Comparing the evolutionary dynamics of predominant SARS-CoV-2 virus lineages co-circulating in Mexico
Curation statements for this article:-
Curated by eLife

eLife assessment
The authors document an in-depth analysis of introduction patterns of 5 variant waves in Mexico. This is an important analysis and dataset since the genomic epidemiology of SARS-CoV-2 in Mexico is generally understudied, and this paper contributes important missing information. The phylogenetic analyses are solid and well-presented, but the lack of detail regarding the collection of samples across Mexican states makes it difficult to evaluate conclusions about the relationship between observed viral lineages and local case counts. Additionally, in its current form, the manuscript is mostly descriptive, without clear hypotheses tested or discussion of implications.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Over 200 different SARS-CoV-2 lineages have been observed in Mexico by November 2021. To investigate lineage replacement dynamics, we applied a phylodynamic approach and explored the evolutionary trajectories of five dominant lineages that circulated during the first year of local transmission. For most lineages, peaks in sampling frequencies coincided with different epidemiological waves of infection in Mexico. Lineages B.1.1.222 and B.1.1.519 exhibited similar dynamics, constituting clades that likely originated in Mexico and persisted for >12 months. Lineages B.1.1.7, P.1 and B.1.617.2 also displayed similar dynamics, characterized by multiple introduction events leading to a few successful extended local transmission chains that persisted for several months. For the largest B.1.617.2 clades, we further explored viral lineage movements across Mexico. Many clades were located within the south region of the country, suggesting that this area played a key role in the spread of SARS-CoV-2 in Mexico.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
Castelán-Sánchez et al. analyzed SARS-CoV-2 genomes from Mexico collected between February 2020 and November 2021. This period spans three major spikes in daily COVID-19 cases in Mexico and the rise of three distinct variants of concern (VOCs; B.1.1.7, P.1., and B.1.617.2). The authors perform careful phylogenetic analyses of these three VOCs, as well as two other lineages that rose to substantial frequency in Mexico, focusing on identifying periods of cryptic transmission (before the lineage was first detected) and introductions to and from the neighboring United States. The figures are well presented and described, and the results add to our understanding of SARS-CoV-2 in Mexico. However, I have some concerns and questions about sampling that could affect the results and conclusions. The …
Author Response
Reviewer #1 (Public Review):
Castelán-Sánchez et al. analyzed SARS-CoV-2 genomes from Mexico collected between February 2020 and November 2021. This period spans three major spikes in daily COVID-19 cases in Mexico and the rise of three distinct variants of concern (VOCs; B.1.1.7, P.1., and B.1.617.2). The authors perform careful phylogenetic analyses of these three VOCs, as well as two other lineages that rose to substantial frequency in Mexico, focusing on identifying periods of cryptic transmission (before the lineage was first detected) and introductions to and from the neighboring United States. The figures are well presented and described, and the results add to our understanding of SARS-CoV-2 in Mexico. However, I have some concerns and questions about sampling that could affect the results and conclusions. The authors do not provide any details on the distribution of samples across the various Mexican States, making it hard to evaluate several key conclusions. Although this information is provided in Supplementary Data 2, it is not presented in a way that enables the reader to evaluate if lineages were truly predominant in certain regions of the country, or if these results are attributable purely to sampling bias. Specifically, each lineage is said to be dominant in a particular state or region, but it was not clear to me if sampling across states was even at all-time points. For example, the authors state that most B.1.1.7 genome sampling is from the state of Chihuahua, but it is not clear if this was due to more sequenced samples from that region during the time that B.1.1.7 was circulating, or if the effects of B.1.1.7 were truly differential across the country. The authors do mention sequencing biases several times, but need to be more specific about the nature of this bias and how it could affect their conclusions. It is surprising to see in this manuscript that the B.1.1.7 lineage did not rise above 25% prevalence in the data presented, despite its rapid rise in prevalence in many other parts of the world. This calls into question if the presented frequencies of each lineage are truly representative of what was circulating in Mexico at the time, especially since the coordinated sampling and surveillance program across Mexico did not start until May 2021.
