Common genetic variations in telomere length genes and lung cancer
Curation statements for this article:-
Curated by eLife

eLife assessment
This study is of interest to epidemiologists and geneticists studying the association between telomere length and lung cancer risk. This work provides useful insight into risk factors for lung cancer. Overall, the results of this study are solid, as the genetic instrument used here is better powered and the battery of MR analysis makes this broad set of results convincing compared to previous work.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Background
Genome-wide association studies (GWAS) have identified genetic susceptibility variants for both leukocyte telomere length (LTL) and lung cancer susceptibility. Recently, 108 novel genetic loci within genes involved in telomere biology and DNA repair have been linked to LTL in UK Biobank. In the current work, we investigated the relationship between genetically predicted LTL and lung cancer.
Methods
To explore the shared genetic basis between LTL and lung cancer, we performed genetic correlation, Mendelian Randomization (MR), and colocalisation analyses using the largest available GWASs of LTL (N=464,716) and lung cancer (29,239 cases; 56,450 controls). To further characterize the molecular mechanisms underlying this relationship, principal component analysis (PCA) was used to summarize gene expression profiles in lung adenocarcinoma tumours from The Cancer Genome Atlas.
Results
Although there was no genome-wide genetic correlation between LTL and lung cancer risk (r g =-0.01, p=0.88), MR analyses using 144 instruments identified a putatively causal association. Longer LTL conferred an increased risk of lung cancer (OR=1.62, 95%CI=1.44-1.83, p=9.9×10 −15 ), lung cancer in never smokers (OR=2.02, 95%CI=1.45-2.83, p=3.78×10 −05 ), and lung adenocarcinoma (OR=2.43, 95%CI=2.02-2.92, p=3.8×10 −21 ). Of these 144 LTL genetic instruments, 12 showed evidence of colocalisation with lung adenocarcinoma risk and revealed novel susceptibility loci, including MPHOSPH6 (rs2303262), PRPF6 (rs80150989), and POLI (rs2276182). A polygenic risk score for LTL was associated with the second principal component (PC2) of gene expression (Beta=0.17, p=1.0×10 −3 ). The aspect of PC2 associated with longer LTL was also associated with being female (p=0.005), never smokers (p=0.04), and earlier tumour stage (p=0.002). PC2 was strongly associated with cell proliferation score (p=3.6×10 −30 ) and genomic features related to genome stability, including copy number changes (p=1.6×10 −5 ) and telomerase activity (p=1.3×10 −5 ) in the multivariate regression analyses.
Conclusions
This study identified an association between longer genetically predicted LTL and lung cancer and sheds light on the potential molecular mechanisms related to LTL in lung adenocarcinomas.
Article activity feed
-

Author Response
Reviewer #1 (Public Review):
This study used GWAS and RNAseq data of TCGA to show a link between telomere length and lung cancer. Authors identified novel susceptibility loci that are associated with lung adenocarcinoma risk. They showed that longer telomeres were associated with being a female nonsmoker and early-stage cancer with a signature of cell proliferation, genome stability, and telomerase activity.
Major comments:
- It is not clear how are the signatures captured by PC2 specific for lung adenocarcinoma compared to other lung subtypes. In other words, why is the association between long telomeres specific to lung adenocarcinoma?
We thank the reviewer for raising this point (similarly mentioned by reviewer #2). Indeed, it is unclear why genetically predicted LTL appears more relevant to lung adenocarcinoma. …
Author Response
Reviewer #1 (Public Review):
This study used GWAS and RNAseq data of TCGA to show a link between telomere length and lung cancer. Authors identified novel susceptibility loci that are associated with lung adenocarcinoma risk. They showed that longer telomeres were associated with being a female nonsmoker and early-stage cancer with a signature of cell proliferation, genome stability, and telomerase activity.
Major comments:
- It is not clear how are the signatures captured by PC2 specific for lung adenocarcinoma compared to other lung subtypes. In other words, why is the association between long telomeres specific to lung adenocarcinoma?
