Interplay between VSD, pore, and membrane lipids in electromechanical coupling in HCN channels
Curation statements for this article:-
Curated by eLife

eLife assessment
Hyperpolarised-activated and Cyclic Nucleotide-gated (HCN) channels are the only mammalian channels to open under hyperpolarisation, being important for their roles in cardiac and neuronal cells. The authors of this study use atomistic simulations to enforce changing interaction distances that have been identified from a cryoEM structure and a homology model based on the hERG channel. The simulations suggest state-dependent interactions involving pore and voltage sensor helices, as well as with lipids, leading the authors to propose a domino-like mechanism of activation. These findings will be of considerable interest to the ion channel community.
This article has been Reviewed by the following groups
Discuss this preprint
Start a discussion What are Sciety discussions?Listed in
- Evaluated articles (eLife)
Abstract
Hyperpolarized-activated and cyclic nucleotide-gated (HCN) channels are the only members of the voltage-gated ion channel superfamily in mammals that open upon hyperpolarization, conferring them pacemaker properties that are instrumental for rhythmic firing of cardiac and neuronal cells. Activation of their voltage-sensor domains (VSD) upon hyperpolarization occurs through a downward movement of the S4 helix bearing the gating charges, which triggers a break in the alpha-helical hydrogen bonding pattern at the level of a conserved Serine residue. Previous structural and molecular simulation studies had however failed to capture pore opening that should be triggered by VSD activation, presumably because of a low VSD/pore electromechanical coupling efficiency and the limited timescales accessible to such techniques. Here, we have used advanced modeling strategies, including enhanced sampling molecular dynamics simulations exploiting comparisons between non-domain swapped voltage-gated ion channel structures trapped in closed and open states to trigger pore gating and characterize electromechanical coupling in HCN1. We propose that the coupling mechanism involves the reorganization of the interfaces between the VSD helices, in particular S4, and the pore-forming helices S5 and S6, subtly shifting the balance between hydrophobic and hydrophilic interactions in a ‘domino effect’ during activation and gating in this region. Remarkably, our simulations reveal state-dependent occupancy of lipid molecules at this emergent coupling interface, suggesting a key role of lipids in hyperpolarization-dependent gating. Our model provides a rationale for previous observations and a possible mechanism for regulation of HCN channels by the lipidic components of the membrane.
Article activity feed
-

-

Author Response
Reviewer #1 (Public Review):
HCN channels are atypically opened by the downward movement of gating charges during hyperpolarisation and have such weak coupling between the VSD and pore domain, and in the absence of an open state structure, extracting mechanistic information has been difficult. This manuscript is a continuation of a previous study on HCN channel gating that revealed how hyperpolarisation causes a downward movement of the VSD's S4, with breakage into two helices. The authors explore gating motions and the coupling between VSD and the pore domain using atomistic simulations. This includes microsecond MD with and without very strong -1V applied potentials to try to drive VSD-TMD changes to open the channel. In the end, however, the authors used a biased simulation approach (adiabatic bias) to enforce …
Author Response
Reviewer #1 (Public Review):
HCN channels are atypically opened by the downward movement of gating charges during hyperpolarisation and have such weak coupling between the VSD and pore domain, and in the absence of an open state structure, extracting mechanistic information has been difficult. This manuscript is a continuation of a previous study on HCN channel gating that revealed how hyperpolarisation causes a downward movement of the VSD's S4, with breakage into two helices. The authors explore gating motions and the coupling between VSD and the pore domain using atomistic simulations. This includes microsecond MD with and without very strong -1V applied potentials to try to drive VSD-TMD changes to open the channel. In the end, however, the authors used a biased simulation approach (adiabatic bias) to enforce conformational change from resting to an open homology model of HCN based on hERG/rEAG. This microsecond simulation followed three interaction distances that were suggested to change between resting and open states based on free MD. This simulation caused pore opening and allowed a description of changes that may occur during gating, including a competition of S5-S6 and S6-S6 contacts and lipid binding locations, which may suggest lipid-dependent function and explain an unexpected closed structure at 0mV in micelles. While I feel the manuscript is written for the HCN expert audience, the mechanistic information in terms of hyperpolarisation-induced voltage gating makes it of much interest. The manuscript is presented at a high level, though there are a couple of points to address, including reproducibility of simulations and potential for more relation to experimental findings.