We thank the reviewer for the constructive comments. We recognize the need to better explain how the sequencing efforts in the country were set up and carried out, and this has now been clarified throughout the main text (L43-51, L95-105). A new figure comparing the overall cumulative proportion of genomes generated per state between 2020-2021 is now available as Supplementary Figure 1 c. The cumulative proportion of genomes sampled across states per lineage of interest, and corresponding to the period of circulation of the given lineage, were originally provided as maps in Figures 2-4. This has been further clarified in the Results section and in the corresponding figure legends. We also now provide additional maps representing the geographic distribution of the clades identified per lineage, integrating in the figures the information previously available in Supplementary Data 2, Supplementary Figures 4 and 5. As a note, for our analyses, we used the total cumulative genome data available from the country (and not only that generated by CoViGen-Mex, representing one third of the SARS-CoV-2 genomes from Mexico). This is expected to improve any sampling biases related to the scheme adopted by CoViGenMex, and is now clearly stated in the main text.
However, we believe that there has been a misunderstanding related to the genome sampling scheme adopted by CoViGen-Mex, as ‘coordinated sampling and surveillance program across Mexico did not start until May 2021’. Although it is true that further improvements were implemented after this date (enabling genome sampling and sequencing to become more homogenous across the country), the overall virus genome sequencing in Mexico was already sufficient from February 2021. This is represented by the cumulative number of viral genomes sequenced throughout 2020-2021 (both by CoViGen-Mex and other contributing institutions) correlating to the number of cases officially reported in the country during this time (see Supplementary Figure 1 a). This has now been clarified in the Results section (L94-105). Therefore, we hold that “SARS-CoV-2 sequencing in Mexico has been sufficient to explore the spatial and temporal frequency of viral lineages across national territory, and now to further investigate the number of lineage-specific introduction events, and to characterize the extension and geographic distribution of associated transmission chains, as we present in this study” (L102-105). In this context, “a more homogenous sampling across the country is unlikely to impact our main findings, but could i) help pinpoint additional clades we are currently unable to detect, ii) provide further details on the geographic distribution of clades across other regions of the country, and iii) deliver a higher resolution for the viral spread reconstructions we present” (discussed in L466-470).
For the B.1.1.7 lineage in Mexico, we have clarified the issue raised as follows: “during its circulation period, most B.1.1.7 genomes from Mexico were generated from the state of Chihuahua, with these representing the earliest B.1.1.7-assigned genomes from the country. However, our phylodynamic analysis revealed that only a small proportion of these grouped within a larger clade denoting an extended transmission chain (C2a), with the rest falling within minor clusters, or representing singleton events. Relative to other states, Chihuahua generated an overall lower proportion of viral genomes throughout 2020-2021. Thus, more viral genomes sequenced from a particular state does not necessarily translate into more well-supported clades denoting extended transmission chains, whilst the geographic distribution of clades is somewhat independent to the genome sampling across the country.” (L202-211). Again, these observations are supported by a sufficient overall genome sampling from Mexico.
We would further like to make clear that “our results confirm that the B.1.1.7 lineage reached an overall lower sampling frequency of up to 25% (relative to other virus lineages circulating in the country), as was noted prior to this study (for example, see Zárate et al. 2022)” (L189-193). As similar observations were independently made for other Latin American countries such as Brazil, Chile, and Peru (some with better genome representation than others, like Brazil https://www.gisaid.org/), it is possible that “the overall epidemiological dynamics of the B.1.1.7 in Latin America may have substantially differed from what was observed in the USA and UK. Such differences could be partly explained by competition between cocirculating lineages, exemplified in Mexico by the regional co-circulation of B.1.1.7, P.1 and B.1.1.519. Nonetheless, the lack of a representative number of viral genomes for most of these countries prevents exploring such hypothesis at a larger scale, and further highlights the need to strengthen genomic epidemiology-based surveillance across the region” (now discussed in L372-379). We hope the reviewer considers that the issues raised have now been resolved.
Reviewer #2 (Public Review):
The authors use a series of subsampling methods based on phylogenetic placement and geographic setting, informed by human movement data to control for differences in sampling of SARS-CoV-2 genomes across countries. Of note, the authors show that 2 variants likely arose in Mexico and spread via multiple introductions globally, while other variant waves were driven by repeat introductions into Mexico from elsewhere. Finally, they use human mobility data to assess the impact of movement on transmission within Mexico. Overall, the study is well done and provides nice data on an under-studied country. The authors take a thoughtful approach to subsampling and provide a very thorough analysis. Because of the care given to subsampling and the great challenge that proper subsampling represents for the field of phylodynamics, the paper would benefit from a more thorough exploration of how their migration-informed subsampling procedure impacts their results. This would not only help strengthen the findings of the paper, but would likely provide a useful reference for others doing similar studies. Additionally, I would suggest the authors provide a bit more discussion of this subsampling approach and how it may be useful to others in the discussion section of the paper.
