Proteome-wide systems genetics identifies UFMylation as a regulator of skeletal muscle function
Curation statements for this article:-
Curated by eLife

eLife assessment
This manuscript will be of broad interest to those working in the genetics of complex diseases, with the results strongly supporting the author's primary claims. Overall, this is an important study that demonstrates the power of proteomics-based systems genetics studies in the mouse.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Improving muscle function has great potential to improve the quality of life. To identify novel regulators of skeletal muscle metabolism and function, we performed a proteomic analysis of gastrocnemius muscle from 73 genetically distinct inbred mouse strains, and integrated the data with previously acquired genomics and >300 molecular/phenotypic traits via quantitative trait loci mapping and correlation network analysis. These data identified thousands of associations between protein abundance and phenotypes and can be accessed online ( https://muscle.coffeeprot.com/ ) to identify regulators of muscle function. We used this resource to prioritize targets for a functional genomic screen in human bioengineered skeletal muscle. This identified several negative regulators of muscle function including UFC1, an E2 ligase for protein UFMylation. We show UFMylation is up-regulated in a mouse model of amyotrophic lateral sclerosis, a disease that involves muscle atrophy. Furthermore, in vivo knockdown of UFMylation increased contraction force, implicating its role as a negative regulator of skeletal muscle function.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
This is a well-executed study using cutting-edge proteomics analysis to characterize muscle tissue from a genetically diverse mouse population. The use of only females in the study is a serious limitation that the authors acknowledge. The statistical methods, including protein quantification, QTL mapping, and trait correlation analysis are appropriate and include corrections for multiple testing. One concern is that missense variants, if they occur in peptides used to quantify proteins, could lead to false-positive signatures of low abundance (see lines 123-127). The experimental validation and deep dive into UFMylation provide some confidence in the reliability of other associations that can be mined from these data. The authors have provided a web-based tool for exploring the data.
We …
Author Response
Reviewer #1 (Public Review):
This is a well-executed study using cutting-edge proteomics analysis to characterize muscle tissue from a genetically diverse mouse population. The use of only females in the study is a serious limitation that the authors acknowledge. The statistical methods, including protein quantification, QTL mapping, and trait correlation analysis are appropriate and include corrections for multiple testing. One concern is that missense variants, if they occur in peptides used to quantify proteins, could lead to false-positive signatures of low abundance (see lines 123-127). The experimental validation and deep dive into UFMylation provide some confidence in the reliability of other associations that can be mined from these data. The authors have provided a web-based tool for exploring the data.
We thank the reviewer for these very positive comments and for reviewing the manuscript.
We agree the quantification of peptides containing missense variants could confound quantification at the protein level. This is an important consideration when there are only a few peptides identified for a specific protein. However, in our data the average number of peptides used to quantify the 14 proteins containing missense-associated pQTLs was ~68 peptides/protein (lowest was 5 peptides for FGB and highest 703 peptides for NEB).
In the case of EPHX1, we quantified 15 peptides (Figure R1A). We identified a peptide adjacent to R338 spanning amino acids 339-347. As such, mutation of R338C would prevent trypsin from cleavage resulting in the missense peptide not being identified and may lead to false-positive signatures of low abundance as suggested by the reviewer. To investigate this, we re-quantified EPHX1 relative protein abundance with or without the peptide spanning 339-347 for each genotype (Figure R1B). This made little difference to protein quantification and EPHX1 abundance was still significantly lower following mutation of R338C (AA genotype). In fact, quantification at the peptide-level revealed 12 out of the remaining 14 peptides were also significantly lower in AA genotype (data not shown).
Although we agree this a very important consideration, we are mindful of the length of the article and feel including these data would not significantly improve the manuscript. We therefore request to not include these data as it would detract from the main findings of the paper focused on phenotypic associations and validation of UFMylation as a regulator of muscle function.
