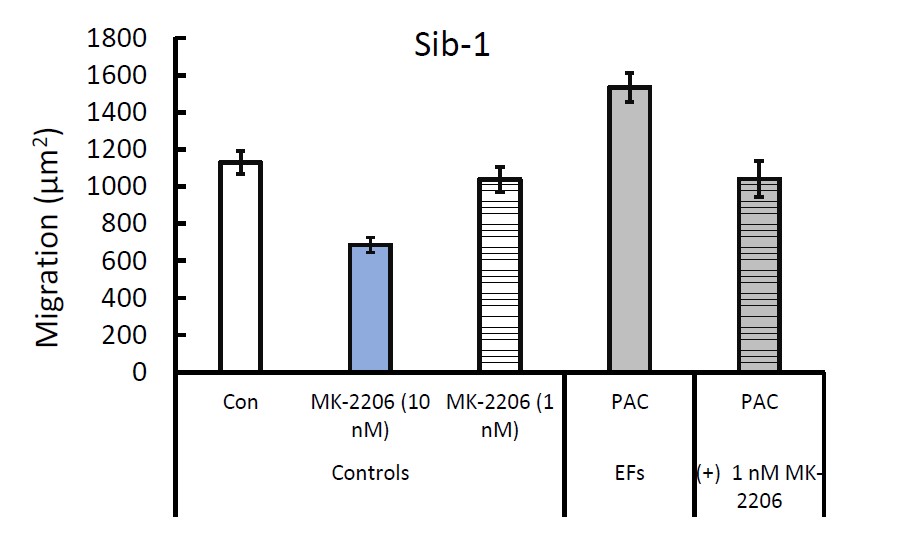
Reviewer #3: Public Review
We appreciate the kind, detailed and very thorough review you provided for us!
The results on the mTOR signaling pathway as a point of convergence in these particular ASD subtypes is interesting, but the discussion should address that this has been demonstrated for other autism syndromes, and in the present manuscript, there should be some recognition that other signaling pathways are also implicated as common factors between the ASD subtypes.
With regards to the mTOR pathway, we had included the other ASD syndromes in which mTOR dysregulation has been seen including tuberous sclerosis, Cowden Syndrome, NF-1, as well as Fragile-X, Angelman, Rett and Phelan McDermid in the final paragraph of the discussion section “mTOR Signaling as a Point of Convergence in ASD”. We have now expanded our discussion to include that other signaling pathways such as MAPK, cyclins, WNT, and reelin which have also been implicated as common factors between the ASD subtypes.
The conclusions of this paper are mostly well supported by data, but for the cell migration assay, it is not clear if the authors control for initial differences in the inner cell mass area of the neurospheres in control vs ASD samples, which would affect the measurement of migration.
Thank you for this thoughtful comment! When we first started our migration data, inner cell mass size was indeed a major concern for which we controlled in our methods. First, when plating the neurospheres, we would only collect spheres when a majority of spheres were approximately a diameter of 100 um. Very large spheres often could not be imaged due to being out of focus and very small spheres would often disperse when plated. Thus, there were some constraints to the variability of inner cell mass size.
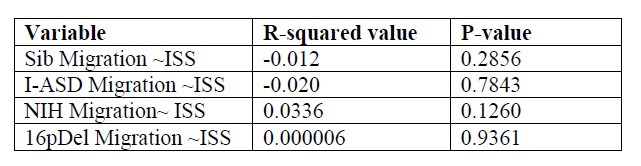
Furthermore, when we initially collected data, we conducted a proof of principal test to see if initial inner cell mass area (henceforth referred to as initial sphere size or ISS) influenced migration data. To do so, we obtained migration and ISS data from each diagnosis (Sib, NIH, I-ASD, 16pASD). Then we utilized R studio to see if there is a relationship between Migration and ISS in each diagnosis category using the equation (lm(Migration~ISS, data=bydiagnosis). In this equation, lm indicates linear modeling and (~) is a term used to ascertain the relationship between Migration and ISS and the term data=bydiagnosis allows the data to be organized by diagnosis
The results were expressed as R-squared values indicating the correlation between ISS and Migration for each diagnosis and the p-value showing statistical significance for each comparison. As shown in Author response table 1, for each data set, there is minimal correlation between Migration and ISS in each data set. Moreover, there are no statistically significant relationships between Migration and ISS indicating that initial sphere size DOES NOT influence migration data in any of our data-sets.