We thank the reviewer for the constructive comments, and appreciate the recognition of our sub-sampling scheme as a valuable tool with potential application in other studies. We acknowledge the need for a ‘more thorough exploration and discussion of how a different migration-informed subsampling approach could impact our results’. To address this issue, “we further sought to validate our migration-informed genome subsampling scheme (applied to B.1.617.2+, representing the best sampled lineage in Mexico). For this, an independent dataset was built using a different migration sub-sampling approach, comprising all countries represented by B.1.617.2+ sequences deposited in GISAID (available up to November 30th 2021). In order to compare the number of introduction events, the new dataset was analysed independently under a time-scaled DTA (as described in Methods Section 4).” (L517-524). In the new dataset, <100 genome sequences from the USA were retained for further analysis (Supplementary Figure 2b), compared to approximately 2000 ‘USA’ genome sequences included in the original B.1.617.2+ alignment. Thus, we expected a lower number of inferred introduction events into Mexico, as an undersampling of viral genome sequences from the USA is likely to result in ‘Mexico’ clades not fully segregating (particularly impacting C5d).
Our original results revealed a minimum number of 142 introduction events into Mexico (95% HPD interval = [125-148]), with 6 clades identified as denoting extended transmission chains. The DTA results derived from the new dataset (subsampling all countries) revealed a minimum number of 84 introduction events into Mexico (95% HPD interval = [81-87]), with again 6 major clades identified. Thus, a significantly lower number of introduction events into Mexico were inferred, as was expected. On the other hand, the number of clades identified were consistent between both datasets, supporting for the robustness of our phylogenetic methodological approach. However, in the new dataset, we observe that C5d displayed a reduced diversity (represented by the AY.113 and AY.100 genomes from Mexico, but excluded the B.1.617.2 genome sampled from the USA). This highlights the relevance of our genome sub-sampling using migration data as a proxy.
In further agreement with these observations, publicly available data on global human mobility (https://migration-demography-tools.jrc.ec.europa.eu/data- hub/index.html?state=5d6005b30045242cabd750a2) shows that migration into Mexico is mostly represented by movements from the USA, followed by Indonesia, Guatemala, Belize and Colombia and Belize. However, the volume of movements from the USA into Mexico is much higher (up to 6 orders of magnitude above the volumes recorded into Mexico from any other country).
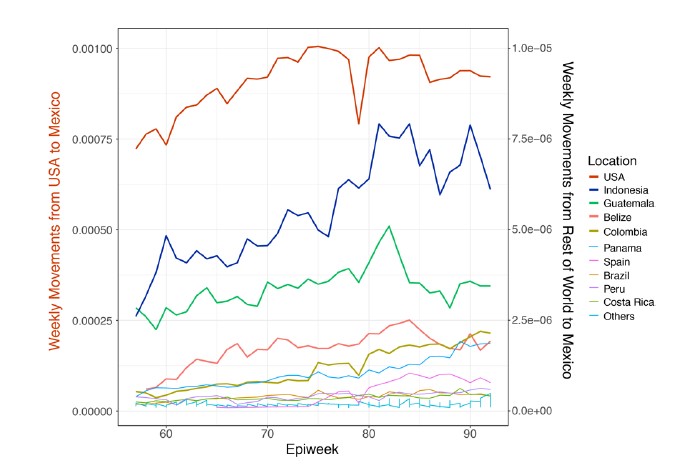
Given time constraints related to performing additional analyses, we decided to exclude the subsampling scheme for ‘top ten countries’ suggested by the reviewer. However, we consider that the results derived from the comparison between the original and the new dataset (top-5 vs all countries) is sufficient to support for our migration-informed subsampling approach. A full description of the methodology and the result obtained, as well as a short discussion, is now available as Supplementary Text 2, and Supplementary Figure 2b and 2c. We hope the reviewer considers that the issues raised has been addressed.
-
eLife assessment
The authors document an in-depth analysis of introduction patterns of 5 variant waves in Mexico. This is an important analysis and dataset since the genomic epidemiology of SARS-CoV-2 in Mexico is generally understudied, and this paper contributes important missing information. The phylogenetic analyses are solid and well-presented, but the lack of detail regarding the collection of samples across Mexican states makes it difficult to evaluate conclusions about the relationship between observed viral lineages and local case counts. Additionally, in its current form, the manuscript is mostly descriptive, without clear hypotheses tested or discussion of implications.