We appreciate the comments, thank you, please find a detailed answer below.
The authors carried out 1μs-MD simulations of the resting, activated, and a Y289D mutant at 0 mV, and then tried to drive the conformational change with a very large -1V voltage (double that studied previously). In 1 us MD, is the membrane stable with such a big voltage, as it would likely not be experimentally? Even with a volt applied, there was incomplete activation of the voltage sensors, despite timescales approaching that of activation.
This reviewer is correct in cautioning against membrane rupturing effects in simulations with a voltage of this magnitude. We have indeed checked that the membrane and the protein remains intact under these conditions and can confirm that no poration occurs. As membrane poration is stochastic, it could indeed occur over microsecond timescales under 1V, but the probability remains low, and we were lucky to not face this situation herein. Note that whereas potentials of this magnitude could not be applied in experiments, they are relatively routinely used in MD simulations to speed up processes that are driven by changes in transmembrane potentials.
Interestingly, other work from our lab (Rems et al. Biophysical Journal 119 (1) 190-205 (2020)) has shown that HCN1 voltage sensor domains are less prone to poration than those from other voltage sensor domains, for reasons that remain to be determined.
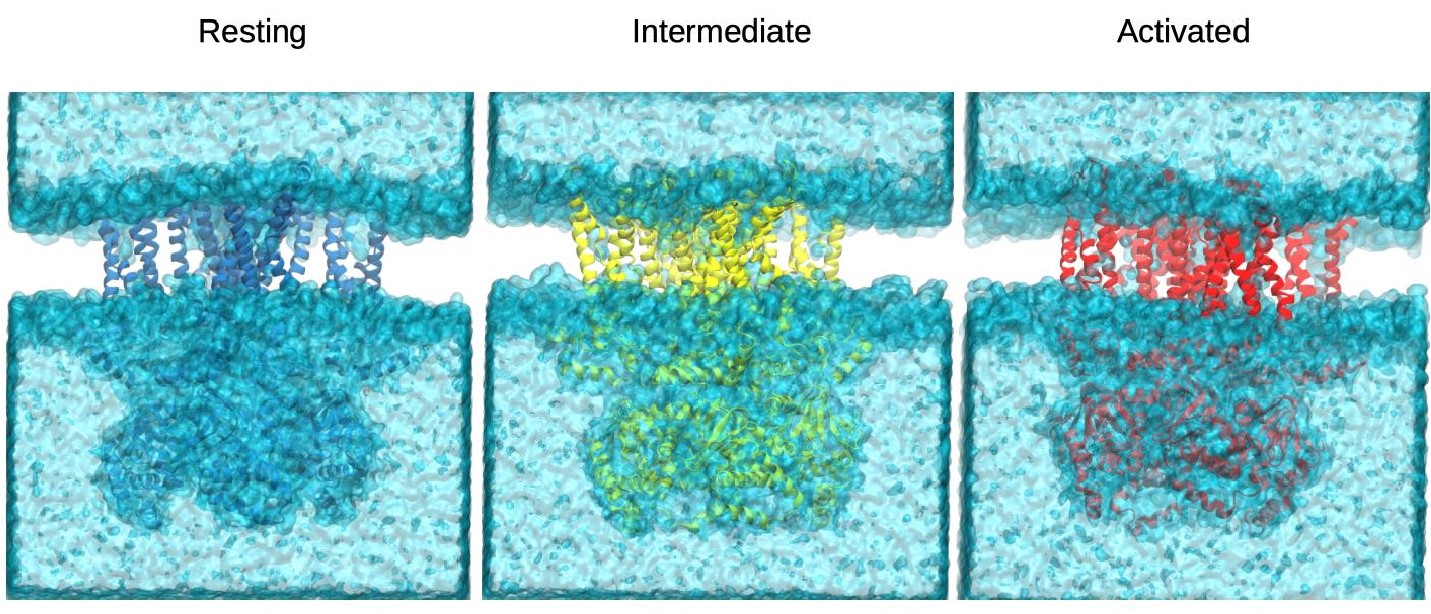
Author Response Figure 1. Final snapshots from the simulations of the resting (blue), intermediate (yellow) and activated (red) states. The representation of the solvent (water+ions) in cyan showed no membrane poration at the end of the 1us simulations.