We thank the reviewer for raising this point (similarly mentioned by reviewer #2). Indeed, it is unclear why genetically predicted LTL appears more relevant to lung adenocarcinoma. We have used LASSO approach to select important features of PC2 in lung adenocarcinoma and inferred PC2 in lung squamous cell carcinomas tumours to better explore the differences between histological subtypes. The new results are presented in Figure 5, as well as being described in the methods and results sections. In addition, we have expanded upon this point in the discussion with the following paragraph (page 11, lines 229-248):
‘An explanation for why long LTL was associated with increased risk of lung cancer might be that individuals with longer telomeres have lower rates of telomere attrition compared to individuals with shorter telomeres. Given a very large population of histologically normal cells, even a very small difference in telomere attrition would change the probability that a given cell is able to escape the telomere-mediated cell death pathways (24). Such inter-individual differences could suffice to explain the modest lung cancer risk observed in our MR analyses. However, it is not clear why longer TL would be more relevant to lung adenocarcinoma compared to other lung cancer subtypes. A suggestion may come from our observation that longer LTL is related to genomic stable lung tumours (such as lung adenocarcinomas in never smokers and tumours with lower proliferation rates) but not genomic unstable lung tumours (such as heavy smoking related, highly proliferating lung squamous carcinomas). One possible hypothesis is that histologic normal cells exposed to highly genotoxic compounds, such as tobacco smoking, might require an intrinsic activation of telomere length maintenance at early steps of carcinogenesis that would allow them to survival, and therefore, genetic differences in telomere length are less relevant in these cells. By contrast, in more genomic stable lung tumours, where TL attrition rate is more modest, the hypothesis related to differences in TL length may be more relevant and potentially explaining the heterogeneity in genetic effects between lung tumours (Figure 2). Alternately, we also note that the cell of origin may also differ, with lung adenocarcinoma is postulated to be mostly derived from alveolar type 2 cells, the squamous cell carcinoma is from bronchiolar epithelium cells (19), possibly suggesting that LTL might be more relevant to the former.
- The manuscript is lacking specific comparisons of gene expression changes across lung cancer subtypes for identified genes such as telomerase etc since all the data is presented as associations embedded within PCs.
The genes associated with telomere maintenance such as TERT and TERC are very low expressed in these tumours (Barthel et al NG 2017). In this context, no sample has more than 5 normalised read counts by RNA-sequencing for TERT within TCGA lung cohorts (TCGA-LUSC, TCGA-LUAD). As such we have not explored the difference by individual telomere related genes. Nevertheless, we have explored an inferred telomerase activity gene signature, developed by Barthel et al and we did explore this in the context of lung adenocarcinoma tumours. We have added a note in the result section to inform the reader regarding why we did not directly test TERT/TERC expression (page 9, lines 184-187).
- It is not clear how novel are the findings given that most of these observations have been made previously i.e. the genetic component of the association between telomere length and cancer.
Others, including ourselves, have studied TL and lung cancer. We have built on that on the most updated TL genetic instrument and the largest lung cancer study available. In addition, we provided insights into the possible mechanisms in which telomere length might affect lung adenocarcinoma development. Using colocalisation analyses, we reported novel shared genetic loci between telomere length and lung adenocarcinoma (MPHOSPH6, PRPF6, and POLI), such genes/loci that have not previously linked to lung adenocarcinoma susceptibility. For MPHOSPH6 locus, we showed that the risk allele of rs2303262 (missense variant annotated for MPHOSPH6 gene) colocalized with increased lung adenocarcinoma risk, lower lung function (FEV1 and FVC), and increased MPHOSPH6 gene expression in lung, as highlighted in the discussion section of the revised manuscript.
In addition, we have used a PRS analysis to identify a gene expression component associated with genetically predicted telomere length in lung adenocarcinoma but not in squamous cell carcinoma subtype. The aspect of this gene expression component associated with longer telomere length are also associated with molecular characteristics related to genome stability (lower accumulation of DNA damage, copy number alterations, and lower proliferation rates), being female, early-stage tumours, and never smokers, which is an interesting but not completely understood lung cancer strata. As far as we are aware, this is the first time an association between a PRS related to an etiological factor, such as telomere length and a particular expression component in the tumour.
We have adjusted the discussion further highlight the novel aspects in the discussion section of the revised manuscript.