Author response table 1.
Lastly, utilizing R, we modeled what predicted migration would be like for Sib, NIH, I-ASD, and 16pASD if we accounted for ISS in each group. Raw migration data was then plotted against the predicted data as in Author response image 3.
Author response image 3.
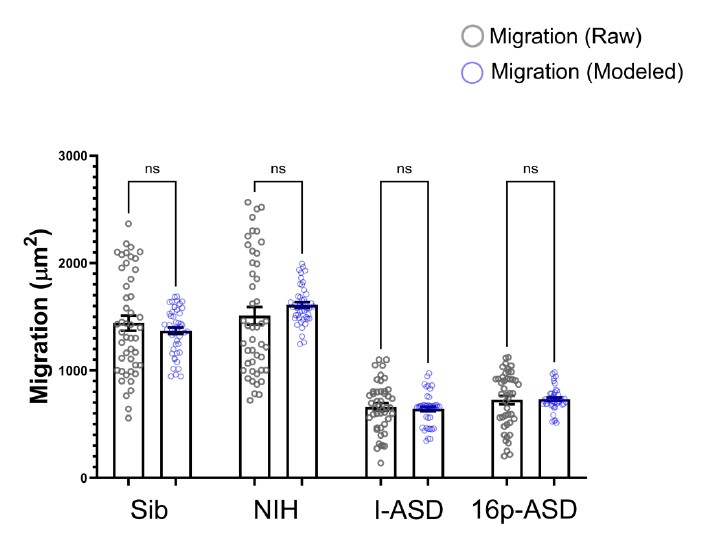
As shown in the graph, there are no statistical differences between the raw migration data (the data that we actually measured in the dish) and the modeled data in which ISS is accounted for as a variable. As such, we chose not to normalize to or account for ISS in our other experiments. We have now included the above R studio analyses in our supplemental figures (Figure S1) as well.
Also, in Fig 5 and 6, panels I and J omit the effects of drug on mTOR phosphorylation as shown for other conditions.
Both SC-79 and MK2206 were selected in our experiments after thorough analysis of their effects on human epithelial cells and other cultured cells (citations in manuscript). However, initially, we did not know whether either of these drugs would modulate the mTOR pathway in human NPCs, thus, in Figures 5A,5D, 6A and 6D we chose to focus on two of our data-sets to establish the effect of these drugs in human NPCs. Our experiments in Family-1 and Family-2 showed us that SC-79 increases PS6 in human NPCs while MK-2206 downregulates it. Once this was established, we knew the drugs would have similar effects in the NPCs from the other families. Thus, we only conducted a proof of principle test to confirm the drug does indeed have the intended effect in I-ASD-3 and 16pDel. We have included these proof of principle westerns in Figure 5I, 5K, 6I and 6K to show that the effects of these drugs are reproducible across all our NPC lines. We did not include quantification since the data is only from our single proof of principle western.
-
eLife assessment:
This important study describes converging cellular phenotypes in human neural progenitor cells derived from individuals with differing genetic forms of autism spectrum disorders. These convincing data demonstrate that altered mTOR signaling occurs in all cases of autism examined in the study, providing a common starting point for understanding the etiology of neuronal deficits in autism. The work will be of broad interest to neurobiologists especially those studying molecular mechanisms of brain development and disease.