For the pulling/ driving simulations (adiabatic bias MD) to change suspected interaction distances (V390-I302, N300-W281, and D290-K412), it seems to be just 1 simulation, without reproducibility. One has to wonder, if the simulation was redone from a very different initial conformation, would the results be the same (in addition to the distances themselves that were enforced by the ABMD). Moreover, the authors had to model the open state, such that the results depend on a homology model based on other CNBD channels, hERG / rEAG. Although the model stayed open for a microsecond, what other measures of accuracy of the homology model are there, such as preserved distances according to mutants/double mutants?
The ABMD simulations were repeated, please refer to the response to essential revisions point 1 for details.
For reasons mentioned by the reviewer as well as a reconsideration of our strategy to model channel opening, we have decided to omit homology models from the revised version of the paper.
The authors find that activation involves hydrophobic forces that strengthen the intra-subunit S4/S5/S6 interface, as well as lipid headgroups that make contact with hydrophilic residues at this interface, with lipid tails also contributing to hydrophobic contacts. The authors see bending and rotation of the lower S4 and a displacement of S1 away from S4 that exposes the VSD-pore interface to lipids, with increased lipid contacts at S4 and S5 during activation. This indicates lipid tails may play a role in coupling in HCN1 and may explain the closed state micelle structure at 0mV. Two sites of lipid contact are identified, one engaging VSD residues and the other polar or charged residues on S5 and S6. No experiments are presented or proposed to test the predicted lipid sites. e.g. Mutation of key residues, such as the arginine and histidine seen binding lipid headgroups could be tested as proof of their involvement, or perhaps experiments with varied phosphate moieties? In the absence of new experiments, is there existing data that could help validate the findings?
We thank this reviewer for this comment. As noted in the response to essential revisions point 3, such experiments are challenging, and have not been reported so far in HCN channels. We do agree that aspects of the mechanism we propose remain hypothetical awaiting further work, but are happy to report that importance of lipid interactions with the crucial salt bridge pair mentioned in the response to essential revisions point 3 has been completely independently validated, thus strengthening our mechanistic hypothesis substantially.
During free MD simulation, the authors see tilting of S5 caused by activation of the Y289D mutation that brings D290 and K412 positions into proximity. How do we know that the adjacent mutant of Y289 to aspartate has not caused this, or was this interaction also seen in wild-type simulation? Fig.3c might suggest the wt activated simulation may see such an interaction, but it is unclear given the large C_alpha distances, as opposed to H-bonding distances.
Indeed, Figure 3 appears to indicate that this interaction between D290 and K412 is present in the activated state when the mutation is reverted to the WT sequence. We have recalculated the interaction propensity using all atoms of the residues and present an updated Figure 3c in response.
The authors predict that a D290-K412 salt bridge may be important for gating and sought to experimentally validate the interaction in the activated-open state using cysteine cross-bridging. As this is the only experimental backing in the paper, it is important to be able to judge its ability to report on the D290-K412 salt bridge. A comparison experiment demonstrating other crosslinks that do not favour the open state would have been helpful in this regard e.g. if crossbridging at similar locations (but not predicted to change interaction during gating) had little effect on I/Imax, then the result may be bolstered. Are there existing mutagenesis experiments that may suggest the importance of these residues (as well as for other key interaction distances identified)?