Reviewer #2 (Public Review):
The manuscript of Penha et al performs genetic correlation, Mendelian randomization (MR), and colocalization studies to determine the role of genetically determined leukocyte telomere length (LTL) and susceptibility to lung cancer. They develop an instrument from the most recent published association of LTL (Codd et al), which here is based on n=144 genetic variants, and the largest association study of lung cancer (including ~29K cases and ~56K controls). They observed no significant genetic correlation between LTL and lung cancer, in MR they observed a strong association that persisted after accounting for smoking status. They performed colocalization to identify a subset of loci where LTL and lung cancer risk coincided, mainly around TERT but also other loci. They also utilized RNA-Seq data from TCGA lung cancer adenocarcinoma, noting that a particular gene expression profile (identified by a PC analysis) seemed to correlate with LTL. This expression component was associated with some additional patient characteristics, genome stability, and telomerase activity.
In general, most of the MR analysis was performed reasonably (with some suggestions and comments below), it seems that most of this has been performed, and the major observations were made in previous work. That said, the instrument is better powered and some sub-analyses are performed, so adds further robustness to this observation. While perhaps beyond the scope here, the mechanism of why longer LTL is associated with (lung) cancer seems like one of the key observations and mechanistically interesting but nothing is added to the discussion on this point to clarify or refute previous speculations listed in the discussion mentioned here (or in other work they cite).
Some broad comments:
- The observations that lung adenocarcinoma carries the lion's share of risk from LTL (relative to other cancer subtypes) could be interesting but is not particularly highlighted. This could potentially be explored or discussed in more detail. Are there specific aspects of the biology of the substrata that could explain this (or lead to testable hypotheses?)
We thank the reviewer for these comments. A similar point was raised by reviewer #1. Please see our response above, as well as the additional analysis described in Figure 5 that considers the differences by histological subtype.
- Given that LTL is genetically correlated (and MR evidence suggests also possibly causal evidence in some cases) across a range of traits (e.g., adiposity) that may also associate with lung cancer, a larger genetic correlation analysis might be in order, followed by a larger set of multivariable MR (MVMR) beyond smoking as a risk factor. Basically, can the observed relationship be explained by another trait (beyond smoking)? For example, there is previous MR literature on adiposity measures, for example (BMI, WHR, or WHRadjBMI) and telomere length, plus literature on adiposity with lung cancer; furthermore, smoking with BMI. A bit more comprehensive set of MVMR analyses within this space would elevate the significance and interpretation compared to previous literature.
Indeed, there are important effects related to BMI and lung cancer (Zhou et al., 2021. Doi:10.1002/ijc.33292; Mariosa et al., 2022. Doi: 10.1093/jnci/djac061). We have tested the potential for influence on our finding using MVMR, modelling LTL and BMI using a BMI genetic instrument of 755 SNPs obtained from UKBB (feature code: ukb-b-19953). This multivariate approach did not result any meaningful changes in the associations between LTL and lung cancer risk.
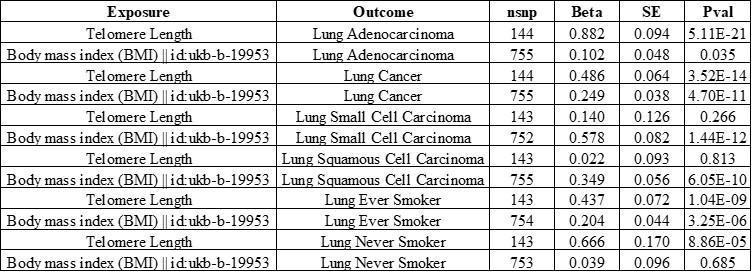
- In the initial LTL paper, the authors constructed an IV for MR analyses, which appears different than what the authors selected here. For example, Codd et al. proposed an n=130 SNP instrument from their n=193 sentinel variants, after filtering for LD (n=193 >>> n=147) and then for multi-trait association (n=147 >> n=130). I don't think this will fundamentally change the author's result, but the authors may want to confirm robustness to slightly different instrument selection procedures or explain why they favor their approach over the previous one.