-
Reviewer #1 (Public Review):
The manuscript by Prem and colleagues uses neural progenitor cells (NPCs) from individuals with different types of autism (either idiopathic or 16p11.2 deletion) to determine whether cells from these individuals show similar or varying phenotypes. An equal number of control cell lines are also used for a total of 6 autism compared to 6 control lines. The results are surprising in that the NPCs from individuals with autism have common cell biology defects (in neurite outgrowth and cell migration) yet have no overlapping genetic defects between the groups. Using proteomics, the authors also show converging biology at the level of the phospho-proteome with mTOR signaling being affected (albeit in differing directions depending on the idiopathic case). Finally, the authors use a number of pharmacological …
Reviewer #1 (Public Review):
The manuscript by Prem and colleagues uses neural progenitor cells (NPCs) from individuals with different types of autism (either idiopathic or 16p11.2 deletion) to determine whether cells from these individuals show similar or varying phenotypes. An equal number of control cell lines are also used for a total of 6 autism compared to 6 control lines. The results are surprising in that the NPCs from individuals with autism have common cell biology defects (in neurite outgrowth and cell migration) yet have no overlapping genetic defects between the groups. Using proteomics, the authors also show converging biology at the level of the phospho-proteome with mTOR signaling being affected (albeit in differing directions depending on the idiopathic case). Finally, the authors use a number of pharmacological approaches to show that the various cell biology phenotypes can be rescued or induced (in controls) through manipulation of mTOR signaling. In summary, it is a remarkable result that would seem to indicate that many forms of autism somehow alter mTOR signaling, at least in the NPC stage. I have only a handful of comments about the study:
A sample size of 3 idiopathic seems underpowered relative to the many types of genetic changes that can occur in ASD. Since the authors carried out WGS, it would be useful to know what potential causative variants were found in these 3 individuals and even if not overlapping if they might expect to be in a similar biological pathway. If the authors randomly selected 3 more idiopathic cell lines from individuals with autism, would these cell lines also have altered mTOR signaling? And could a line have the same cell biology defects without a change in mTOR signaling? The authors argue that the sample size could be the reason for lack of overlap of the proteomic changes (unlike the phosphor-proteomic overlaps), which makes the overlapping cell biology findings even more remarkable. Or is the phenotyping simply too crude to know if the phenotypes truly are the same?
-
Reviewer #2 (Public Review):
Convergence of cellular/molecular phenotypes across seemingly different types and subtypes of neurodevelopmental disorder represent an exciting frontier to identify effective drug targets. In this study, authors compared early neurodevelopmental processes, namely neurite outgrowth and migration, between a form of autism caused by copy number variation mutation in 16p11.2 (16pdel), which deletes 28 genes, to idopathic autism (I-ASD), in which genetic causes are unknown. All experiments involved neurospheres or neural precursor cells (NPCs) derived from human iPSCs. Three patients with I-ASD and their unaffected siblings as well as three patients with 16pdel were included in the study. Authors then go on to show perturbed migration and neurite length phenotypes shared between I-ASD and 16p11 NPCs. …
Reviewer #2 (Public Review):
Convergence of cellular/molecular phenotypes across seemingly different types and subtypes of neurodevelopmental disorder represent an exciting frontier to identify effective drug targets. In this study, authors compared early neurodevelopmental processes, namely neurite outgrowth and migration, between a form of autism caused by copy number variation mutation in 16p11.2 (16pdel), which deletes 28 genes, to idopathic autism (I-ASD), in which genetic causes are unknown. All experiments involved neurospheres or neural precursor cells (NPCs) derived from human iPSCs. Three patients with I-ASD and their unaffected siblings as well as three patients with 16pdel were included in the study. Authors then go on to show perturbed migration and neurite length phenotypes shared between I-ASD and 16p11 NPCs. Interestingly they identified two subclasses of I-ASD patients, either with high or low levels of mTOR, and show that change in mTOR level in either direction result in similar migration and neurite extension phenotypes, either at baseline or induced by pro-migratory factors. The study design is particularly strong and communication of sample structure throughout the manuscript is commendable. This study effectively points to the potential of patient-derived in vitro models to uncover novel molecular drivers of disease. I only have a few comments below:
(1) I found that interpreting how differential EF sensitivity is connected to the rest of the story difficult at times. First, it is unclear why these extracellular factors were picked. These are seemingly different in nature (a neuropeptide, a growth factor and a neuromodulator) targeting largely different pathways. This limits the interpretation of the ASD subtype-specific rescue results. One way of reframing that could help is that these are pro-migratory factors instead of EFs broadly defined that fail to promote migration in I-ASD lines due to a shared malfunctioning of the intracellular migration machinery or cell-cell interactions (possibly through tight junction signaling, Fig S2A). Yet, this doesn't explain the migration/neurite phenotypes in 16p11 lines where EF sensitivity is not altered, overall implying that divergent EF sensitivity independent of underlying mTOR state. What is the proposed model that connects all three findings (divergent EF sensitivity based on ASD subtypes, 2 mTOR classes, convergent cellular phenotypes)?