Negative results in cross bridging and cysteine accessibility studies in general are difficult to interpret as the lack of a cadmium-specific effect may be due to inaccessibility of the site to cadmium, pairwise distance too far to bridge by cadmium, or bridging or the specified site without a functional effect. However, as reviewer 2 pointed out below, the Yellen group has performed extensive cross bridging experiments in the S4-S5 to Clinker region in spHCN and in most of these positions, the pairs favoring the open state are closer together in our models than pairs favoring the closed state or those without functional effect. We have added Videos 1-6 to highlight this comparison on our open state models and describe in our updated discussion section.
Rotation of the V390 side chain from a position facing the pore lumen to a position facing I302 on S5 is coupled to an increase of the pore radius at V390, an increased hydration of the pore intracellular gate, and K+ ion movement. Perhaps 5 or 6 ions cross in that single simulation. As K channel ion permeation can depend critically on starting ion configs (as well as the model/force field), reproducibility of this finding is important but does not appear to have been tested. How can we be sure that periods of permeation or no permeation in individual simulations are reliable?
As mentioned in our response to essential revisions point 1, we have modified the collective variable set used in ABMD, and repeated the simulations in 4 replicates. Whereas the number of permeation events is low in each simulation (Figure 4 S1), the consistency across repeats indicates that these open pore models indeed represent conductive states. Given how short the simulations are, however, it appears unreasonable to infer conductance values from these observations.
Reviewer #3 (Public Review):
In this work, Elbahnsi and colleagues use enhanced sampling MD simulation, to recapitulate step by step, the electromechanical coupling between VSD and the pore in HCN1 channels. Building on the available cryoEM structures of HCN1 with the VSD in resting and active state, the authors characterize by MD a subset of interactions that seemingly stabilize the open channel. This subset is, in turn, used in enhanced-sampling simulations to guide channel opening. The main findings are that S4 movement induces a rearrangement of the hydrophobic interaction at the level of S1- S4- and S5 interfaces. Occupancy of lipids seems therefore statedependent and highlights their regulatory role in HCN gating.
The approach is rather innovative, and it apparently allows the reconstruction of the whole mechanism of gating, pushing the predictive power of MD simulation well beyond its actual temporal limitations. At the same time, the initial choice of interactions is crucial for this approach, because the result cannot differ from the inputs. And reading the paper it does not emerge clearly how the correctness of the reconstructed gating pathway can be verified, if not by functional validation.
We thank the reviewer for this thoughtful review. It has pushed us to reconsider our approach to enhance the sampling of channel activation and gating. Please refer to the detailed response below as well as the response in particular to essential revisions point 1.
Here are my comments on the main interactions that were used to feed the final MD simulation:
- W281-N300: this interaction, previously identified and studied in SpH channels (Ramentol et al, 2020; Wu et al, 2021), has been elegantly confirmed in this paper. Its inclusion in the initial subset seems appropriate. In the other two cases, the choice of interactions requires further explanations and experimental validation.
- D290 and K412: the validation of this interaction shown in Figure 3 and suppl Figure 1 is missing a control, i.e., the effect of the addition of Cd++ on the wt channel. Please add.
We have performed the control suggested. Please also refer to the answer to essential revisions point 2.
- Modelling the open state of HCN1 pore (page 18), is done on the structure of the distantly related hERG rather than on the available open pore structure of HCN4. This choice is justified as follows by the authors:
a) "Available structures in the CNBD channel family for which representative structures have been solved in closed and open states".
b) "The structural mechanism of pore gating (i.e. the ⍺ to 𝜋 helix occurring at the glycine657 hinge in hERG) observed in rEAG/hERG may be a conserved gating transition in the CNBD family of channels"
I encourage the authors to consider the following:
a) The structure of hERG channel is not available in the closed/open configuration, indeed the comparison must be done with the closed configuration of the related channel rEAG. On the contrary, HCN4 is available in the closed/open configurations. Moreover, one of the open pore structures shows S4-S5-S6 in a very similar conformation to the lock open mutant (F186C/S264C) of HCN1 (Saponaro et al, 2021). With an available HCN4 open structure, forcing HCN1 to the open pore structure of hERG channel (which opens in depolarization and is not regulated by cAMP) seems not necessary.