-
Reviewer #1 (Public Review):
Castelán-Sánchez et al. analyzed SARS-CoV-2 genomes from Mexico collected between February 2020 and November 2021. This period spans three major spikes in daily COVID-19 cases in Mexico and the rise of three distinct variants of concern (VOCs; B.1.1.7, P.1., and B.1.617.2). The authors perform careful phylogenetic analyses of these three VOCs, as well as two other lineages that rose to substantial frequency in Mexico, focusing on identifying periods of cryptic transmission (before the lineage was first detected) and introductions to and from the neighboring United States. The figures are well presented and described, and the results add to our understanding of SARS-CoV-2 in Mexico. However, I have some concerns and questions about sampling that could affect the results and conclusions:
The authors do not …
Reviewer #1 (Public Review):
Castelán-Sánchez et al. analyzed SARS-CoV-2 genomes from Mexico collected between February 2020 and November 2021. This period spans three major spikes in daily COVID-19 cases in Mexico and the rise of three distinct variants of concern (VOCs; B.1.1.7, P.1., and B.1.617.2). The authors perform careful phylogenetic analyses of these three VOCs, as well as two other lineages that rose to substantial frequency in Mexico, focusing on identifying periods of cryptic transmission (before the lineage was first detected) and introductions to and from the neighboring United States. The figures are well presented and described, and the results add to our understanding of SARS-CoV-2 in Mexico. However, I have some concerns and questions about sampling that could affect the results and conclusions:
The authors do not provide any details on the distribution of samples across the various Mexican States, making it hard to evaluate several key conclusions. Although this information is provided in Supplementary Data 2, it is not presented in a way that enables the reader to evaluate if lineages were truly predominant in certain regions of the country, or if these results are attributable purely to sampling bias. Specifically, each lineage is said to be dominant in a particular state or region, but it was not clear to me if sampling across states was even at all time points. For example, the authors state that most B.1.1.7 genome sampling is from the state of Chihuahua, but it is not clear if this was due to more sequenced samples from that region during the time that B.1.1.7 was circulating, or if the effects of B.1.1.7 were truly differential across the country. The authors do mention sequencing biases several times but need to be more specific about the nature of this bias and how it could affect their conclusions.
It is surprising to see in this manuscript that the B.1.1.7 lineage did not rise above 25% prevalence in the data presented, despite its rapid rise in prevalence in many other parts of the world. This calls into question if the presented frequencies of each lineage are truly representative of what was circulating in Mexico at the time, especially since the coordinated sampling and surveillance program across Mexico did not start until May 2021.
-
Reviewer #2 (Public Review):
The authors use a series of subsampling methods based on phylogenetic placement and geographic setting, informed by human movement data to control for differences in sampling of SARS-CoV-2 genomes across countries. Of note, the authors show that 2 variants likely arose in Mexico and spread via multiple introductions globally, while other variant waves were driven by repeat introductions into Mexico from elsewhere. Finally, they use human mobility data to assess the impact of movement on transmission within Mexico.
Overall, the study is well done and provides nice data on an under-studied country. The authors take a thoughtful approach to subsampling and provide a very thorough analysis. Because of the care given to subsampling and the great challenge that proper subsampling represents for the field of …
Reviewer #2 (Public Review):
The authors use a series of subsampling methods based on phylogenetic placement and geographic setting, informed by human movement data to control for differences in sampling of SARS-CoV-2 genomes across countries. Of note, the authors show that 2 variants likely arose in Mexico and spread via multiple introductions globally, while other variant waves were driven by repeat introductions into Mexico from elsewhere. Finally, they use human mobility data to assess the impact of movement on transmission within Mexico.
Overall, the study is well done and provides nice data on an under-studied country. The authors take a thoughtful approach to subsampling and provide a very thorough analysis. Because of the care given to subsampling and the great challenge that proper subsampling represents for the field of phylodynamics, the paper would benefit from a more thorough exploration of how their migration-informed subsampling procedure impacts their results. This would not only help strengthen the findings of the paper but would likely provide a useful reference for others doing similar studies. Additionally, I would suggest the authors provide a bit more discussion of this subsampling approach and how it may be useful to others in the discussion section of the paper.
-