(2) A similar bidirectional migration phenotype has been described in hiSPC-derived human cortical interneurons generated from individuals with Timothy Syndrome (Birey et al 2022, Cell Stem Cell). Here, authors show that the intracellular calcium influx that is excessive in Timothy Syndrome or pharmacologically dampened in controls results in similar migration phenotypes. Authors can consider referring to this report in support of the idea that bimodal perturbations of cardinal signaling pathways can converge upon common cellular migration deficits.
(3) Given that authors have access to 8 I-ASD hiPSC lines, it'd very informative to assay the mTOR state (e.g. pS6 westerns) in NPCs derived from all 8 lines instead of the 3 presented, even without assessing any additional cellular phenotypes, which authors have shown to be robust and consistent. This can help the readers better get a sense of the proportion of high-mTOR vs low-mTOR classes in a larger cohort.
(4) Does the mTOR modulation rescue EF-specific responses to migration as well (Figure 7)?
-
Reviewer #3 (Public Review):
This study examines cellular deficits in human neural precursor cells derived from ASD individuals and unaffected controls. The ASD cases include idiopathic ASD individuals and 16p Deletion ASD individuals. These are rigorous studies employing multiple differentiations and multiple clones for all experiments. The authors report common deficits in neurite outgrowth and migration in these distinct ASD samples, and further demonstrate common deficits in downstream mTOR signaling. Outgrowth and migration deficits could be mimicked or rescued by manipulating mTOR signaling, further supporting a role for mTOR in these deficits. Differences in EF responsiveness identify ASD-subtype specific deficits, and the effects of subthreshold concentrations of EFs represent novel and interesting findings.
The results on the …
Reviewer #3 (Public Review):
This study examines cellular deficits in human neural precursor cells derived from ASD individuals and unaffected controls. The ASD cases include idiopathic ASD individuals and 16p Deletion ASD individuals. These are rigorous studies employing multiple differentiations and multiple clones for all experiments. The authors report common deficits in neurite outgrowth and migration in these distinct ASD samples, and further demonstrate common deficits in downstream mTOR signaling. Outgrowth and migration deficits could be mimicked or rescued by manipulating mTOR signaling, further supporting a role for mTOR in these deficits. Differences in EF responsiveness identify ASD-subtype specific deficits, and the effects of subthreshold concentrations of EFs represent novel and interesting findings.
The results on the mTOR signaling pathway as a point of convergence in these particular ASD subtypes is interesting, but the discussion should address that this has been demonstrated for other autism syndromes, and in the present manuscript, there should be some recognition that other signaling pathways are also implicated as common factors between the ASD subtypes.
The conclusions of this paper are mostly well supported by data, but for the cell migration assay, it is not clear if the authors control for initial differences in the inner cell mass area of the neurospheres in control vs ASD samples, which would affect the measurement of migration. Also, in Fig 5 and 6, panels I and J omit the effects of drug on mtTOR phosphorylation as shown for other conditions.
-