In response to this point, we reconsidered our approach and chose to instead use a biasing distance that is consistently increased in CNBD channels of resolved structures, that between neighboring and cross-subunits V390. We have detailed our rationale in the response to essential revisions point 1.
To my knowledge, hERG is the only channel of the CNBD family for which the transition ⍺ to 𝜋 helix reported by the Authors, occurs in S6. It is not reported for other CNBD family members, in particular for the CNG channels mentioned by the Authors (Zheng et al., 2020; Xue et al., 2021, 2022). Task 4 (Zheng et al) does not show it. Its pore opens by a right-handed twist of S6 at glycine 399, a conserved glycine in all CNG. Human CNGA1 too, opens the pore by a rotational movement of S6 hinged at the equivalent glycine (glycine 385) (Xue et al, 2021). And the same occurs in the non-symmetrical channel CNGA1/B1 (Xue te al, 2022). So, it seems that CNG channels do not show the ⍺ to 𝜋 helix transition in the open pore. Moreover, hERG excluded, all other members of the CNBD family, CNG, EAG, and HCN4 included, do not bend at the hinge glycine 657 of hERG, but at another glycine (gly 648 in hERG numbering) located upstream. Further, their opening is due to a rotation of S6 associated with an outward movement, rather than to the lifting of the lower part of S6, as in hERG.
After considering this reviewer’s comment, we were surprised to see that HCN1 is apparently prone to secondary structure deformation in S6, even when biasing the aforementioned distances, and thus enforcing no rotation at all in S6. We are intrigued by this observation and eagerly await experimental validation or disproval.
In the meantime, we have made clear in the text that this hypothesis remains based exclusively on modeling work.- V390-I302: this interaction is predicted to stabilize the open pore configuration and was included in the subset. The contact between V390 on S6 and I302 on S5 is observed in the homology model discussed above when the S6 is twisted at the glycine hinge, rotating the preceding residue (V390) out of its pore-lining position and is.
Again, I can only disagree with this hypothesis because it has been experimentally demonstrated (Cheng et al, J Pharmacol Exp Ther. 2007 Sep;322(3):931-9) that the side chain of Valine390 is inside the cavity of the open pore of HCN1 channels as it controls the affinity for the pore blocker ZD7288.
In accordance with other comments above, we have eliminated the bias applied to the V390I302 distance. However, the new ABMD simulations with bias applied to encourage dilation at position 390 still involve rotation of V390 away from the central pore axis, albeit with bending of S6 at the upper glycine mentioned by this reviewer. The degree of rotation is lower than in our previous simulations so that V390 still lines the inner vestibule in the open state, consistent with the observation that this position influences the apparent affinity of open pore blockers.
In conclusion, modelling the open state pore of HCN1 on hERG rather than on that of HCN4 seems not justified based on accumulated evidence in the published literature. Therefore, the choice of the authors to use it as the open pore model of HCN1 channels needs to be experimentally validated. One possibility is to mutate the glycine hinge, gly391 in HCN1, into an Alanine in order to remove the flexible hinge. If this mutation alters pore gating, it will support the choice of the Authors.
Once more, we thank the reviewer for the comments, which have led us to reconsider a larg part of our modeling work.