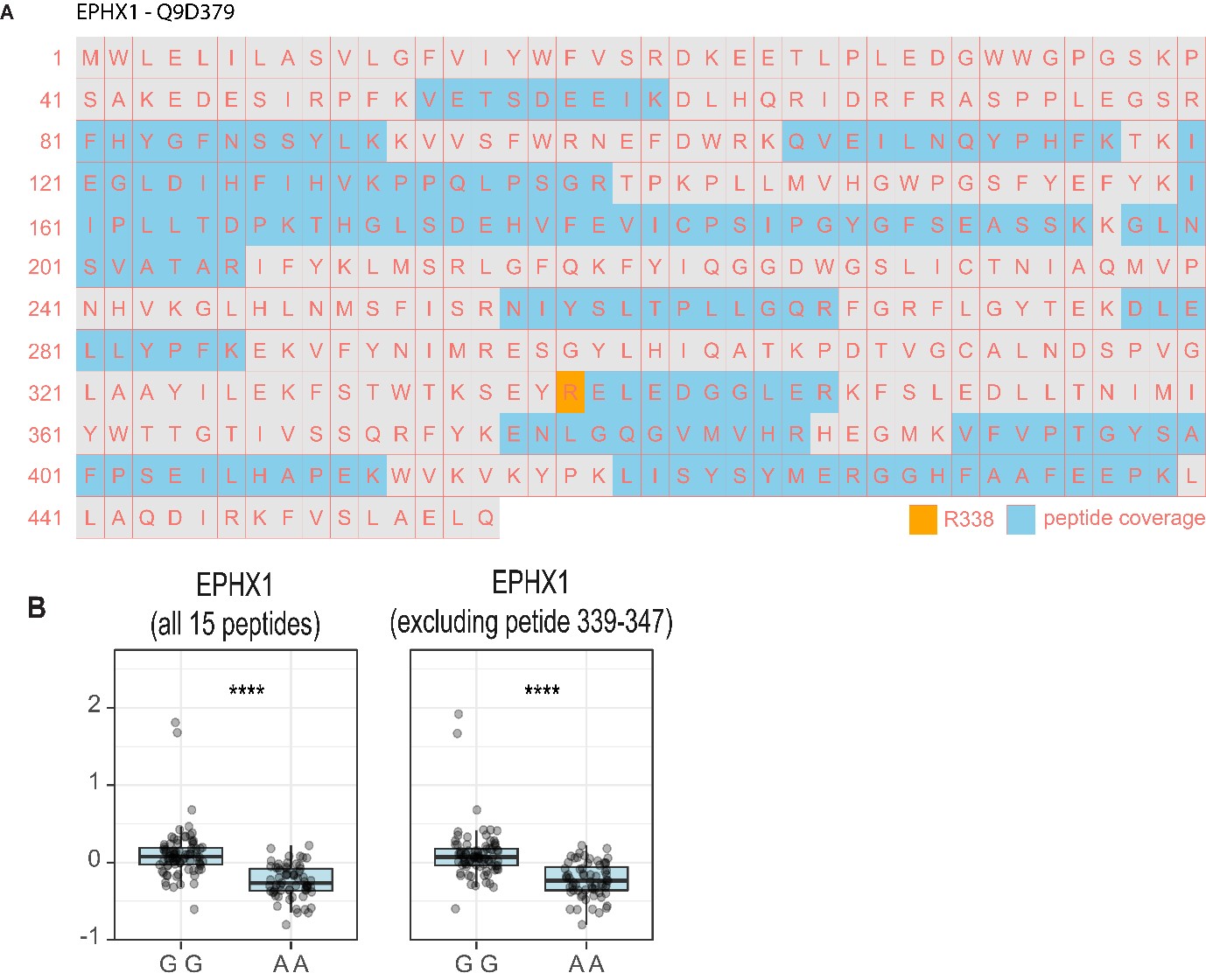
Figure 1R. (A) Identified peptides from EPHX1 mapped onto primary amino acid sequence highlighting the missense mutation induced by SNP rs32746574 that was associated to EPHX1 protein levels by pQTL analysis. (B) Relative quantification of EPHX1 between the two genotypes of SNP rs32746574 with and without the peptide neighboring the missense mutation (amino acids 339-347) (****p<0.001, students t-test)
-
eLife assessment
This manuscript will be of broad interest to those working in the genetics of complex diseases, with the results strongly supporting the author's primary claims. Overall, this is an important study that demonstrates the power of proteomics-based systems genetics studies in the mouse.
-
Reviewer #1 (Public Review):
This is a well-executed study using cutting-edge proteomics analysis to characterize muscle tissue from a genetically diverse mouse population. The use of only females in the study is a serious limitation that the authors acknowledge. The statistical methods, including protein quantification, QTL mapping, and trait correlation analysis are appropriate and include corrections for multiple testing. One concern is that missense variants, if they occur in peptides used to quantify proteins, could lead to false-positive signatures of low abundance (see lines 123-127). The experimental validation and deep dive into UFMylation provide some confidence in the reliability of other associations that can be mined from these data. The authors have provided a web-based tool for exploring the data.
-
Reviewer #2 (Public Review):
Molendijk et al. have performed muscle proteomics on a large population of genetically-variable mice - 161 mice from 73 different inbred strains out of the HMDP set, typically in duplicate but with a handful of strains with 3 or 4 biological replicates. Proteomics was run by TMT-based DDA, with around ~2000 proteins quantified in the entire cohort and an additional ~2000 quantified in at least 30% of the cohort. They have identified a few dozen of genes of interest that were detected through QTL mapping to be possibly associated with muscle phenotypes, of which a couple of dozen were designed to be targeted with AAVs and checked in vitro. Potentially two - and definitely one - of the knockdowns were successful in cell lines, and the successful one, on the gene UFC1, was tested in mice. The knockdown of UFC1 …
Reviewer #2 (Public Review):
Molendijk et al. have performed muscle proteomics on a large population of genetically-variable mice - 161 mice from 73 different inbred strains out of the HMDP set, typically in duplicate but with a handful of strains with 3 or 4 biological replicates. Proteomics was run by TMT-based DDA, with around ~2000 proteins quantified in the entire cohort and an additional ~2000 quantified in at least 30% of the cohort. They have identified a few dozen of genes of interest that were detected through QTL mapping to be possibly associated with muscle phenotypes, of which a couple of dozen were designed to be targeted with AAVs and checked in vitro. Potentially two - and definitely one - of the knockdowns were successful in cell lines, and the successful one, on the gene UFC1, was tested in mice. The knockdown of UFC1 in mice had a very striking phenotypic effect on muscle function, providing new insight into the physiology and opening new avenues of research for how this mechanism may work.
-