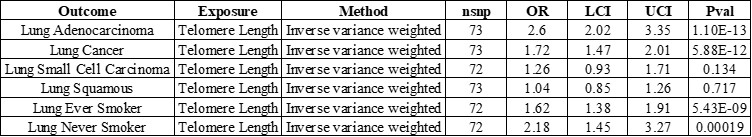
We appreciate the reviewer’s suggestion. Our study is designed for a Mendelian Randomization framework and chose to be conservative in the construction of our instrumental variable (IV). We therefore applied more stringent filters to the LTL variants relative to Codd et al’s approach. We applied a wider LD window (10MB vs. 1MB) centered around the LTL variants that were significant at genome-wide level (p<5e-08) and we restricted our analyses to biallelic common SNPs (MAF>1% and r2<0.01 in European population from 1000 genomes). Nevertheless, the LTL genetic instrument based on our study (144 LTL variants) is highly correlated with the PRS based on the 130 variants described by Codd et al. (correlation estimate=0.78, p<2.2e-16). The MR analyses based on the 130 LTL instrument described by Codd et al showed similar results to our study.
- Colocalization analysis suggests that a /subset/ of LTL signals map onto lung cancer signals. Does this mean that the MR relationships are driven entirely by this small subset, or is there evidence (polygenic) from other loci? Rather than do a "leave one out" the authors could stratify their instrument into "coloc +ve / coloc -ve" and redo the MR analyses.
Mainly here, the goal is to interpret if the subset of signals at the top (looks like n=14, the bump of non-trivial PP4 > 0.6, say) which map predominantly to TERT, TERC, and OBFC1 explain the observed effect here. I.e., it is biology around these specific mechanisms or generally LTL (polygenicity) but exemplified by extreme examples (TERT, etc.). I appreciate that statistical power is a consideration to keep in mind with interpretation.
We appreciate the reviewer’s comment and, indeed, we considered this idea. However, the analytical approach used the lung cancer GWAS to identify variants that colocalise. To validate this hypothesis that a subset of colocalised variants would be driving all the MR associations, we would need an independent lung cancer case control study to act as an out-of-sample validation set. This is not available to us at this point. Nevertheless, we slightly re-worded the discussion to highlight that the colocalised loci tend to be near genes related to telomere length biology and are also exploring the colocalisation approach to select variants for PRS analysis elsewhere.
-
eLife assessment
This study is of interest to epidemiologists and geneticists studying the association between telomere length and lung cancer risk. This work provides useful insight into risk factors for lung cancer. Overall, the results of this study are solid, as the genetic instrument used here is better powered and the battery of MR analysis makes this broad set of results convincing compared to previous work.
-
Reviewer #1 (Public Review):
This study used GWAS and RNAseq data of TCGA to show a link between telomere length and lung cancer. Authors identified novel susceptibility loci that are associated with lung adenocarcinoma risk. They showed that longer telomeres were associated with being a female nonsmoker and early-stage cancer with a signature of cell proliferation, genome stability, and telomerase activity.
Major comments:
1. It is not clear how are the signatures captured by PC2 specific for lung adenocarcinoma compared to other lung subtypes. In other words, why is the association between long telomeres specific to lung adenocarcinoma?
2. The manuscript is lacking specific comparisons of gene expression changes across lung cancer subtypes for identified genes such as telomerase etc since all the data is presented as associations …
Reviewer #1 (Public Review):
This study used GWAS and RNAseq data of TCGA to show a link between telomere length and lung cancer. Authors identified novel susceptibility loci that are associated with lung adenocarcinoma risk. They showed that longer telomeres were associated with being a female nonsmoker and early-stage cancer with a signature of cell proliferation, genome stability, and telomerase activity.
Major comments:
1. It is not clear how are the signatures captured by PC2 specific for lung adenocarcinoma compared to other lung subtypes. In other words, why is the association between long telomeres specific to lung adenocarcinoma?
2. The manuscript is lacking specific comparisons of gene expression changes across lung cancer subtypes for identified genes such as telomerase etc since all the data is presented as associations embedded within PCs.
3. It is not clear how novel are the findings given that most of these observations have been made previously i.e. the genetic component of the association between telomere length and cancer.