-
eLife assessment
Hyperpolarised-activated and Cyclic Nucleotide-gated (HCN) channels are the only mammalian channels to open under hyperpolarisation, being important for their roles in cardiac and neuronal cells. The authors of this study use atomistic simulations to enforce changing interaction distances that have been identified from a cryoEM structure and a homology model based on the hERG channel. The simulations suggest state-dependent interactions involving pore and voltage sensor helices, as well as with lipids, leading the authors to propose a domino-like mechanism of activation. These findings will be of considerable interest to the ion channel community.
-
Reviewer #1 (Public Review):
HCN channels are atypically opened by the downward movement of gating charges during hyperpolarisation and have such weak coupling between the VSD and pore domain, and in the absence of an open state structure, extracting mechanistic information has been difficult. This manuscript is a continuation of a previous study on HCN channel gating that revealed how hyperpolarisation causes a downward movement of the VSD's S4, with breakage into two helices. The authors explore gating motions and the coupling between VSD and the pore domain using atomistic simulations. This includes microsecond MD with and without very strong -1V applied potentials to try to drive VSD-TMD changes to open the channel. In the end, however, the authors used a biased simulation approach (adiabatic bias) to enforce conformational change …
Reviewer #1 (Public Review):
HCN channels are atypically opened by the downward movement of gating charges during hyperpolarisation and have such weak coupling between the VSD and pore domain, and in the absence of an open state structure, extracting mechanistic information has been difficult. This manuscript is a continuation of a previous study on HCN channel gating that revealed how hyperpolarisation causes a downward movement of the VSD's S4, with breakage into two helices. The authors explore gating motions and the coupling between VSD and the pore domain using atomistic simulations. This includes microsecond MD with and without very strong -1V applied potentials to try to drive VSD-TMD changes to open the channel. In the end, however, the authors used a biased simulation approach (adiabatic bias) to enforce conformational change from resting to an open homology model of HCN based on hERG/rEAG. This microsecond simulation followed three interaction distances that were suggested to change between resting and open states based on free MD. This simulation caused pore opening and allowed a description of changes that may occur during gating, including a competition of S5-S6 and S6-S6 contacts and lipid binding locations, which may suggest lipid-dependent function and explain an unexpected closed structure at 0mV in micelles. While I feel the manuscript is written for the HCN expert audience, the mechanistic information in terms of hyperpolarisation-induced voltage gating makes it of much interest. The manuscript is presented at a high level, though there are a couple of points to address, including reproducibility of simulations and potential for more relation to experimental findings.
The authors carried out 1μs-MD simulations of the resting, activated, and a Y289D mutant at 0 mV, and then tried to drive the conformational change with a very large -1V voltage (double that studied previously). In 1 us MD, is the membrane stable with such a big voltage, as it would likely not be experimentally? Even with a volt applied, there was incomplete activation of the voltage sensors, despite timescales approaching that of activation. For the pulling/ driving simulations (adiabatic bias MD) to change suspected interaction distances (V390-I302, N300-W281, and D290-K412), it seems to be just 1 simulation, without reproducibility. One has to wonder, if the simulation was redone from a very different initial conformation, would the results be the same (in addition to the distances themselves that were enforced by the ABMD). Moreover, the authors had to model the open state, such that the results depend on a homology model based on other CNBD channels, hERG / rEAG. Although the model stayed open for a microsecond, what other measures of accuracy of the homology model are there, such as preserved distances according to mutants/double mutants?
The authors find that activation involves hydrophobic forces that strengthen the intra-subunit S4/S5/S6 interface, as well as lipid headgroups that make contact with hydrophilic residues at this interface, with lipid tails also contributing to hydrophobic contacts. The authors see bending and rotation of the lower S4 and a displacement of S1 away from S4 that exposes the VSD-pore interface to lipids, with increased lipid contacts at S4 and S5 during activation. This indicates lipid tails may play a role in coupling in HCN1 and may explain the closed state micelle structure at 0mV. Two sites of lipid contact are identified, one engaging VSD residues and the other polar or charged residues on S5 and S6. No experiments are presented or proposed to test the predicted lipid sites. e.g. Mutation of key residues, such as the arginine and histidine seen binding lipid headgroups could be tested as proof of their involvement, or perhaps experiments with varied phosphate moieties? In the absence of new experiments, is there existing data that could help validate the findings?
During free MD simulation, the authors see tilting of S5 caused by activation of the Y289D mutation that brings D290 and K412 positions into proximity. How do we know that the adjacent mutant of Y289 to aspartate has not caused this, or was this interaction also seen in wild-type simulation? Fig.3c might suggest the wt activated simulation may see such an interaction, but it is unclear given the large C_alpha distances, as opposed to H-bonding distances.
The authors predict that a D290-K412 salt bridge may be important for gating and sought to experimentally validate the interaction in the activated-open state using cysteine cross-bridging. As this is the only experimental backing in the paper, it is important to be able to judge its ability to report on the D290-K4512 salt bridge. A comparison experiment demonstrating other cross-links that do not favour the open state would have been helpful in this regard e.g. if cross-bridging at similar locations (but not predicted to change interaction during gating) had little effect on I/Imax, then the result may be bolstered. Are there existing mutagenesis experiments that may suggest the importance of these residues (as well as for other key interaction distances identified)?
Rotation of the V390 side chain from a position facing the pore lumen to a position facing I302 on S5 is coupled to an increase of the pore radius at V390, an increased hydration of the pore intracellular gate, and K+ ion movement. Perhaps 5 or 6 ions cross in that single simulation. As K channel ion permeation can depend critically on starting ion configs (as well as the model/force field), reproducibility of this finding is important but does not appear to have been tested. How can we be sure that periods of permeation or no permeation in individual simulations are reliable?
-
Reviewer #2 (Public Review):
The authors here study the electromechanical coupling in HCN1 channels using molecular dynamics simulations and electrophysiological data. They proposed that the interfaces between S4, S5, S6, and lipids contribute to the coupling mechanism. Their simulations showed state-dependent interactions at the S4-S5 and S5-S6 interfaces, as well as at the interface between the S4-S5 linker and the C-linker. These later interactions were also shown with Cd2+ crosslinking experiments. Furthermore, lipids were also shown to have state-dependent interactions in their simulations and were proposed to be crucial for hyperpolarization-dependent gating. Finally, they propose a domino-like mechanism of activation of HCN channels.
This is a well-written manuscript on a hot topic. The study would attract many readers.
-
Reviewer #3 (Public Review):
In this work, Elbahnsi and colleagues use enhanced sampling MD simulation, to recapitulate step by step, the electromechanical coupling between VSD and the pore in HCN1 channels. Building on the available cryoEM structures of HCN1 with the VSD in resting and active state, the authors characterize by MD a subset of interactions that seemingly stabilize the open channel. This subset is, in turn, used in enhanced-sampling simulations to guide channel opening.
The main findings are that S4 movement induces a rearrangement of the hydrophobic interaction at the level of S1- S4- and S5 interfaces. Occupancy of lipids seems therefore state-dependent and highlights their regulatory role in HCN gating.The approach is rather innovative, and it apparently allows the reconstruction of the whole mechanism of gating, …
Reviewer #3 (Public Review):
In this work, Elbahnsi and colleagues use enhanced sampling MD simulation, to recapitulate step by step, the electromechanical coupling between VSD and the pore in HCN1 channels. Building on the available cryoEM structures of HCN1 with the VSD in resting and active state, the authors characterize by MD a subset of interactions that seemingly stabilize the open channel. This subset is, in turn, used in enhanced-sampling simulations to guide channel opening.
The main findings are that S4 movement induces a rearrangement of the hydrophobic interaction at the level of S1- S4- and S5 interfaces. Occupancy of lipids seems therefore state-dependent and highlights their regulatory role in HCN gating.The approach is rather innovative, and it apparently allows the reconstruction of the whole mechanism of gating, pushing the predictive power of MD simulation well beyond its actual temporal limitations. At the same time, the initial choice of interactions is crucial for this approach, because the result cannot differ from the inputs. And reading the paper it does not emerge clearly how the correctness of the reconstructed gating pathway can be verified, if not by functional validation.