-
Reviewer #2 (Public Review):
The manuscript of Penha et al performs genetic correlation, Mendelian randomization (MR), and colocalization studies to determine the role of genetically determined leukocyte telomere length (LTL) and susceptibility to lung cancer. They develop an instrument from the most recent published association of LTL (Codd et al), which here is based on n=144 genetic variants, and the largest association study of lung cancer (including ~29K cases and ~56K controls). They observed no significant genetic correlation between LTL and lung cancer, in MR they observed a strong association that persisted after accounting for smoking status. They performed colocalization to identify a subset of loci where LTL and lung cancer risk coincided, mainly around TERT but also other loci. They also utilized RNA-Seq data from TCGA lung …
Reviewer #2 (Public Review):
The manuscript of Penha et al performs genetic correlation, Mendelian randomization (MR), and colocalization studies to determine the role of genetically determined leukocyte telomere length (LTL) and susceptibility to lung cancer. They develop an instrument from the most recent published association of LTL (Codd et al), which here is based on n=144 genetic variants, and the largest association study of lung cancer (including ~29K cases and ~56K controls). They observed no significant genetic correlation between LTL and lung cancer, in MR they observed a strong association that persisted after accounting for smoking status. They performed colocalization to identify a subset of loci where LTL and lung cancer risk coincided, mainly around TERT but also other loci. They also utilized RNA-Seq data from TCGA lung cancer adenocarcinoma, noting that a particular gene expression profile (identified by a PC analysis) seemed to correlate with LTL. This expression component was associated with some additional patient characteristics, genome stability, and telomerase activity.
In general, most of the MR analysis was performed reasonably (with some suggestions and comments below), it seems that most of this has been performed, and the major observations were made in previous work. That said, the instrument is better powered and some sub-analyses are performed, so adds further robustness to this observation. While perhaps beyond the scope here, the mechanism of why longer LTL is associated with (lung) cancer seems like one of the key observations and mechanistically interesting but nothing is added to the discussion on this point to clarify or refute previous speculations listed in the discussion mentioned here (or in other work they cite).
Some broad comments:
1. The observations that lung adenocarcinoma carries the lion's share of risk from LTL (relative to other cancer subtypes) could be interesting but is not particularly highlighted. This could potentially be explored or discussed in more detail. Are there specific aspects of the biology of the substrata that could explain this (or lead to testable hypotheses?)
2. Given that LTL is genetically correlated (and MR evidence suggests also possibly causal evidence in some cases) across a range of traits (e.g., adiposity) that may also associate with lung cancer, a larger genetic correlation analysis might be in order, followed by a larger set of multivariable MR (MVMR) beyond smoking as a risk factor. Basically, can the observed relationship be explained by another trait (beyond smoking)? For example, there is previous MR literature on adiposity measures, for example (BMI, WHR, or WHRadjBMI) and telomere length, plus literature on adiposity with lung cancer; furthermore, smoking with BMI. A bit more comprehensive set of MVMR analyses within this space would elevate the significance and interpretation compared to previous literature.
3. In the initial LTL paper, the authors constructed an IV for MR analyses, which appears different than what the authors selected here. For example, Codd et al. proposed an n=130 SNP instrument from their n=193 sentinel variants, after filtering for LD (n=193 >>> n=147) and then for multi-trait association (n=147 >> n=130). I don't think this will fundamentally change the author's result, but the authors may want to confirm robustness to slightly different instrument selection procedures or explain why they favor their approach over the previous one.
4. Colocalization analysis suggests that a /subset/ of LTL signals map onto lung cancer signals. Does this mean that the MR relationships are driven entirely by this small subset, or is there evidence (polygenic) from other loci? Rather than do a "leave one out" the authors could stratify their instrument into "coloc +ve / coloc -ve" and redo the MR analyses.
Mainly here, the goal is to interpret if the subset of signals at the top (looks like n=14, the bump of non-trivial PP4 > 0.6, say) which map predominantly to TERT, TERC, and OBFC1 explain the observed effect here. I.e., it is biology around these specific mechanisms or generally LTL (polygenicity) but exemplified by extreme examples (TERT, etc.). I appreciate that statistical power is a consideration to keep in mind with interpretation.
-