Here are my comments on the main interactions that were used to feed the final MD simulation:
1. W281-N300: this interaction, previously identified and studied in SpH channels (Ramentol et al, 2020; Wu et al, 2021), has been elegantly confirmed in this paper. Its inclusion in the initial subset seems appropriate.
In the other two cases, the choice of interactions requires further explanations and experimental validation.2. D290 and K412: the validation of this interaction shown in Figure 3 and suppl Figure 1 is missing a control, i.e., the effect of the addition of Cd++ on the wt channel. Please add.
3. Modelling the open state of HCN1 pore (page 18), is done on the structure of the distantly related hERG rather than on the available open pore structure of HCN4. This choice is justified as follows by the authors:
a) "Available structures in the CNBD channel family for which representative structures have been solved in closed and open states".
b) "The structural mechanism of pore gating (i.e. the ⍺ to 𝜋 helix occurring at the glycine657 hinge in hERG) observed in rEAG/hERG may be a conserved gating transition in the CNBD family of channels"
I encourage the authors to consider the following:a) The structure of hERG channel is not available in the closed/open configuration, indeed the comparison must be done with the closed configuration of the related channel rEAG. On the contrary, HCN4 is available in the closed/open configurations. Moreover, one of the open pore structures shows S4-S5-S6 in a very similar conformation to the lock open mutant (F186C/S264C) of HCN1 (Saponaro et al, 2021). With an available HCN4 open structure, forcing HCN1 to the open pore structure of hERG channel (which opens in depolarization and is not regulated by cAMP) seems not necessary.
To my knowledge, hERG is the only channel of the CNBD family for which the transition ⍺ to 𝜋 helix reported by the Authors, occurs in S6. It is not reported for other CNBD family members, in particular for the CNG channels mentioned by the Authors (Zheng et al., 2020; Xue et al., 2021, 2022). Task 4 (Zheng et al) does not show it. Its pore opens by a right-handed twist of S6 at glycine 399, a conserved glycine in all CNG. Human CNGA1 too, opens the pore by a rotational movement of S6 hinged at the equivalent glycine (glycine 385) (Xue et al, 2021). And the same occurs in the non-symmetrical channel CNGA1/B1 (Xue te al, 2022). So, it seems that CNG channels do not show the ⍺ to 𝜋 helix transition in the open pore. Moreover, hERG excluded, all other members of the CNBD family, CNG, EAG, and HCN4 included, do not bend at the hinge glycine 657 of hERG, but at another glycine (gly 648 in hERG numbering) located upstream. Further, their opening is due to a rotation of S6 associated with an outward movement, rather than to the lifting of the lower part of S6, as in hERG.
4- V390-I302: this interaction is predicted to stabilize the open pore configuration and was included in the subset. The contact between V390 on S6 and I302 on S5 is observed in the homology model discussed above when the S6 is twisted at the glycine hinge, rotating the preceding residue (V390) out of its pore-lining position and is.
Again, I can only disagree with this hypothesis because it has been experimentally demonstrated (Cheng et al, J Pharmacol Exp Ther. 2007 Sep;322(3):931-9) that the side chain of Valine390 is inside the cavity of the open pore of HCN1 channels as it controls the affinity for the pore blocker ZD7288.In conclusion, modelling the open state pore of HCN1 on hERG rather than on that of HCN4 seems not justified based on accumulated evidence in the published literature. Therefore, the choice of the authors to use it as the open pore model of HCN1 channels needs to be experimentally validated. One possibility is to mutate the glycine hinge, gly391 in HCN1, into an Alanine in order to remove the flexible hinge. If this mutation alters pore gating, it will support the choice of the Authors.
-
